The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases


Abstract
:1. Introduction
2. Glutamate Transporters in the Brain
2.1. Excitatory Amino Acid Transporters
2.2. Location of Excitatory Amino Acid Transporters
2.3. Structure and Function of Excitatory Amino Acid Transporters—The Case for Astrocytic EAAT1 and EAAT2
3. Astrocytic EAAT Regulation
3.1. Regulation of EAAT Expression by Soluble Factors
3.1.1. Cyclic AMP Signalling
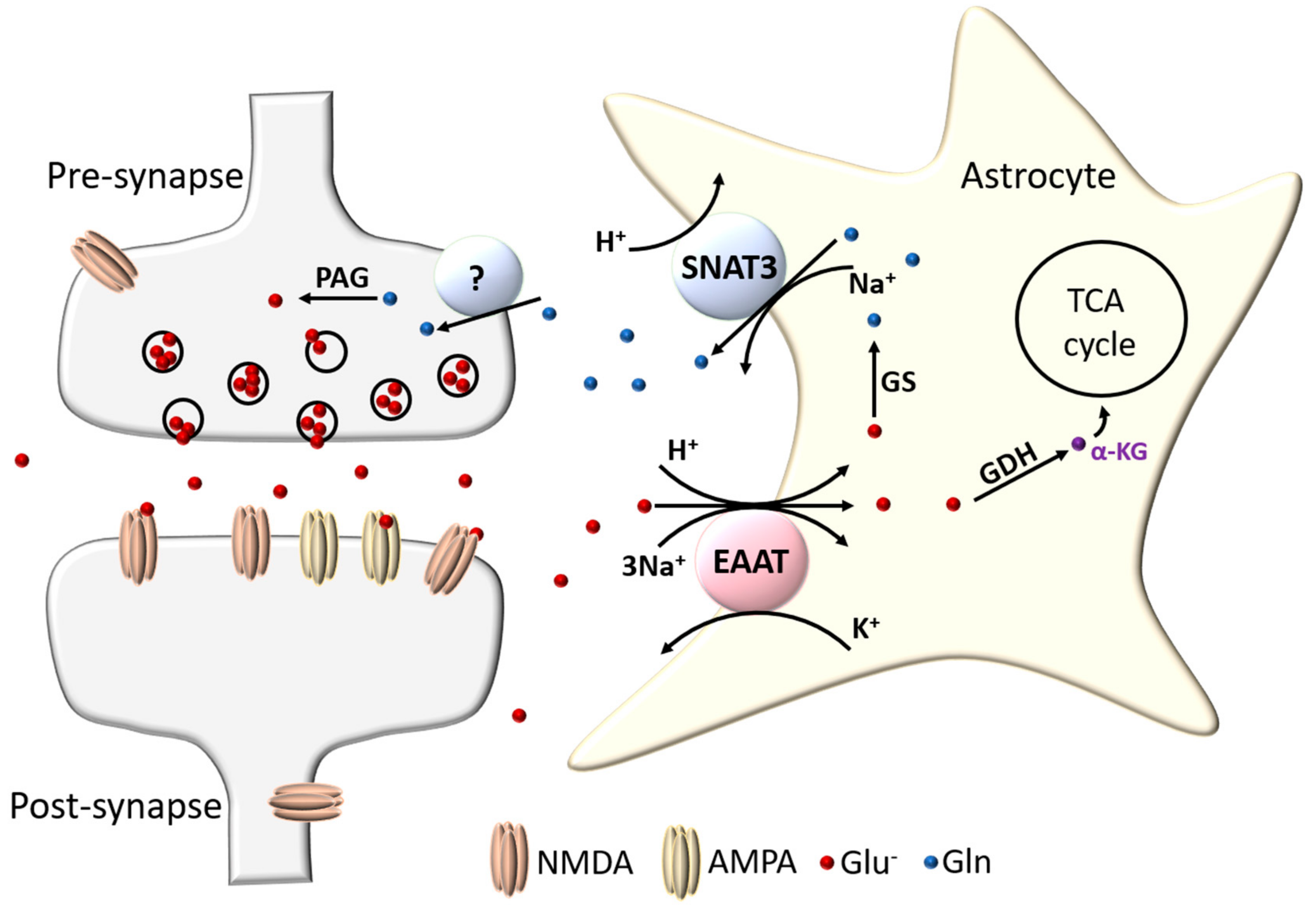
3.1.2. Glutamatergic Signalling and EAAT Trafficking
3.1.3. Other Secreted Signals
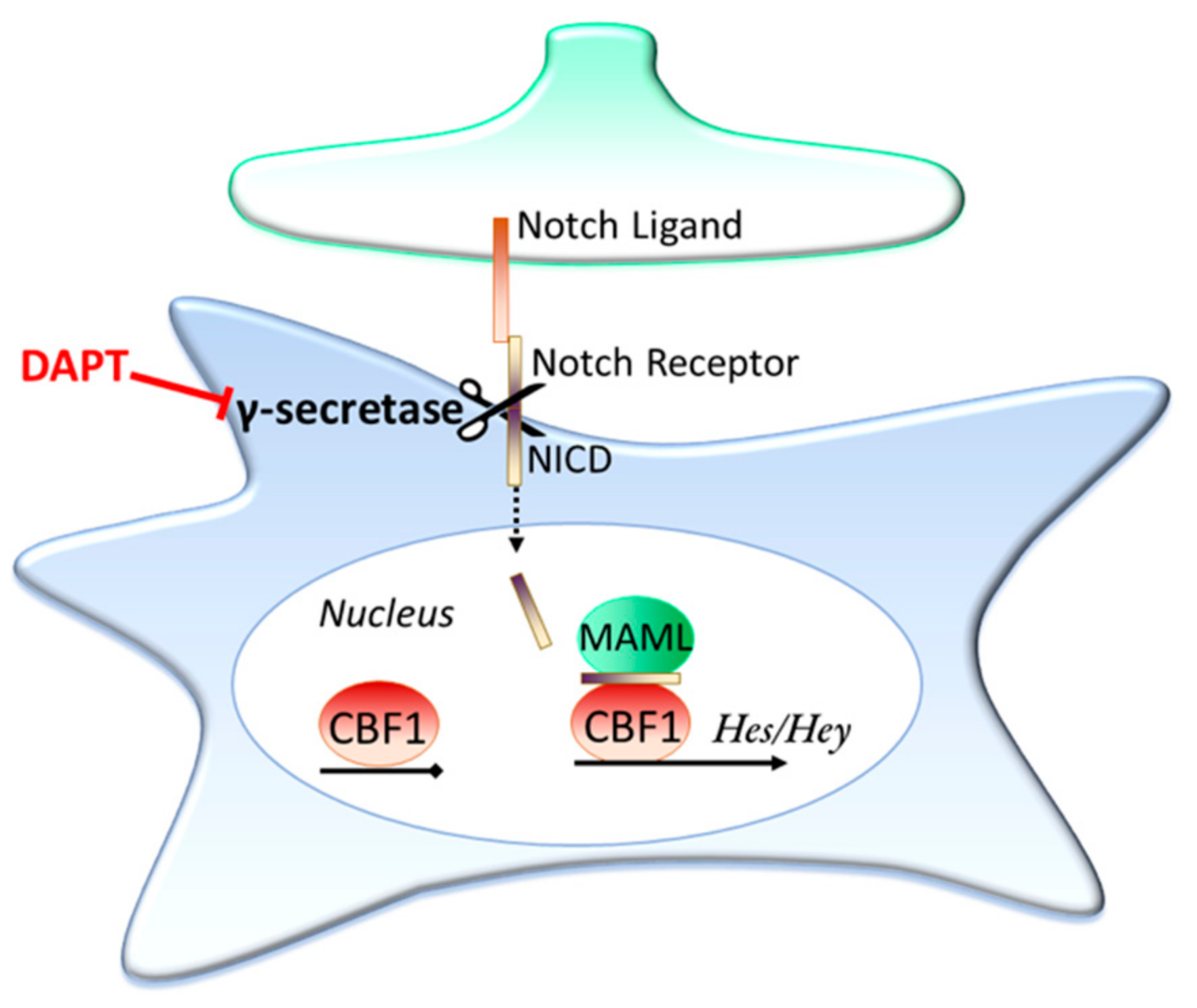
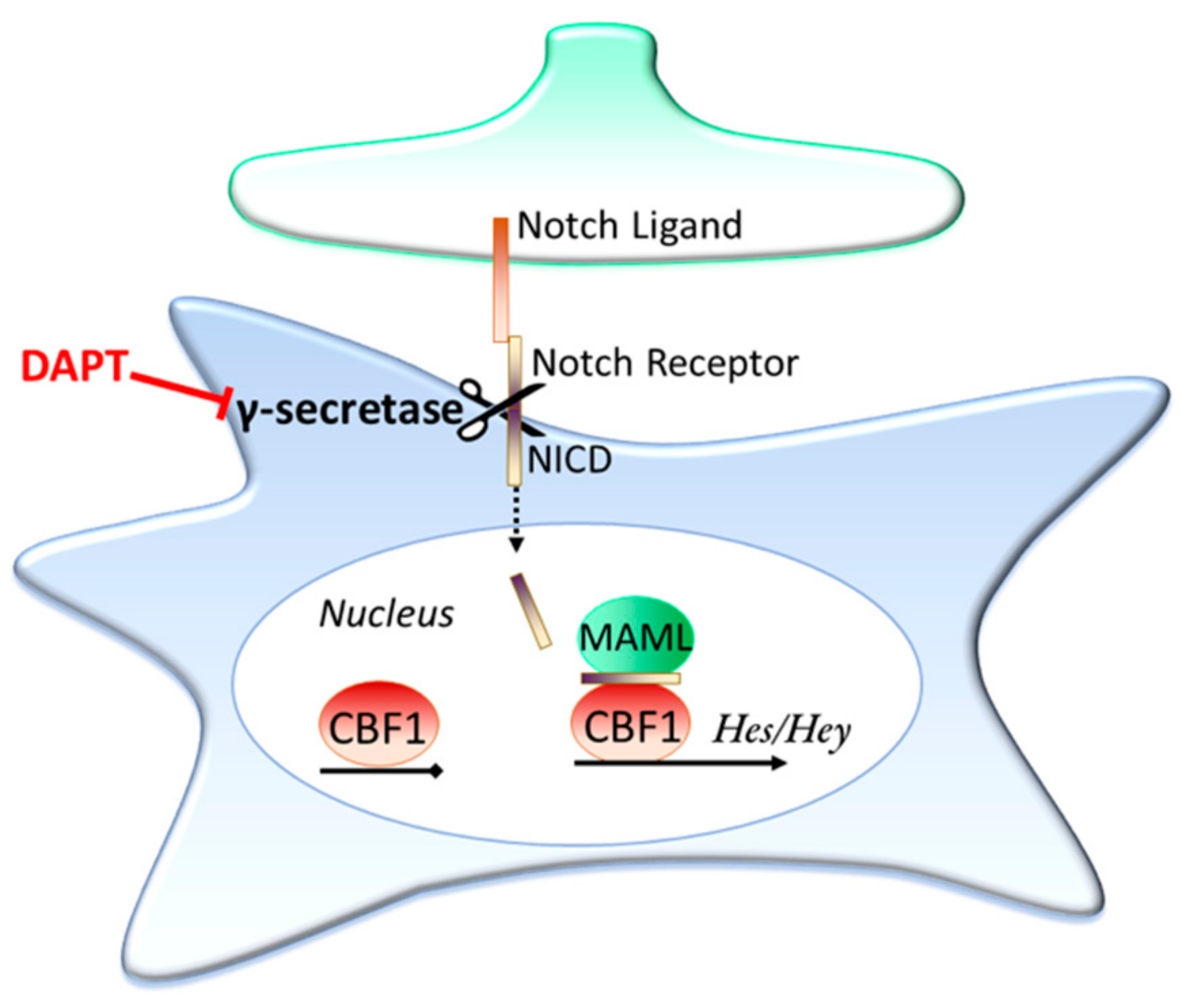
3.2. Contact Dependent Regulation: Notch Signalling
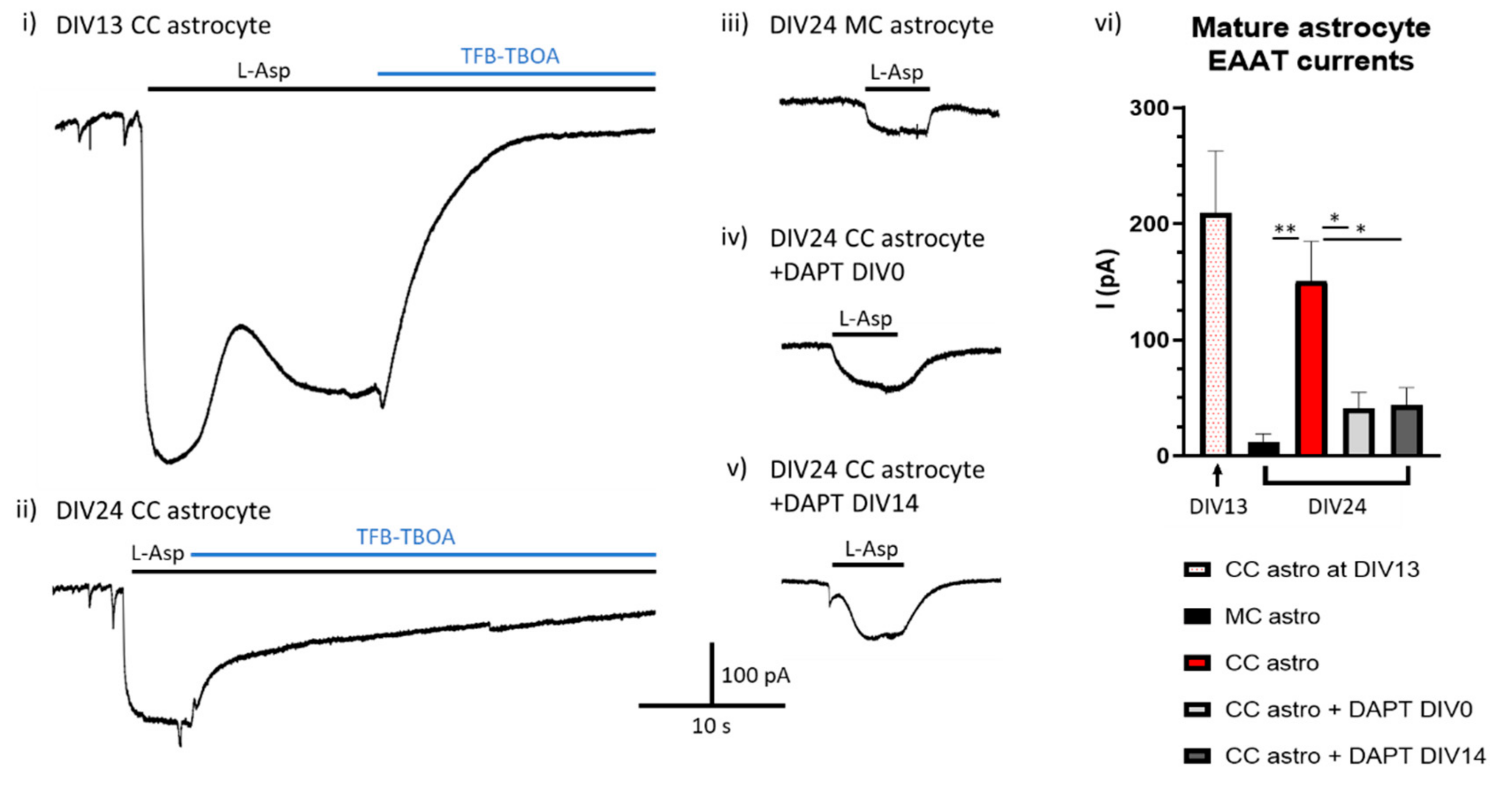
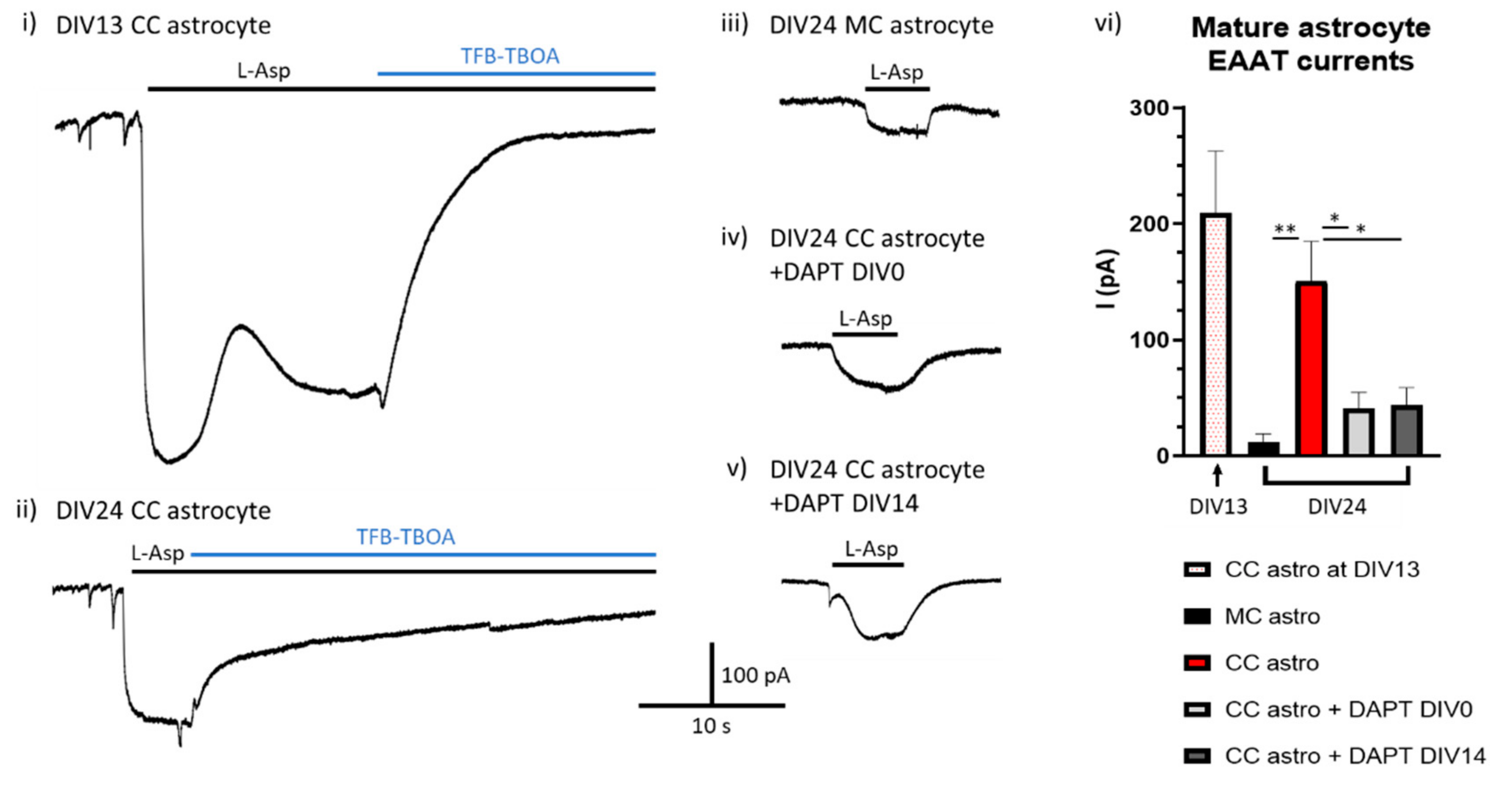
Is Ongoing Notch Signalling Required to Maintain Glutamate Uptake Capacity?
3.3. Epigenetic Regulation of EAATs
4. Astrocytic EAAT in Ageing and Neurodegenerative Disease
4.1. Epilepsy and EAATs
4.2. Neurodegenerative Diseases and EAATs
4.3. Ageing and EAATs
5. Notch Signalling in Ageing and Disease
6. Concluding Remarks
7. Materials and Methods
7.1. Tissue Cultures and Stimulations
7.2. Electrophysiological Recordings
7.3. Statistical Analysis
Funding
Conflicts of Interest
References
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, A.-S.; Buchthal, B.; Rode, F.; Bomholt, S.; Freitag, H.; Hardingham, G.E.; Rønn, L.; Bading, H. Hypoxic/ischemic conditions induce expression of the putative pro-death gene Clca1 via activation of extrasynaptic N-methyl-d-aspartate receptors. Neuroscience 2009, 158, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M. Adult rat brain astrocytes support survival of both NGF-dependent and NGF-insensitive neurones. Nat. Cell Biol. 1979, 282, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Banker, G.A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 1980, 209, 809–810. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.A.; Amin, S.; Leitner, M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J. Neurosci. 1992, 12, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.A.; Aizenman, E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Lipton, S.A. Regulation of Neuronal Oxidative and Nitrosative Stress by Endogenous Protective Pathways and Disease Processes. Antioxid. Redox Signal. 2011, 14, 1421–1424. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Rodrigues, T.B. Astrocytic—Neuronal—Astrocytic Pathway Selection for Formation and Degradation of Glutamate/GABA. Front. Endocrinol. 2014, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate–Glutamine Cycle. In The Glutamate/GABA-Glutamine Cycle: Amino Acid Neurotransmitter Homeostasis; Schousboe, A., Sonnewald, U., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–7. [Google Scholar]
- Billups, D.; Marx, M.-C.; Mela, I.; Billups, B. Inducible Presynaptic Glutamine Transport Supports Glutamatergic Transmission at the Calyx of Held Synapse. J. Neurosci. 2013, 33, 17429–17434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A Local Glutamate-Glutamine Cycle Sustains Synaptic Excitatory Transmitter Release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.C.; Marx, M.-C.; Hulme, S.R.; Bröer, S.; Billups, B. SNAT3-mediated glutamine transport in perisynaptic astrocytesin situis regulated by intracellular sodium. Glia 2017, 65, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Uwechue, N.M.; Marx, M.-C.; Chevy, Q.; Billups, B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J. Physiol. 2012, 590, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Freidman, N.; Chen, I.; Wu, Q.; Briot, C.; Holst, J.; Font, J.; Vandenberg, R.J.; Ryan, R. Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer. Neurochem. Res. 2020, 45, 1268–1286. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflügers Arch. Eur. J. Physiol. 2013, 466, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Ho, M.S.; Zorec, R.; Parpura, V. Physiology of Astroglia. In Neuroglia in Neurodegenerative Diseases; Springer: Singapore, 2019; pp. 45–91. [Google Scholar]
- Hädel, S.; Wirth, C.; Rapp, M.; Gallinat, J.; Schubert, F. Effects of age and sex on the concentrations of glutamate and glutamine in the human brain. J. Magn. Reson. Imaging 2013, 38, 1480–1487. [Google Scholar] [CrossRef]
- Schubert, F.; Gallinat, J.; Seifert, F.; Rinneberg, H. Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 Tesla. NeuroImage 2004, 21, 1762–1771. [Google Scholar] [CrossRef]
- Lehmann, A.; Isacsson, H.; Hamberger, A. Effects of In Vivo Administration of Kainic Acid on the Extracellular Amino Acid Pool in the Rabbit Hippocampus. J. Neurochem. 1983, 40, 1314–1320. [Google Scholar] [CrossRef]
- Hamberger, A.; Nyström, B. Extra- and intracellular amino acids in the hippocampus during development of hepatic encephalopathy. Neurochem. Res. 1984, 9, 1181–1192. [Google Scholar] [CrossRef]
- Erecinska, M. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 1990, 35, 245–296. [Google Scholar] [CrossRef]
- Helms, H.C.C.; Nielsen, C.U.; Waagepetersen, H.S.; Brodin, B. Glutamate Transporters in the Blood-Brain Barrier. In Advances in Neurobiology; Ortega, A., Schousboe, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 297–314. [Google Scholar]
- Fotiadis, D.; Kanai, Y.; Palacín, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Asp. Med. 2013, 34, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc- cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2011, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottestad-Hansen, S.; Hu, Q.X.; Follin-Arbelet, V.V.; Bentea, E.; Sato, H.; Massie, A.; Zhou, Y.; Danbolt, N.C. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia 2018, 66, 951–970. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.-A.; Halatsch, M.-E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-Kinases Upregulate System xc− via Eukaryotic Initiation Factor 2α and Activating Transcription Factor 4—A Pathway Active in Glioblastomas and Epilepsy. Antioxid. Redox Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [Green Version]
- Logan, W.J.; Snyder, S.H. Unique High Affinity Uptake Systems for Glycine, Glutamic and Aspartic Acids in Central Nervous Tissue of the Rat. Nature 1971, 234, 297–299. [Google Scholar] [CrossRef]
- Balcar, V.J.; Johnston, G.R. The Structural Specificity of the High Affinity Uptake of l-glutamate and l-Aspartate by Rat Brain Slices. J. Neurochem. 1972, 19, 2657–2666. [Google Scholar] [CrossRef]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain l-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcar, V.J.; Borg, J.; Mandel, P. High affinity uptake of l-glutamate and l-Aspartate by glial cells. J. Neurochem. 1977, 28, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Cloning and expression of a glutamate transporter from mouse brain. Neurosci. Lett. 1993, 159, 183–186. [Google Scholar] [CrossRef]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaught, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef] [Green Version]
- Arriza, J.L.; Fairman, W.A.; Wadiche, J.I.; Murdoch, G.H.; Kavanaugh, M.P.; Amara, S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 1994, 14, 5559–5569. [Google Scholar] [CrossRef] [Green Version]
- Wadiche, J.I.; Kavanaugh, M.P. Macroscopic and Microscopic Properties of a Cloned Glutamate Transporter/Chloride Channel. J. Neurosci. 1998, 18, 7650–7661. [Google Scholar] [CrossRef] [Green Version]
- Wadiche, J.I.; Amara, S.G.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Jensen, A.A.; Bräuner-Osborne, H. Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem. Pharmacol. 2004, 67, 2115–2127. [Google Scholar] [CrossRef]
- Grewer, C.; Watzke, N.; Wiessner, M.; Rauen, T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc. Natl. Acad. Sci. USA 2000, 97, 9706–9711. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, A.; Braams, S.; Rauen, T.; Grewer, C. The Discovery of Slowness: Low-Capacity Transport and Slow Anion Channel Gating by the Glutamate Transporter EAAT5. Biophys. J. 2011, 100, 2623–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mim, C.; Balani, P.; Rauen, T.; Grewer, C. The Glutamate Transporter Subtypes EAAT4 and EAATs 1-3 Transport Glutamate with Dramatically Different Kinetics and Voltage Dependence but Share a Common Uptake Mechanism. J. Gen. Physiol. 2005, 126, 571–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellocchio, E.E.; Reimer, R.J.; Fremeau, R.T.; Edwards, R.H. Uptake of Glutamate into Synaptic Vesicles by an Inorganic Phosphate Transporter. Science 2000, 289, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.; Bellenchi, G.C.; Gras, C.; Bernard, V.; Ravassard, P.; Bedet, C.; Gasnier, B.; Giros, B.; El Mestikawy, S. The Existence of a Second Vesicular Glutamate Transporter Specifies Subpopulations of Glutamatergic Neurons. J. Neurosci. 2001, 21, RC181. [Google Scholar] [CrossRef]
- Fremeau, R.T.; Burman, J.; Qureshi, T.; Tran, C.H.; Proctor, J.; Johnson, J.; Zhang, H.; Sulzer, D.; Copenhagen, D.R.; Storm-Mathisen, J.; et al. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc. Natl. Acad. Sci. USA 2002, 99, 14488–14493. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc−: An obligate exchanger of l-glutamate and L-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the System x−C cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219–226. [Google Scholar] [CrossRef]
- Al Awabdh, S.; Gupta-Agarwal, S.; Sheehan, D.F.; Muir, J.; Norkett, R.; Twelvetrees, A.E.; Griffin, L.D.; Kittler, J.T. Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 2016, 64, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Šerý, O.; Sultana, N.; Kashem, M.A.; Pow, D.V.; Balcar, V.J. GLAST But Not Least—Distribution, Function, Genetics and Epigenetics of l-glutamate Transport in Brain—Focus on GLAST/EAAT1. Neurochem. Res. 2015, 40, 2461–2472. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. GABA and Glutamate Transporters in Brain. Front. Endocrinol. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, F.A.; Lehre, K.P.; Campagne, M.V.L.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Lehre, K.P.; Danbolt, N.C. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehre, K.; Levy, L.; Ottersen, O.; Storm-Mathisen, J.; Danbolt, N. Differential expression of two glial glutamate transporters in the rat brain: Quantitative and immunocytochemical observations. J. Neurosci. 1995, 15, 1835–1853. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Asan, E.; Püschel, B.; Kugler, P. Cellular and Regional Distribution of the Glutamate Transporter GLAST in the CNS of Rats: Nonradioactive In Situ Hybridization and Comparative Immunocytochemistry. J. Neurosci. 1997, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Derouiche, A.; Rauen, T. Coincidence of l-glutamate/l-Aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: Evidence for coupling of GLAST and GS in transmitter clearance. J. Neurosci. Res. 1995, 42, 131–143. [Google Scholar] [CrossRef]
- Lehre, K.P.; Davanger, S.; Danbolt, N.C. Localization of the glutamate transporter protein GLAST in rat retina. Brain Res. 1997, 744, 129–137. [Google Scholar] [CrossRef]
- Pow, D.V.; Barnett, N.L. Changing patterns of spatial buffering of glutamate in developing rat retinae are mediated by the Müller cell glutamate transporter GLAST. Cell Tissue Res. 1999, 297, 57–66. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Kitchen, R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Sestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Ziemens, D.; Untiet, V.; Fahlke, C. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 2018, 136, 3–16. [Google Scholar] [CrossRef]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The Density of EAAC1 (EAAT3) Glutamate Transporters Expressed by Neurons in the Mammalian CNS. J. Neurosci. 2012, 32, 6000–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashidharan, P.; Huntley, G.W.; Murray, J.M.; Buku, A.; Moran, T.; Walsh, M.J.; Morrison, J.H.; Plaitakis, A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res. 1997, 773, 139–148. [Google Scholar] [CrossRef]
- Dehnes, Y.; Chaudhry, F.A.; Ullensvang, K.; Lehre, K.P.; Storm-Mathisen, J.; Danbolt, N.C. The Glutamate Transporter EAAT4 in Rat Cerebellar Purkinje Cells: A Glutamate-Gated Chloride Channel Concentrated near the Synapse in Parts of the Dendritic Membrane Facing Astroglia. J. Neurosci. 1998, 18, 3606–3619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, A.; Cnops, L.; Smolders, I.; McCullumsmith, R.; Kooijman, R.; Kwak, S.; Arckens, L.; Michotte, Y. High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J. Comp. Neurol. 2008, 511, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Pow, D.V.; Barnett, N.L. Developmental expression of excitatory amino acid transporter 5: A photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci. Lett. 2000, 280, 21–24. [Google Scholar] [CrossRef]
- Eliasof, S.; Arriza, J.L.; Leighton, B.H.; Kavanaugh, M.P.; Amara, S.G. Excitatory Amino Acid Transporters of the Salamander Retina: Identification, Localization, and Function. J. Neurosci. 1998, 18, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Wersinger, E.; Schwab, Y.; Sahel, J.-A.; Rendon, A.; Pow, D.V.; Picaud, S.; Roux, M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006, 577, 221–234. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Schousboe, A. Cellular Distribution and Kinetic Properties of High-Affinity Glutamate Transporters. Brain Res. Bull. 1998, 45, 233–238. [Google Scholar] [CrossRef]
- Levy, L.M.; Warr, O.; Attwell, D. Stoichiometry of the Glial Glutamate Transporter GLT-1 Expressed Inducibly in a Chinese Hamster Ovary Cell Line Selected for Low Endogenous Na+-Dependent Glutamate Uptake. J. Neurosci. 1998, 18, 9620–9628. [Google Scholar] [CrossRef] [Green Version]
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nature 1996, 383, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Nussberger, S.; Romero, M.F.; Boron, W.F.; Hebert, S.C.; Hediger, M.A. Electrogenic Properties of the Epithelial and Neuronal High Affinity Glutamate Transporter. J. Biol. Chem. 1995, 270, 16561–16568. [Google Scholar] [CrossRef] [Green Version]
- Yernool, D.; Boudker, O.; Jin, Y.; Gouaux, E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004, 431, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.; Guskov, A.; Rempel, S.; Hänelt, I.; Slotboom, D.J.; Hänelt, I. Crystal structure of a substrate-free aspartate transporter. Nat. Struct. Mol. Biol. 2013, 20, 1224–1226. [Google Scholar] [CrossRef] [Green Version]
- Arkhipova, V.; Trinco, G.; Ettema, T.W.; Jensen, S.; Slotboom, D.J.; Guskov, A. Binding and transport of D-aspartate by the glutamate transporter homolog GltTk. eLife 2019, 8. [Google Scholar] [CrossRef]
- Kortzak, D.; Alleva, C.; Weyand, I.; Ewers, D.; Zimmermann, M.I.; Franzen, A.; Machtens, J.-P.; Fahlke, C. Allosteric gate modulation confers K + coupling in glutamate transporters. EMBO J. 2019, 38, e101468. [Google Scholar] [CrossRef] [PubMed]
- Brew, H.; Attwell, D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature 1987, 327, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Arriza, J.L.; Amara, S.G.; Kavanaugh, M.P. Constitutive Ion Fluxes and Substrate Binding Domains of Human Glutamate Transporters. J. Biol. Chem. 1995, 270, 17668–17671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, D.J.A.; Mathie, A.; Symonds, C.J.; Cull-Candy, S.G. Activation of glutamate receptors and glutamate uptake in identified macroglial cells in rat cerebellar cultures. J. Physiol. 1991, 432, 235–258. [Google Scholar] [CrossRef] [Green Version]
- Machtens, J.-P.; Kortzak, D.; Lansche, C.; Leinenweber, A.; Kilian, P.; Begemann, B.; Zachariae, U.; Ewers, D.; De Groot, B.L.; Briones, R.; et al. Mechanisms of Anion Conduction by Coupled Glutamate Transporters. Cell 2015, 160, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Fahlke, C.; Kortzak, D.; Machtens, J.-P. Molecular physiology of EAAT anion channels. Pflügers Arch. Eur. J. Physiol. 2016, 468, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Cordeiro, S.; Machtens, J.-P.; Braams, S.; Rauen, T.; Fahlke, C. Functional Properties of the Retinal Glutamate Transporters GLT-1c and EAAT. J. Biol. Chem. 2013, 289, 1815–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, D.Y.; Chung, I.Y.; Wu, S.M. Possible roles of glutamate transporter EAAT5 in mouse cone depolarizing bipolar cell light responses. Vis. Res. 2014, 103, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLennan, H. The autoradiographic localization of L-[3H]glutamate in rat brain tissue. Brain Res. 1976, 115, 139–144. [Google Scholar] [CrossRef]
- Wilkin, G.P.; Garthwaite, J.; Bala’zs, R. Putative acidic amino acid transmitters in the cerebellum. II. Electron microscopic localization of transport sites. Brain Res. 1982, 244, 69–80. [Google Scholar] [CrossRef]
- Bergles, D.E.; Jahr, C.E. Glial Contribution to Glutamate Uptake at Schaffer Collateral–Commissural Synapses in the Hippocampus. J. Neurosci. 1998, 18, 7709–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergles, D.E.; Jahr, C.E. Synaptic Activation of Glutamate Transporters in Hippocampal Astrocytes. Neuron 1997, 19, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and Exacerbation of Brain Injury in Mice Lacking the Glutamate Transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.-I.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef]
- Matsugami, T.R.; Tanemura, K.; Mieda, M.; Nakatomi, R.; Yamada, K.; Kondo, T.; Ogawa, M.; Obata, K.; Watanabe, M.; Hashikawa, T.; et al. Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc. Natl. Acad. Sci. USA 2006, 103, 12161–12166. [Google Scholar] [CrossRef] [Green Version]
- Peghini, P.; Janzen, J.; Stoffel, W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997, 16, 3822–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petr, G.T.; Sun, Y.; Frederick, N.M.; Zhou, Y.; Dhamne, S.C.; Hameed, M.Q.; Miranda, C.; Bedoya, E.A.; Fischer, K.D.; Armsen, W.; et al. Conditional Deletion of the Glutamate Transporter GLT-1 Reveals That Astrocytic GLT-1 Protects against Fatal Epilepsy While Neuronal GLT-1 Contributes Significantly to Glutamate Uptake into Synaptosomes. J. Neurosci. 2015, 35, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Drejer, J.; Meier, E.; Schousboe, A. Novel neuron-related regulatory mechanisms for astrocytic glutamate and GABA high affinity uptake. Neurosci. Lett. 1983, 37, 301–306. [Google Scholar] [CrossRef]
- Levy, L.M.; Lehre, K.P.; Walaas, S.I.; Storm-Mathisen, J.; Danbolt, N.C. Down-regulation of Glial Glutamate Transporters after Glutamatergic Denervation in the Rat Brain. Eur. J. Neurosci. 1995, 7, 2036–2041. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Civenni, G.; Racagni, G.; Danbolt, N.C.; Schousboe, I.; Schousboe, A. Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. NeuroReport 1996, 8, 261–265. [Google Scholar] [CrossRef]
- Swanson, R.A.; Liu, J.; Miller, J.W.; Rothstein, J.D.; Farrell, K.; Stein, B.A.; Longuemare, M.C. Neuronal Regulation of Glutamate Transporter Subtype Expression in Astrocytes. J. Neurosci. 1997, 17, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Gegelashvili, G.; Danbolt, N.C.; Schousboe, A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J. Neurochem. 1997, 69, 2612–2615. [Google Scholar] [CrossRef] [Green Version]
- Hasel, P.; Dando, O.R.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef]
- Schlag, B.D.; Vondrasek, J.R.; Munir, M.; Kalandadze, A.; Zelenaia, O.A.; Rothstein, J.D.; Robinson, M.B. Regulation of the Glial Na+-Dependent Glutamate Transporters by Cyclic AMP Analogs and Neurons. Mol. Pharmacol. 1998, 53, 355–369. [Google Scholar] [CrossRef]
- Zelenaia, O.; Schlag, B.D.; Gochenauer, G.E.; Ganel, R.; Song, W.; Beesley, J.S.; Grinspan, J.B.; Rothstein, J.D.; Robinson, M.B. Epidermal Growth Factor Receptor Agonists Increase Expression of Glutamate Transporter GLT-1 in Astrocytes through Pathways Dependent on Phosphatidylinositol 3-Kinase and Transcription Factor NF-κB. Mol. Pharmacol. 2000, 57, 667–678. [Google Scholar] [CrossRef]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear Factor- B Contributes to Neuron-Dependent Induction of Glutamate Transporter-1 Expression in Astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef] [PubMed]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of EAAT2 by NF-κB: A role for N-myc in TNFα-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Lane, M.; Krizman, E.; Sattler, R.; Rothstein, J.D.; Robinson, M.B. The transcription factor Pax6 contributes to the induction of GLT-1 expression in astrocytes through an interaction with a distal enhancer element. J. Neurochem. 2016, 136, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.M.; Muraro, N.I.; Peco, E.; Labbe, A.; Thomas, G.B.; Baines, R.A.; Van Meyel, D.J. Drosophila Glial Glutamate Transporter Eaat1 Is Regulated by Fringe-Mediated Notch Signaling and Is Essential for Larval Locomotion. J. Neurosci. 2010, 30, 14446–14457. [Google Scholar] [CrossRef]
- Lee, M.L.; Martinez-Lozada, Z.; Krizman, E.N.; Robinson, M.B. Brain endothelial cells induce astrocytic expression of the glutamate transporter GLT-1 by a Notch-dependent mechanism. J. Neurochem. 2017, 143, 489–506. [Google Scholar] [CrossRef]
- Martinez-Lozada, Z.; Robinson, M.B. Reciprocal communication between astrocytes and endothelial cells is required for astrocytic glutamate transporter 1 (GLT-1) expression. Neurochem. Int. 2020, 139, 104787. [Google Scholar] [CrossRef]
- Hertz, L.; Bock, E.; Schousboe, A. GFA Content, Glutamate Uptake and Activity of Glutamate Metabolizing Enzymes in Differentiating Mouse Astrocytes in Primary Cultures. Dev. Neurosci. 1978, 1, 226–238. [Google Scholar] [CrossRef]
- Goldman, J.E.; Chiu, F.-C. Dibutyryl cyclic AMP causes intermediate filament accumulation and actin reorganization in astrocytes. Brain Res. 1984, 306, 85–95. [Google Scholar] [CrossRef]
- Pisano, P.; Samuel, D.; Nieoullon, A.; Goff, L.K.-L. Activation of the Adenylate Cyclase-dependent Protein Kinase Pathway Increases High Affinity Glutamate Uptake into Rat Striatal Synaptosomes. Neuropharmacology 1996, 35, 541–547. [Google Scholar] [CrossRef]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and GLAST Glutamate Transporters Are Expressed on Morphologically Distinct Astrocytes and Regulated by Neuronal Activity in Primary Hippocampal Cocultures. J. Neurochem. 2002, 75, 1076–1084. [Google Scholar] [CrossRef]
- Simantov, R.; Crispino, M.; Hoe, W.; Broutman, G.; Tocco, G.; Rothstein, J.D.; Baudry, M. Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity. Mol. Brain Res. 1999, 65, 112–123. [Google Scholar] [CrossRef]
- Duan, S.; Anderson, C.M.; Stein, B.A.; Swanson, R.A. Glutamate Induces Rapid Upregulation of Astrocyte Glutamate Transport and Cell-Surface Expression of GLAST. J. Neurosci. 1999, 19, 10193–10200. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, I.; Díez-Guerra, F.J.; Giménez, C.; Zafra, F. Activity dependent internalization of the glutamate transporter GLT-1 mediated by β-arrestin 1 and ubiquitination. Neuropharmacology 2016, 107, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, I.; Bartolomé-Martín, D.; Piniella, D.; Giménez, C.; Zafra, F. Activity dependent internalization of the glutamate transporter GLT-1 requires calcium entry through the NCX sodium/calcium exchanger. Neurochem. Int. 2019, 123, 125–132. [Google Scholar] [CrossRef]
- Gamboa, C.; Ortega, A. Insulin-like growth factor-1 increases activity and surface levels of the GLAST subtype of glutamate transporter. Neurochem. Int. 2002, 40, 397–403. [Google Scholar] [CrossRef]
- Boehmer, C.; Henke, G.; Schniepp, R.; Palmada, M.; Rothstein, J.D.; Bröer, S.; Lang, F. Regulation of the glutamate transporter EAAT1 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid-inducible kinase isoforms SGK1/3 and protein kinase B. J. Neurochem. 2003, 86, 1181–1188. [Google Scholar] [CrossRef]
- Götz, M.; Stoykova, A.; Gruss, P. Pax6 Controls Radial Glia Differentiation in the Cerebral Cortex. Neuron 1998, 21, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Osumi, N.; Aarts, E.; Roelofs, A.; Van Turennout, M. The Neurogenesis-Controlling Factor, Pax6, Inhibits Proliferation and Promotes Maturation in Murine Astrocytes. J. Neurosci. 2008, 28, 4604–4612. [Google Scholar] [CrossRef]
- Gegelashvili, G. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem. Int. 2000, 37, 163–170. [Google Scholar] [CrossRef]
- Namihira, M.; Kohyama, J.; Semi, K.; Sanosaka, T.; Deneen, B.; Taga, T.; Nakashima, K. Committed Neuronal Precursors Confer Astrocytic Potential on Residual Neural Precursor Cells. Dev. Cell 2009, 16, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Gessler, M. Delta Notch and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007, 35, 4583–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo-Rojo, C.; Manning-Cela, R.; Aguirre, A.; Ortega, A.; Lopez-Bayghen, E. Involvement of the Notch Pathway in Terminal Astrocytic Differentiation: Role of PKA. ASN Neuro 2013, 5, AN20130023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, X.; Kusumo, H.; Sakharkar, A.J.; Pandey, S.C.; Guizzetti, M. Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 2014, 128, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Karki, P.; Webb, A.; Smith, K.; Johnson, J.; Lee, K.; Son, D.-S.; Aschner, M.; Lee, E. Yin Yang 1 Is a Repressor of Glutamate Transporter EAAT2, and It Mediates Manganese-Induced Decrease of EAAT2 Expression in Astrocytes. Mol. Cell. Biol. 2014, 34, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschocke, J.; Allritz, C.; Engele, J.; Rein, T. DNA methylation dependent silencing of the human glutamate transporterEAAT2 gene in glial cells. Glia 2007, 55, 663–674. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.F.; Liu, T.J.; Fuller, G.N.; Yung, W.A. The Excitatory Amino Acid Transporter-2 Induces Apoptosis and Decreases Glioma GrowthIn vitroandIn vivo. Cancer Res. 2005, 65, 1934–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.A.; Datta, P.K. Epigenetic Regulation of Excitatory Amino Acid Transporter 2 in Neurological Disorders. Front. Pharmacol. 2019, 10, 1510. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, N.R.; Hardingham, G.E.; Fox, K.D.; Jack, J.J.B. Presynaptic Efficacy Directs Normalization of Synaptic Strength in Layer 2/3 Rat Neocortex After Paired Activity. J. Neurophysiol. 2007, 97, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Turrigiano, G. Homeostatic Synaptic Plasticity: Local and Global Mechanisms for Stabilizing Neuronal Function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [Green Version]
- Larkman, A.U.; Jack, J.J.B. Synaptic plasticity: Hippocampal LTP. Curr. Opin. Neurobiol. 1995, 5, 324–334. [Google Scholar] [CrossRef]
- Hardingham, N.; Read, J.C.A.; Trevelyan, A.J.; Nelson, J.C.; Jack, J.J.B.; Bannister, N.J. Quantal analysis reveals a functional correlation between presynaptic and postsynaptic efficacy in excitatory connections from rat neocortex. J. Neurosci. 2010, 30, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Valtcheva, S.; Venance, L. Astrocytes gate Hebbian synaptic plasticity in the striatum. Nat. Commun. 2016, 7, 13845. [Google Scholar] [CrossRef] [Green Version]
- Rajatileka, S.; Odd, D.; Robinson, M.T.; Spittle, A.C.; Dwomoh, L.; Williams, M.; Harding, D.; Wagstaff, M.; Owen, M.; Crosby, C.; et al. Variants of the EAAT2 Glutamate Transporter Gene Promoter Are Associated with Cerebral Palsy in Preterm Infants. Mol. Neurobiol. 2017, 55, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Poletti, S.; Radaelli, D.; Bosia, M.; Buonocore, M.; Pirovano, A.; Lorenzi, C.; Cavallaro, R.; Smeraldi, E.; Benedetti, F. Effect of glutamate transporter EAAT2 gene variants and gray matter deficits on working memory in schizophrenia. Eur. Psychiatry 2014, 29, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Gibbons, A.; Udawela, M.; Dean, B. Excitatory amino acid transporter (EAAT)1 and EAAT2 mRNA levels are altered in the prefrontal cortex of subjects with schizophrenia. J. Psychiatr. Res. 2020, 123, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Gupta, D.S.; Harotunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr. Res. 2008, 104, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Amen-Hellebrekers, C.J.; Jansen, S.; Pfundt, R.; Schuurs-Hoeijmakers, J.H.; Koolen, D.; Marcelis, C.L.; De Leeuw, N.; De Vries, B.B. Duplications of SLC1A3: Associated with ADHD and autism. Eur. J. Med Genet. 2016, 59, 373–376. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Q.; Chen, X.; Gu, X.; Wang, M.; Wu, J. A functional variant in SLC1A3 influences ADHD risk by disrupting a hsa-miR-3171 binding site: A two-stage association study. Genes Brain Behav. 2019, 18, e12574. [Google Scholar] [CrossRef]
- Higashimori, H.; Schin, C.S.; Chiang, M.S.R.; Morel, L.; Shoneye, T.A.; Nelson, D.L.; Yang, Y. Selective Deletion of Astroglial FMRP Dysregulates Glutamate Transporter GLT1 and Contributes to Fragile X Syndrome Phenotypes In Vivo. J. Neurosci. 2016, 36, 7079–7094. [Google Scholar] [CrossRef]
- Blacker, C.J.; Millischer, V.; Webb, L.M.; Ho, A.M.C.; Schalling, M.; Frye, M.A.; Veldic, M. EAAT2 as a Research Target in Bipolar Disorder and Unipolar Depression: A Systematic Review. Mol. Neuropsychiatry 2019, 5, 1–16. [Google Scholar] [CrossRef]
- Bernard, R.; Kerman, I.A.; Thompson, R.C.; Jones, E.G.; Bunney, W.E.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Akil, H.; Watson, S.J. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 2011, 16, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Hardingham, G.E. Compartmentalized NMDA receptor signalling to survival and death. J. Physiol. 2007, 584, 381–387. [Google Scholar] [CrossRef]
- Bell, K.F.S.; Hardingham, G.E. The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 2011, 21, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, A.; Aalbers, M.; Frigerio, F.; Fiordaliso, F.; Salio, M.; Gobbi, M.; Cagnotto, A.; Gardoni, F.; Battaglia, G.S.; Hoogland, G.; et al. Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol. Dis. 2011, 43, 507–515. [Google Scholar] [CrossRef]
- Stark, D.T.; Bazan, N.G. Synaptic and Extrasynaptic NMDA Receptors Differentially Modulate Neuronal Cyclooxygenase-2 Function, Lipid Peroxidation, and Neuroprotection. J. Neurosci. 2011, 31, 13710–13721. [Google Scholar] [CrossRef]
- Milnerwood, A.; Gladding, C.; Pouladi, M.; Kaufman, A.; Hines, R.; Boyd, J.; Ko, R.; Vasuta, O.; Graham, R.; Hayden, M.; et al. Early increase in extrasynaptic NMDA receptor signalling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.-I.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.-I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [Green Version]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of Extrasynaptic, But Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid-β Production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef]
- Molokanova, E.; Akhtar, M.W.; Sanz-Blasco, S.; Tu, S.; Piña-Crespo, J.C.; McKercher, S.R.; Lipton, S.A. Differential Effects of Synaptic and Extrasynaptic NMDA Receptors on Aβ-Induced Nitric Oxide Production in Cerebrocortical Neurons. J. Neurosci. 2014, 34, 5023–5028. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D. Soluble A Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.-S.V.; Zhang, D.; Lipton, S.A. Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.; Bengtson, C.P.; Buchthal, B.; Bading, H. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep. 2015, 12, 1353–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddifoot, C.; Martel, M.-A.; Soriano, F.X.; Camacho, A.; Vidal-Puig, A.; Wyllie, D.J.A.; Hardingham, G.E. PGC-1 Negatively Regulates Extrasynaptic NMDAR Activity and Excitotoxicity. J. Neurosci. 2012, 32, 6995–7000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpova, A.; Emikhaylova, M.; Bera, S.; Bär, J.; Reddy, P.P.; Behnisch, T.; Rankovic, V.; Spilker, C.; Bethge, P.; Sahin, J.; et al. Encoding and Transducing the Synaptic or Extrasynaptic Origin of NMDA Receptor Signals to the Nucleus. Cell 2013, 152, 1119–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370, eaay3302. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.E.; Wang, Y.; Pedigo, N.W.; Hensley, K.; Butterfield, D.A.; Carney, J.M. Amyloid ? Peptide (25–35) Inhibits Na+?Dependent Glutamate Uptake in Rat Hippocampal Astrocyte Cultures. J. Neurochem. 1996, 67, 277–286. [Google Scholar] [CrossRef]
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The inhibitory effects of β-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997, 754, 65–71. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Oliveira, C.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Scott, H.L.; Pow, D.V.; Tannenberg, A.E.; Dodd, P.R. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer’s disease. J. Neurosci. 2002, 22, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grunblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Hansen, L.; Alford, M.; DeTeresa, R.; Mallory, M. Deficient glutamate tranport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 628–638. [Google Scholar] [CrossRef]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid- 1-42 Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Ba, M.V.K.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic Lateral Sclerosis-linked Glutamate Transporter Mutant Has Impaired Glutamate Clearance Capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Gibb, S.L.; Boston-Howes, W.; Lavina, S.Z.; Gustincich, S.; Brown, R.H.; Pasinelli, P.; Trotti, D. A Caspase-3-cleaved Fragment of the Glial Glutamate Transporter EAAT2 Is Sumoylated and Targeted to Promyelocytic Leukemia Nuclear Bodies in Mutant SOD1-linked Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2007, 282, 32480–32490. [Google Scholar] [CrossRef] [Green Version]
- Boston-Howes, W.; Gibb, S.L.; Williams, E.O.; Pasinelli, P.; Brown, R.H.; Trotti, D. Caspase-3 Cleaves and Inactivates the Glutamate Transporter EAAT. J. Biol. Chem. 2006, 281, 14076–14084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 2018, 497, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.A.; Schrama, L.H.; Van Veelen, C.W.M.; Van Rijen, P.C.; Van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002, 125, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Mathern, G.W.; Mendoza, D.; Lozada, A.; Pretorius, J.K.; Dehnes, Y.; Danbolt, N.C.; Nelson, N.; Leite, J.P.; Chimelli, L.; Born, D.E.; et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 1999, 52, 453. [Google Scholar] [CrossRef]
- Sarac, S.; Afzal, S.; Broholm, H.; Madsen, F.F.; Ploug, T.; Laursen, H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 2009, 117, 291–301. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Domercq, M.; Pérez-Cerdá, F.; Ravid, R.; Matute, C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol. Dis. 2006, 21, 154–164. [Google Scholar] [CrossRef]
- Mitosek-Szewczyk, K.; Sulkowski, G.; Stelmasiak, Z.; Strużyńska, L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neurosci. 2008, 155, 45–52. [Google Scholar] [CrossRef]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered Glutamate Reuptake in Relapsing-Remitting and Secondary Progressive Multiple Sclerosis Cortex: Correlation With Microglia Infiltration, Demyelination, and Neuronal and Synaptic Damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Diniz, L.P.; Araujo, A.P.B.; Matias, I.; Garcia, M.N.; Barros-Aragão, F.G.; Reis, R.A.D.M.; Foguel, D.; Braga, C.; Figueiredo, C.P.; Romão, L.; et al. Astrocyte glutamate transporters are increased in an early sporadic model of synucleinopathy. Neurochem. Int. 2020, 138, 104758. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Chen, L.; Chan, Y.; Yung, K.K.L. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 2008, 511, 421–437. [Google Scholar] [CrossRef] [PubMed]
- El Arfani, A.; Albertini, G.; Bentea, E.; Demuyser, T.; Van Eeckhaut, A.; Smolders, I.; Massie, A. Alterations in the motor cortical and striatal glutamatergic system and d-serine levels in the bilateral 6-hydroxydopamine rat model for Parkinson’s disease. Neurochem. Int. 2015, 88, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Ferrarese, C.; Tremolizzo, L.; Rigoldi, M.; Sala, G.; Brighina, L.; Ricci, G.; Albizzati, M.G.; Piolti, R.; Crosti, F.; Frattola, L.; et al. Decreased platelet glutamate uptake and genetic risk factors in patients with Parkinson’s disease. Neurol. Sci. 2001, 22, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Cha, S.-H.; Choi, Y.R.; Jou, I.; Joe, E.-H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef] [PubMed]
- Arzberger, T.; Krampfl, K.; Leimgruber, S.; Weindl, A. Changes of NMDA Receptor Subunit (NR1, NR2B) and Glutamate Transporter (GLT1) mRNA Expression in Huntington’s Disease—An In Situ Hybridization Study. J. Neuropathol. Exp. Neurol. 1997, 56, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [Green Version]
- Scarr, E.; Udawela, M.; Thomas, E.A.; Dean, B. Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predicts disrupted upstream pathways interacting with that receptor. Mol. Psychiatry 2018, 23, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Illarramendi, A.; Melone, M.; Conti, F.; Matute, C. Clozapine reduces GLT-1 expression and glutamate uptake in astrocyte cultures. Glia 2005, 50, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Medina, A.; Burke, S.; Thompson, R.C.; Bunney, W.; Myers, R.M.; Schatzberg, A.; Akil, H.; Watson, S.J. Glutamate transporters: A key piece in the glutamate puzzle of major depressive disorder. J. Psychiatr. Res. 2013, 47, 1150–1156. [Google Scholar] [CrossRef]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Imai, S.; Zou, S.; Yang, J.; Watanabe, M.; Wang, J.; Dubner, R.; Wei, F.; Ren, K. Altered glial glutamate transporter expression in descending circuitry and the emergence of pain chronicity. Mol. Pain 2019, 15, 1744806918825044. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Bianco, M.R.; Colangelo, A.M.; Cavaliere, C.; Daniele, D.L.; Zaccaro, L.; Alberghina, L.; Papa, M. Reactive astrocytosis-induced perturbation of synaptic homeostasis is restored by nerve growth factor. Neurobiol. Dis. 2011, 41, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Cirillo, G.; Bianco, M.R.; Rossi, F.; De Novellis, V.; Maione, S.; Papa, M. Gliosis alters expression and uptake of spinal glial amino acid transporters in a mouse neuropathic pain model. Neuron Glia Biol. 2007, 3, 141–153. [Google Scholar] [CrossRef]
- Aguirre, G.; Rosas, S.; Lopez-Bayghen, E.; Ortega, A. Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: Involvement of Ying-Yang. Neurochem. Int. 2008, 52, 1322–1331. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Bjerrum, O.J. Glutamate Transport System as a Novel Therapeutic Target in Chronic Pain: Molecular Mechanisms and Pharmacology. In Advances in Neurobiology; Ortega, A., Schousboe, A., Eds.; Springer: Cham, Switzerland, 2017; pp. 225–253. [Google Scholar]
- Chivukula, A.S.; Suslova, M.; Kortzak, D.; Kovermann, P.; Fahlke, C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum. Mutat. 2020, 41, 1892–1905. [Google Scholar] [CrossRef]
- During, M. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhäuser, C. Functional changes in astroglial cells in epilepsy. Glia 2006, 54, 358–368. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.R.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C.; et al. Abundant Tau Filaments and Nonapoptotic Neurodegeneration in Transgenic Mice Expressing Human P301S Tau Protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef] [Green Version]
- Potier, B.; Billard, J.-M.; Rivière, S.; Sinet, P.-M.; Denis, I.; Champeil-Potokar, G.; Grintal, B.; Jouvenceau, A.; Kollen, M.; Dutar, P. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell 2010, 9, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Bardou, I.; Hopp, S.C.; Kaercher, R.M.; Corona, A.W.; Fenn, A.M.; Godbout, J.P.; Wenk, G.L. Riluzole Partially Rescues Age-Associated, but not LPS-Induced, Loss of Glutamate Transporters and Spatial Memory. J. Neuroimmune Pharmacol. 2013, 8, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.C.; Gray, J.D.; Kogan, J.F.; Davidson, R.L.; Rubin, T.G.; Okamoto, M.; Morrison, J.H.; McEwen, B.S. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [Green Version]
- Hutton, M. The presenilins and Alzheimer’s disease. Hum. Mol. Genet. 1997, 6, 1639–1646. [Google Scholar] [CrossRef]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Pickering-Brown, S.; Medway, C.; et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2422.e13–2422.e16. [Google Scholar] [CrossRef]
- De Strooper, B. Lessons from a Failed γ-Secretase Alzheimer Trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Henley, D.B.; Sundell, K.L.; Sethuraman, G.; Dowsett, S.A.; May, P.C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med Res. Opin. 2014, 30, 2021–2032. [Google Scholar] [CrossRef]
- Moehlmann, T.; Winkler, E.; Xia, X.; Edbauer, D.; Murrell, J.; Capell, A.; Kaether, C.; Zheng, H.; Ghetti, B.; Haass, C.; et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on A 42 production. Proc. Natl. Acad. Sci. USA 2002, 99, 8025–8030. [Google Scholar] [CrossRef] [Green Version]
- Brai, E.; Raio, N.A.; Auber, L.A. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Nadeau, P.; Yuan, M.; Yang, X.; Shen, J.; Yankner, B.A. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 6959–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Gu, Y.; Hasegawa, H.; Ruan, X.; Arawaka, S.; Fraser, P.E.; Westaway, D.; Mount, H.; George-Hyslop, P.S. Presenilin 1 Mutations Activate γ42-Secretase but Reciprocally Inhibit ε-Secretase Cleavage of Amyloid Precursor Protein (APP) and S3-Cleavage of Notch. J. Biol. Chem. 2002, 277, 36521–36526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and Predictors of Seizures in Patients with Alzheimer’s Disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Sun, X.; Thomas, R.G.; Aisen, P.S.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Watanabe, H.; Wines-Samuelson, M.; Zhao, H.; Gridley, T.; Kopan, R.; Shen, J. Conditional Deletion ofNotch1andNotch2Genes in Excitatory Neurons of Postnatal Forebrain Does Not Cause Neurodegeneration or Reduction of Notch mRNAs and Proteins. J. Biol. Chem. 2012, 287, 20356–20368. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Turkoz, M.; Dearborn, J.T.; Wozniak, D.F.; Kopan, R.; Hass, M.R. Loss of RBPj in Postnatal Excitatory Neurons Does Not Cause Neurodegeneration or Memory Impairments in Aged Mice. PLoS ONE 2012, 7, e48180. [Google Scholar] [CrossRef]
- He, G.; Luo, W.; Li, P.; Remmers, C.; Netzer, W.J.; Hendrick, J.P.; Bettayeb, K.; Flajolet, M.; Gorelick, F.S.; Wennogle, L.P.; et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 2010, 467, 95–98. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, G.J.; Stevenson, P.; Ward, G.; Papadia, S.; Bading, H.; Chawla, S.; Privalsky, M.; Hardingham, G.E. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005, 93, 171–185. [Google Scholar] [CrossRef]
- Needham, L.A.; Davidson, A.H.; Bawden, L.J.; Belfield, A.; Bone, E.A.; Brotherton, D.H.; Bryant, S.; Charlton, M.H.; Clark, V.L.; Davies, S.J.; et al. Drug Targeting to Monocytes and Macrophages Using Esterase-Sensitive Chemical Motifs. J. Pharmacol. Exp. Ther. 2011, 339, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mubarak, B.; Soriano, F.X.; Hardingham, G.E. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels 2009, 3, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Cl− Conduct. | Kinetics | Location | Protein Abundance |
|---|---|---|---|---|---|
| EAAT1 | SLC1A3 | Mod | KM = 22–48 μM Cycle time = 62 ms | Astrocytes (incl. Bergmann & Müller glia); Predominant EAAT subtype in cerebellum (1.8 mg/g of protein) and retina | 99th percentile of protein found in human CBC; ≥95th in V1C, DFC, MD, STR and HIP; F794th in AMY |
| EAAT2 | SLC1A2 | Low | KM = 25–97 μM Cycle time = 70 ms | Astrocytes (and some sparse neurons); Predominant EAAT subtype in hippocampus (1.3 mg/g of protein) and cortex (0.8 mg/g) | 99th percentile of protein found in human V1C; ≥95th in DFC, HIP, AMY, STR and MD; 93rd in CBC |
| EAAT3 | SLC1A1 | Mod | KM = 42–62 μM Cycle time = 10 ms | Neurons (typically on spines); Highest concentration in hippocampus (0.013 mg/g of protein) | 26th percentile of protein found in human MD; 21st in HIP; ≤20th in AMY, STR, CBC, DFC and V1C |
| EAAT4 | SLC1A6 | High | KM = 2.5 μM Cycle time >166 ms | Cerebellar Purkinje cells (0.2 mg/g of protein in cerebellar molecular layer) | 89th percentile of protein in human CBC; 9th in MD and AMY; <15th in STR, DFC and HIP |
| EAAT5 | SLC1A7 | High | KM = 61–62 μM Cycle time >1000 ms | Retina (rod photo receptors, bipolar cells) |
| Treatment | Slc1a3/EAAT1 | Slc1a2/EAAT2 | Species |
|---|---|---|---|
| Neuronal coculture | Increased expression | Robust induction of expression | Mouse and rat; in vitro |
| Neuronal Conditioned Media | No | Yes | Mouse; in vitro |
| cAMP | Increases expression and function | Robust increases in expression and function | Mouse; in vitro |
| Glutamate | Downregulated by glutamaterigic denervation; Upregulated by AMPA receptor activation | Downregulated by glutamaterigic denervation | Mouse and Rat; in situ |
| Epidermal growth factor/NF-κB | No | Increased expression. Overlap with NCM and cAMP pathway | Mouse, rat and human; in vitro |
| Pax6 | No | Induces expression in pure astrocytes; knockdown represses neuron coculture induction | Mouse; in vitro |
| Notch (Neuron/endothelial cell to astrocyte contact dependant) | Increases expression; inhibition decreases expression | Increased expression; inhibition decreases expression | Mouse, rat and drosophila; in vitro and in vivo |
| Disease | Transporter | Observed Association | Refs |
|---|---|---|---|
| Alzheimer’s Disease (AD) | EAAT1, EAAT2 | EAAT1/2 function and expression reduced by amyloid β; Aberrant EAAT1 expression in AD patient neurons; Reduced function and expression of EAAT1 and 2 in hippocampal and cortical AD tissue | [171,172,173,174,175,176,177,178,179] |
| Amyotrophic lateral sclerosis (ALS)/motor neuron disease | EAAT2 | Impaired Glu uptake in patients with sporadic ALS; Reduced EAAT2 protein in tissue from motor regions; One reported case of a patient with a mutation in SLC1A2 causing reduced EAAT2 activity; Familial ALS with SOD1 mutations expected to reduce functional EAAT2 protein; Deletion of slc1a2 in mice spinal cord leads to motor neuron degeneration; Reduction in slc1a2 in P301S tauopathy mouse model | [101,180,181,182,183,184,185,186,187] |
| Epilepsy/temporal lobe epilepsy (TLE) | EAAT1, EAAT2 | Reduced EAAT2 in TLE patients with hippocampal sclerosis; Reduced EAAT1 & 2 in treatment resistant TLE patients; Mouse EAAT2 KO → lethal epilepsy | [91,188,189,190] |
| Multiple sclerosis (MS) | EAAT1, EAAT2 | Increased EAAT1 and EAAT2 mRNA and protein in MS optic nerve, with increased glutamate uptake; Loss of EAAT1 and EAAT2 in areas surrounding cortical lesions of MS patients; In rat EAE model cortex, increased EAAT2 mRNA and protein, increased EAAT1 mRNA but decreased protein. In rat EAE model cerebellum, increased EAAT1 and EAAT2 mRNA, but decreased EAAT1 and EAAT2 protein. | [191,192,193] |
| Synucleinop-athies (including Parkinson’s disease -PD) | EAAT1, EAAT2 | Increased EAAT1 and EAAT2 expression following injection of α-synuclein oligomers in mouse striatum; Decreased EAAT1 and EAAT2 in rat striatum following dopaminergic denervation via MPTP treatment or 6-ODHA induced lesion rat models; Reduced glutamate uptake in platelets from PD patients; PD-related mutation DJ-1 mouse model showed reduced EAAT2 function | [194,195,196,197,198] |
| Huntington’s disease (HD) | EAAT2 | Decrease in EAAT2 mRNA expression in neostriatum of HD patients, decrease corresponding to disease severity; Decease in EAAT2 mRNA and protein in mice expressing mutant huntingtin; | [199,200] |
| Schizophrenia (SCZ) | EAAT1, EAAT2 | Increased mRNA expression of EAAT1 in Brodmann’s area (BA)9; Increased mRNA expression of EAAT1 and EAAT2 in BA10; Increased EAAT1 mRNA and decreased protein in post mortem SCZ CNS tissue; Clozapine (used to treat SCZ) decreased EAAT2 expression; | [145,146,201,202] |
| Major depressive disorder (MDD) | EAAT1, EAAT2 | Reduced mRNA expression of EAAT1 and EAAT2 in anterior cingulate, dorsolateral prefrontal cortex, locus coeruleus and hippocampus of human MDD patients; Decreased protein in orbitofrontal cortex of MDD patients; | [150,151,152,203] |
| Autism | EAAT1,F7EAAT2 | EAAT1 mRNA expression upregulated; Decreased functional EAAT2 in conditional Fmr1 KO mouse astrocytes (mouse model of fragile-X) | [149,204] |
| Attention deficit hyperactivity disorder (ADHD) | EAAT1 | Duplication of SLC1A3 gene observed in clinical case of ADHD; SLC1A3 rs1049522 allele significantly associated with ADHD; Increased EAAT1 mRNA expression in cerebellar cortex | [147,148] |
| Chronic pain | EAAT2 | Decreased EAAT2 mRNA in rostral ventromedial medulla and spinal cord in rodent chronic pain models; Administration of EAAT2 antagonist alleviates hyperalgesia in rats; Analgesic effects of valproic acid suggested to be due to increasing EAAT1 expression. | [205,206,207,208,209] |
| Episodic ataxia type 6 | EAAT1 | Caused by mutations in SLC1A3 altering properties of EAAT1 | [210] |
| Gene | Mean Human Expression in <40 y.o. (FPKM) | Mean Human Expression in >40 y.o. (FPKM) | Relative Expression with Older Age |
|---|---|---|---|
| Notch genes | |||
| HES1 | 9.31 | 6.57 | 0.71 |
| HES6 | 2.91 | 1.28 | 0.44 |
| HES5 | 3.47 | 0.92 | 0.26 |
| HEY2 | 1.40 | 1.57 | 1.12 |
| HEY1 | 10.43 | 9.76 | 0.94 |
| BCL2 | 5.41 | 4.75 | 0.88 |
| Total FPKM | 32.93 | 24.84 | 0.75 |
| Notch receptors | |||
| NOTCH2 | 22.36 | 14.39 | 0.64 |
| NOTCH1 | 0.84 | 0.41 | 0.49 |
| NOTCH3 | 0.53 | 0.20 | 0.38 |
| NOTCH4 | 0.11 | 0.13 | 1.22 |
| Total FPKM | 23.83 | 15.14 | 0.64 |
| γ-secretase genes | |||
| PSEN1 | 7.87 | 6.99 | 0.89 |
| PSEN2 | 1.23 | 0.67 | 0.55 |
| NCSTN | 24.59 | 11.58 | 0.47 |
| APH1A | 1.63 | 1.27 | 0.78 |
| APH1B | 5.07 | 4.72 | 0.93 |
| PSENEN | 0.26 | 0.65 | 2.51 |
| Total FPKM | 40.64 | 25.89 | 0.64 |
| Notch effectors/activators | |||
| MAML1 | 1.83 | 0.90 | 0.49 |
| MED8 | 4.42 | 2.77 | 0.63 |
| RBPJ | 6.52 | 7.22 | 1.11 |
| FURIN | 0.46 | 0.17 | 0.38 |
| Total FPKM | 13.23 | 11.07 | 0.84 |
| Sum Notch related genes (FPKM) | 110.64 | 76.93 | 0.70 |
| Glutamate transporters | |||
| SLC1A2 | 2454.47 | 1521.31 | 0.62 |
| SLC1A3 | 1146.57 | 797.77 | 0.70 |
| Total EAAT (FPKM) | 3601.05 | 2319.07 | 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todd, A.C.; Hardingham, G.E. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9607. https://doi.org/10.3390/ijms21249607
Todd AC, Hardingham GE. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(24):9607. https://doi.org/10.3390/ijms21249607
Chicago/Turabian StyleTodd, Alison C., and Giles E. Hardingham. 2020. "The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 24: 9607. https://doi.org/10.3390/ijms21249607
APA StyleTodd, A. C., & Hardingham, G. E. (2020). The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(24), 9607. https://doi.org/10.3390/ijms21249607