Opening a Novel Biosynthetic Pathway to Dihydroxyacetone and Glycerol in Escherichia coli Mutants through Expression of a Gene Variant (fsaAA129S) for Fructose 6-Phosphate Aldolase †



Abstract
:1. Introduction
2. Results
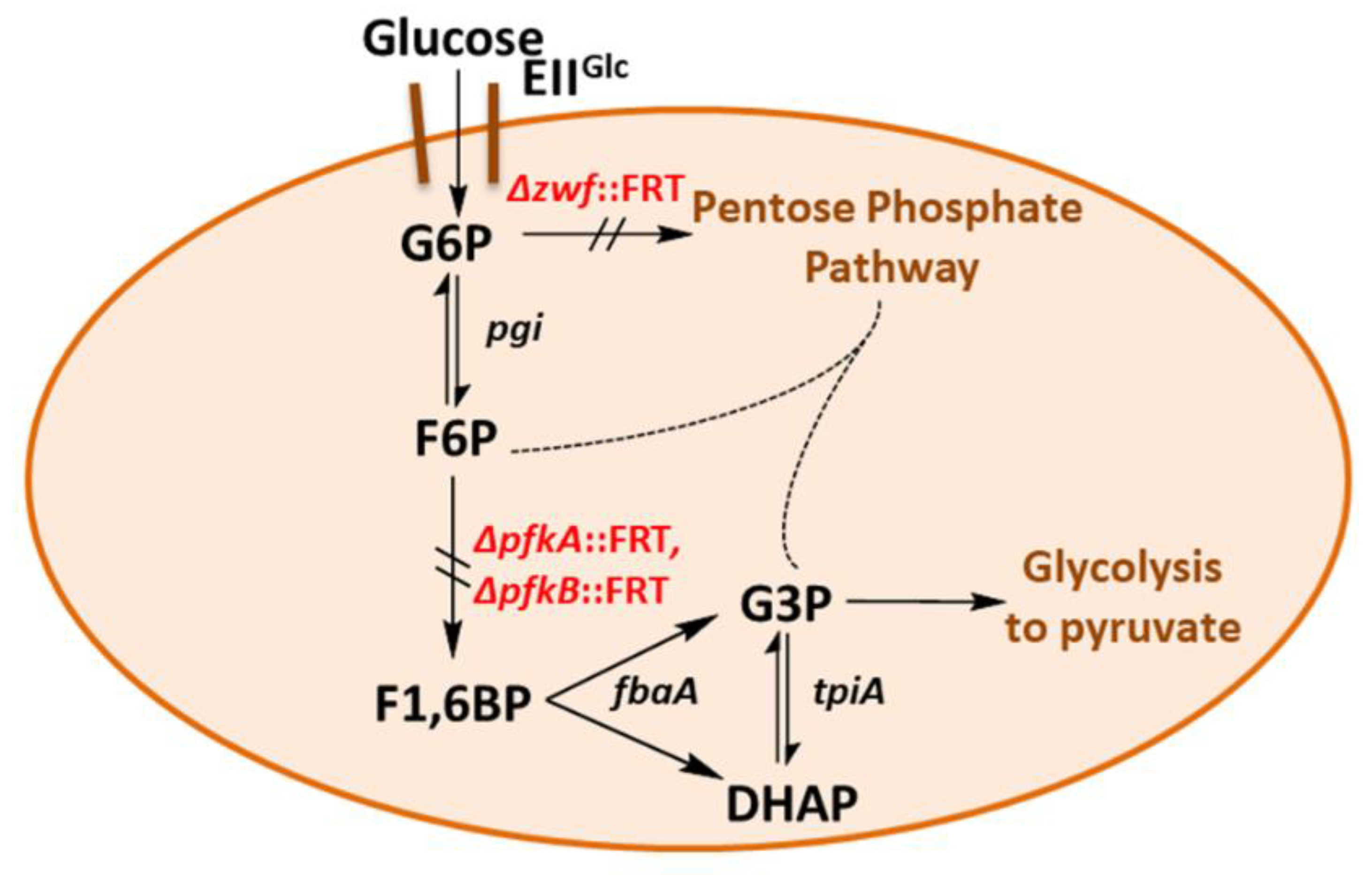
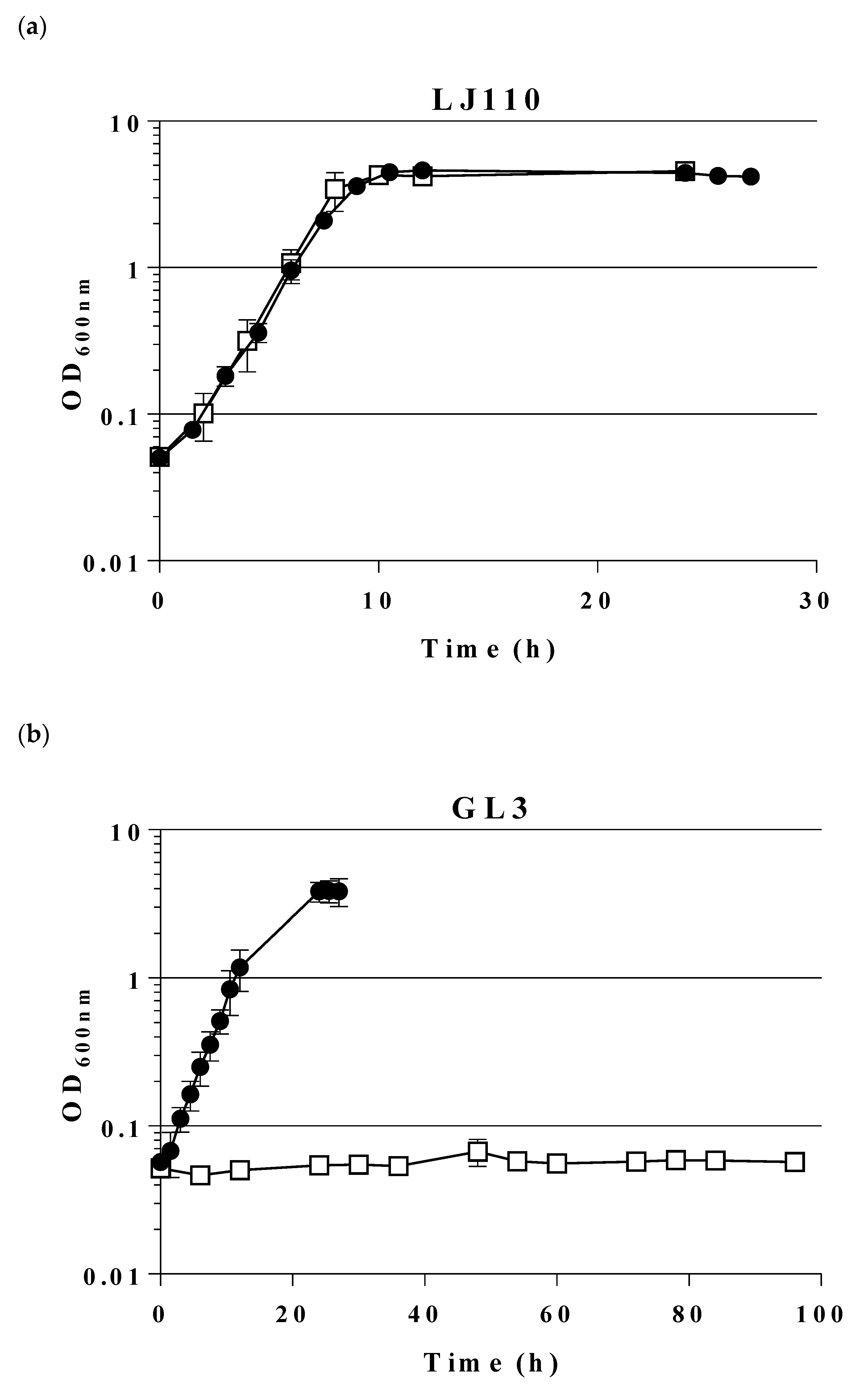
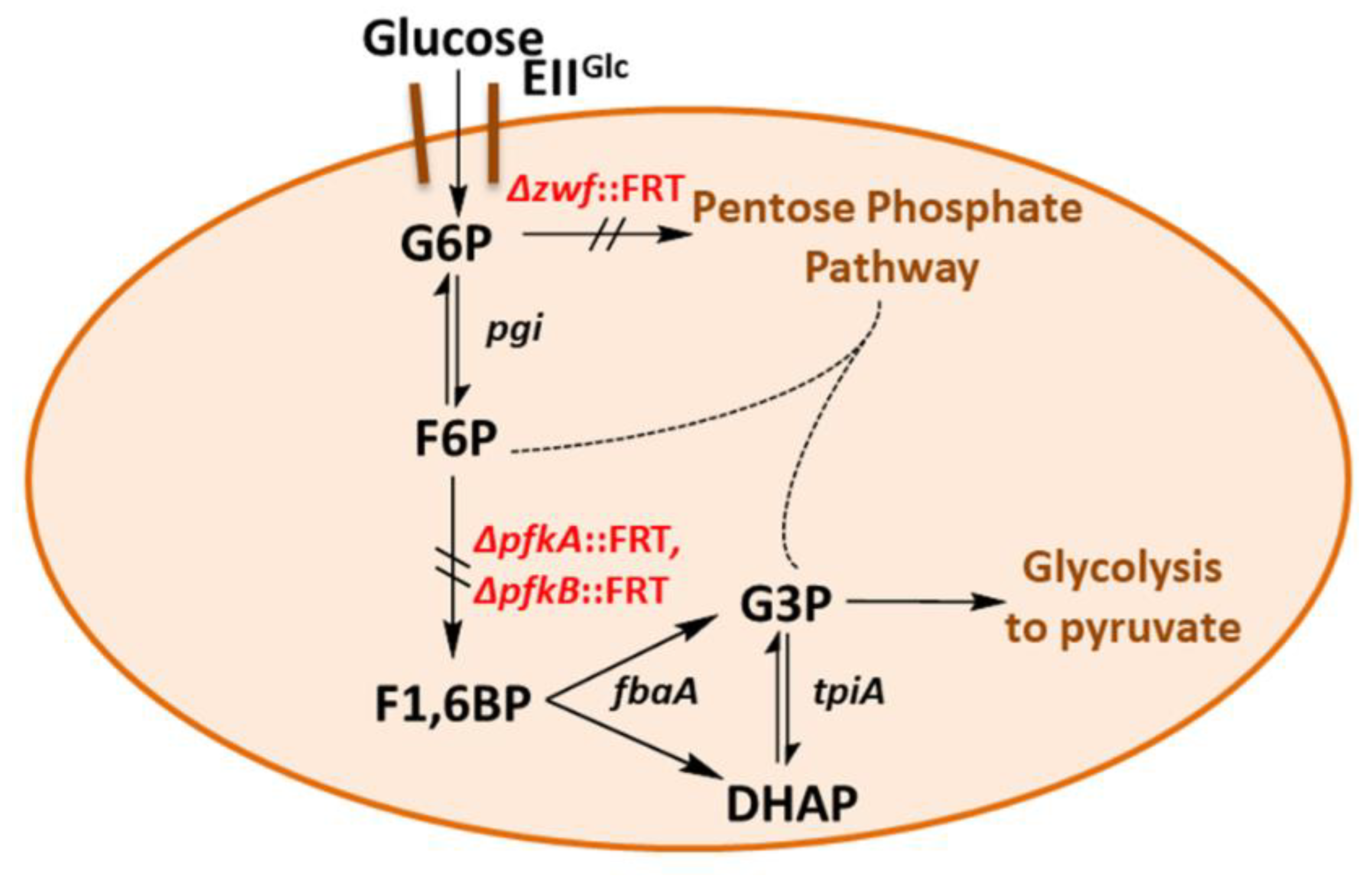
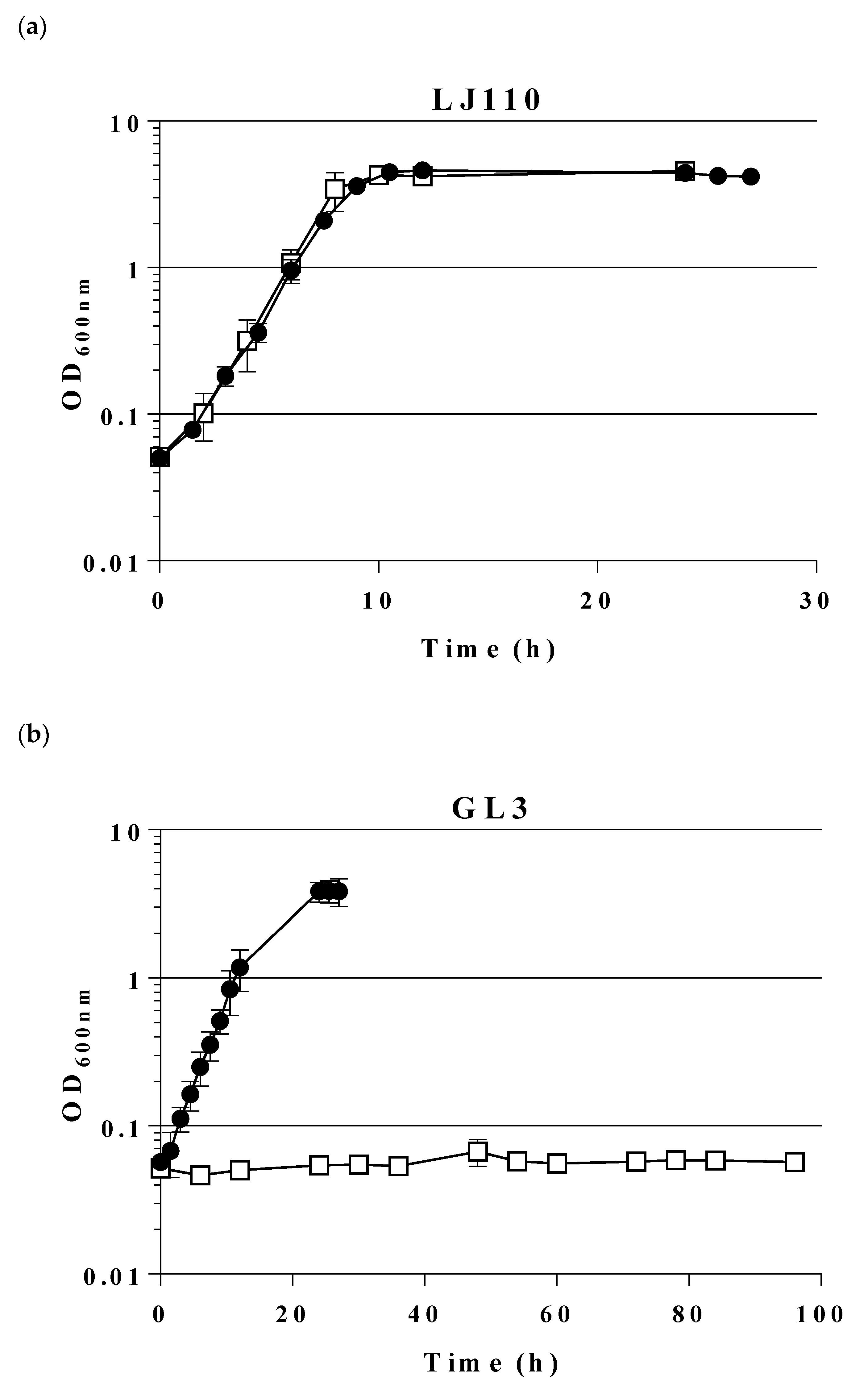
2.1. Construction and Characterization of an E. coli Triple Mutant Strain Deficient in Phosphofructokinase (Genes pfkA, pfkB) and Glucose 6-Phosphate Dehydrogenase (zwf)
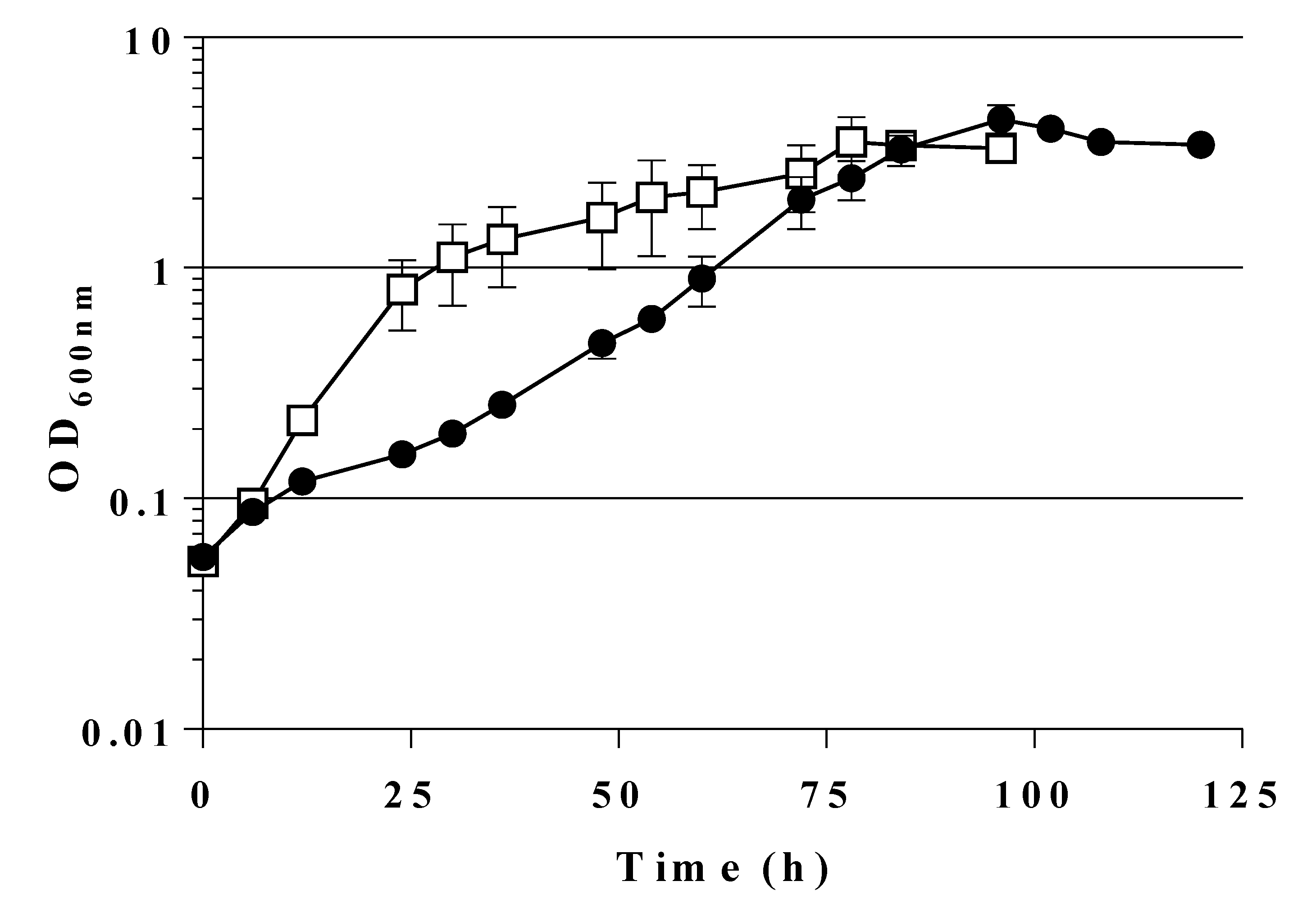
2.2. Expression of a Mutant Gene (fsaAA129S) Restores Growth of GL3 on Glucose
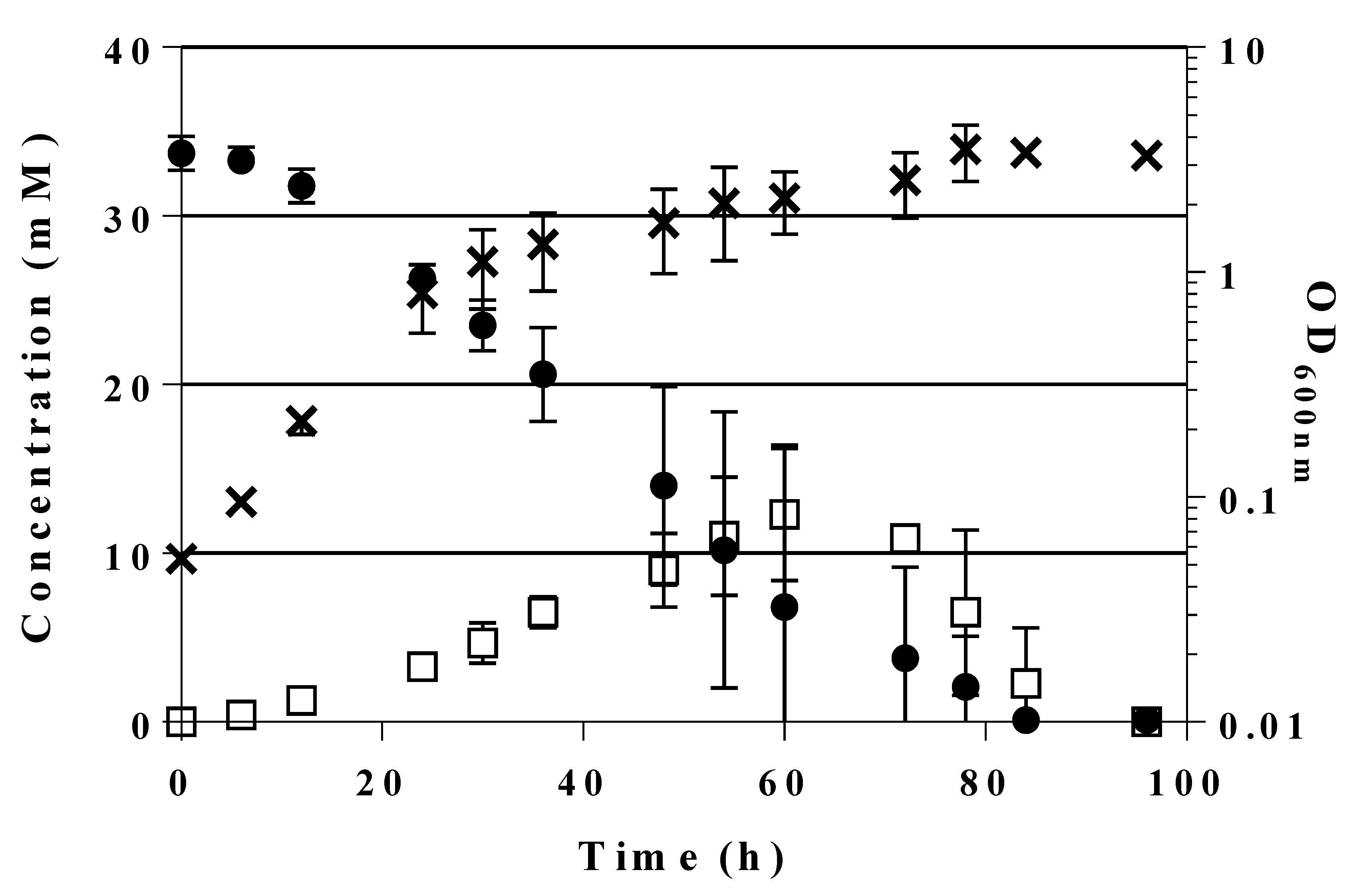
2.3. Transient Formation of Dihydroxyacetone from Glucose
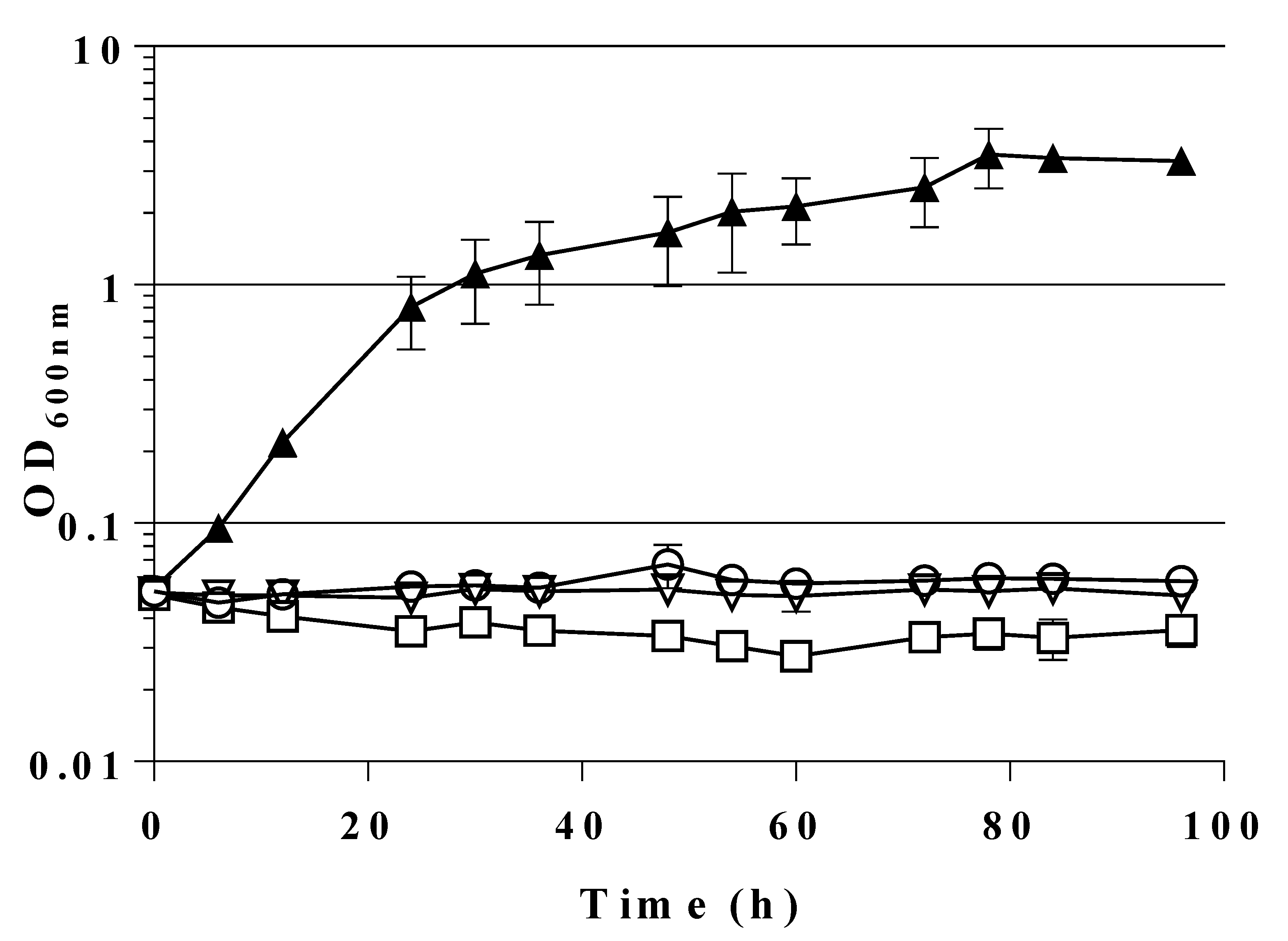
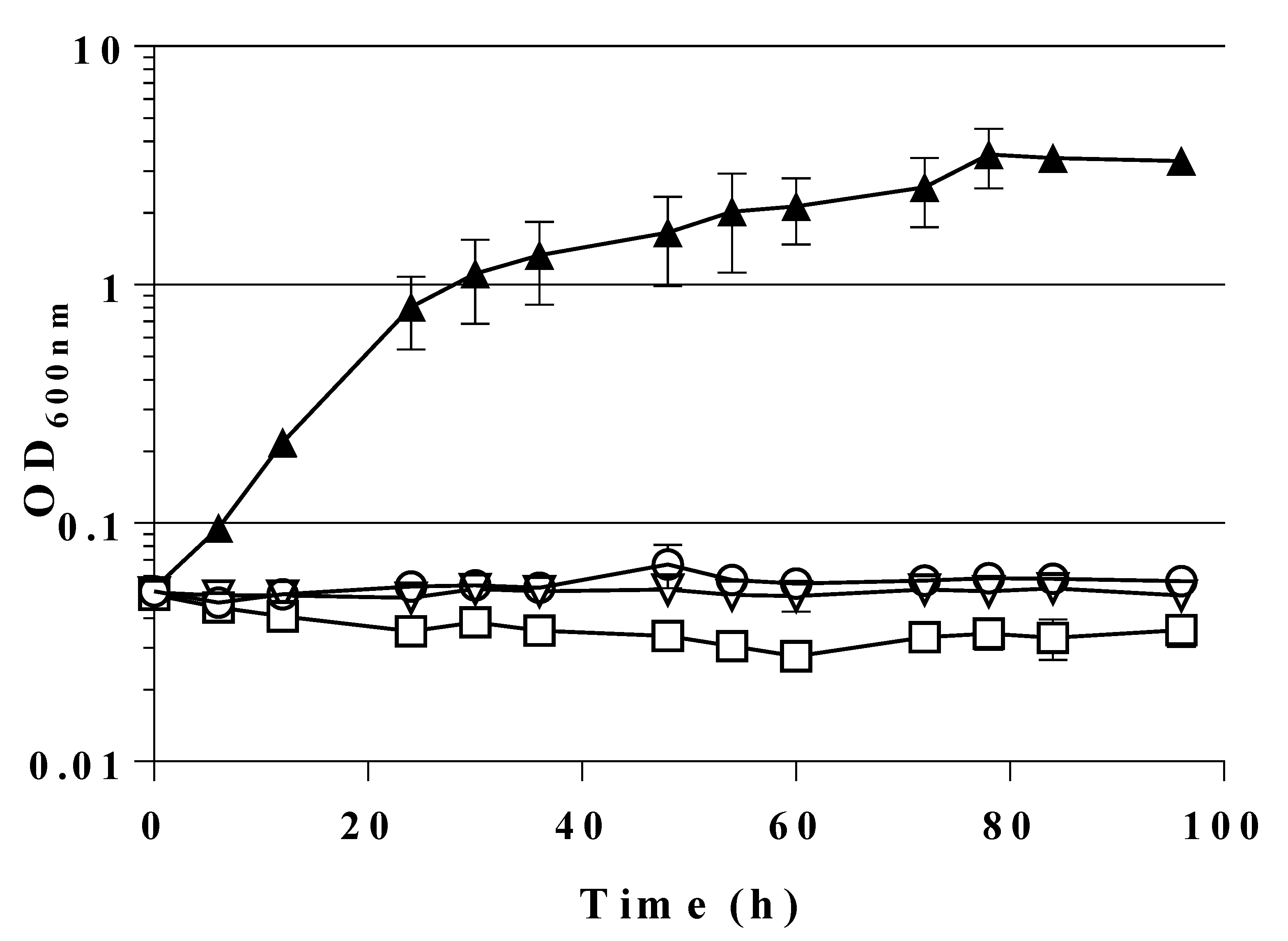
2.4. A Single Chromosomal Copy of fsaAA129S Is Sufficient for Growth of the Triple Mutant
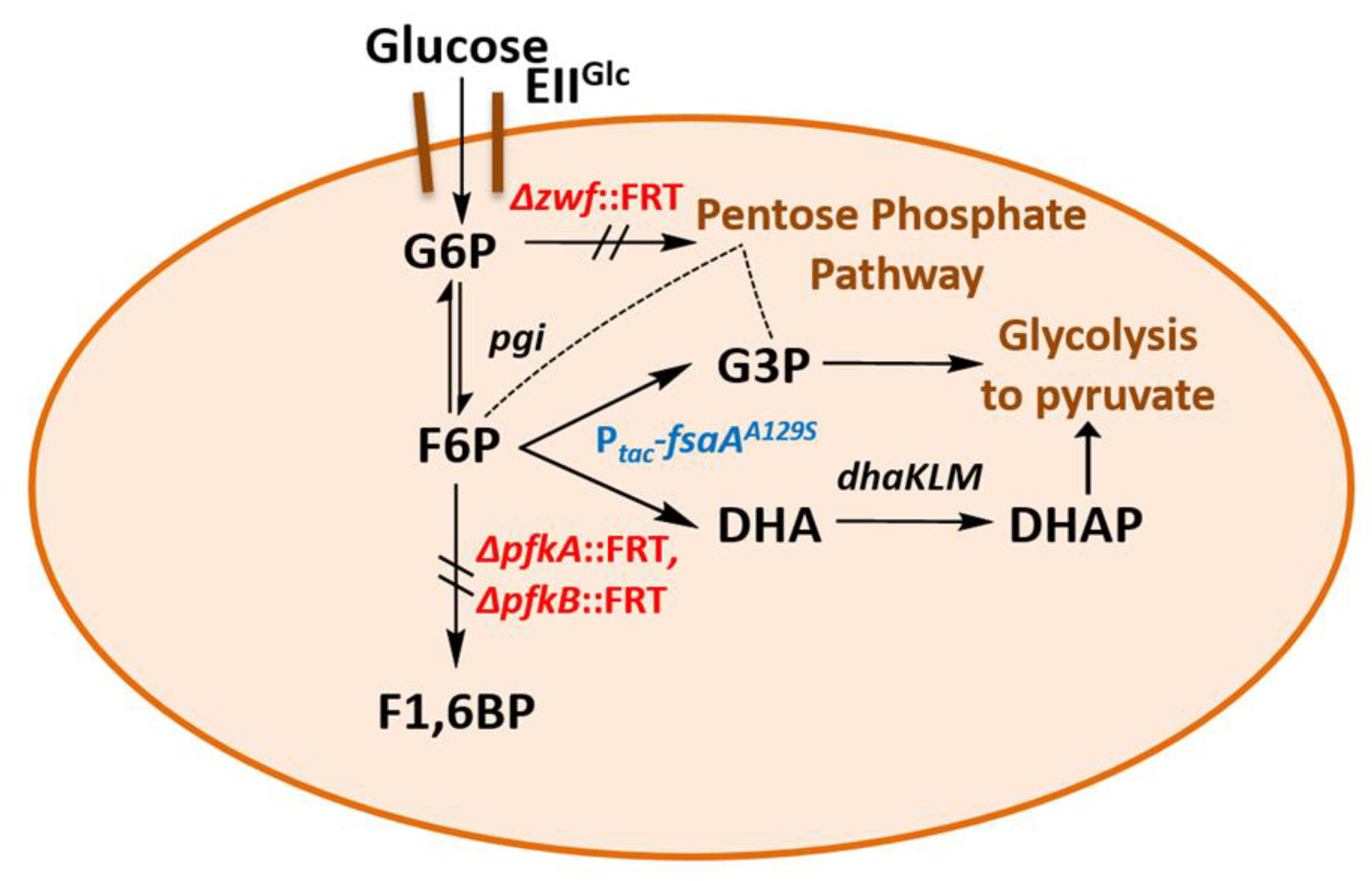
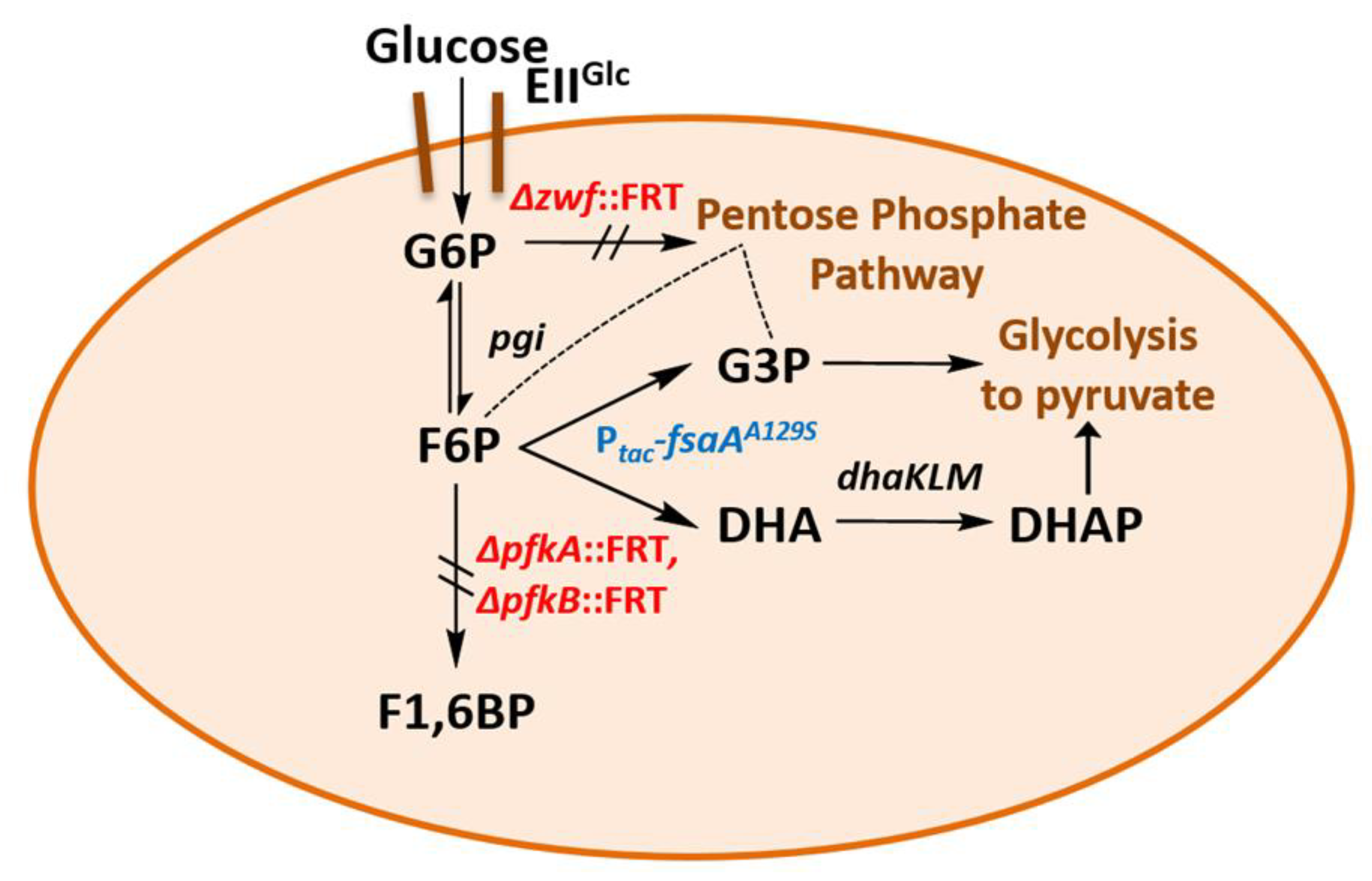
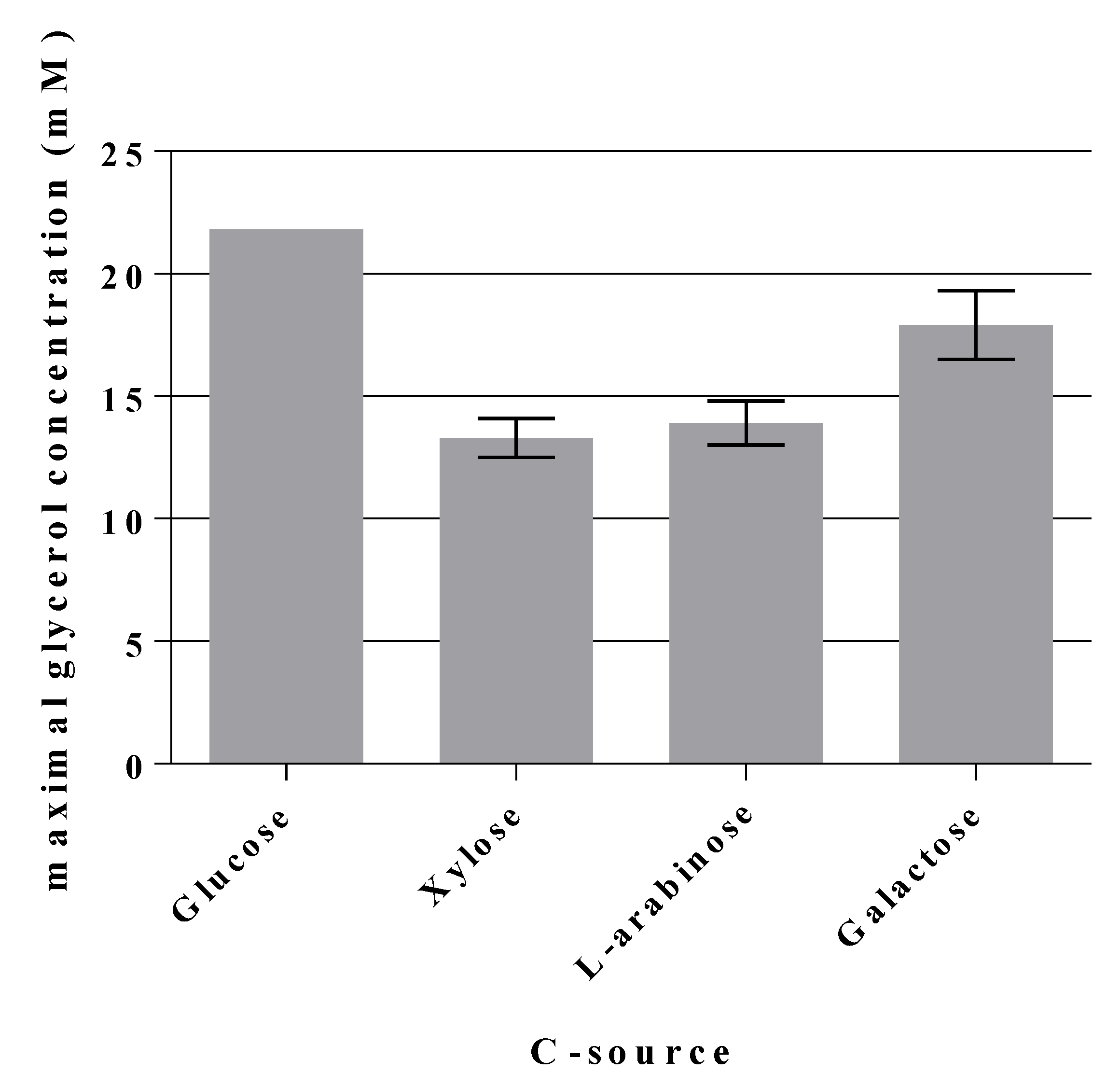
2.5. A Novel Pathway of Glycerol Formation in E. coli
3. Discussion
- (a)
- We have shown here that a bypass of the PFK block mutation is possible if a suitable enzyme activity of FSA is present in E. coli cells. Cleavage of F6P into G3P and DHA with subsequent phosphorylation of DHA would lead to the same couple of triose phosphates as in EMP, and no extra energy expenditure is necessary (see Figure 4). Therefore, one might ask why did Nature not choose this pathway as a general alternative to EMP? The reason could be that DHA may not be funneled completely or fast enough into DHAP. DHA is a short chain sugar which is well-known to readily react with proteins as it forms adducts with lysine and arginine residues [67] leading to browning; this effect is exploited for skin tanning [68], where DHA is an ingredient in many self-tanning compounds. We also have observed that during the course of incubation, only the cultures of the strains that produce DHA became yellow. Still, DHA can also lead to protein inactivation and it is known as a mutagen for E. coli under certain circumstances [67]. Both consequences would be detrimental for cell metabolism and genetic stability. This might be the reason why an elaborate high affinity PTS for DHA phosphorylation (DHAKLM) is present in E. coli [5,58,69] whose main function arguably lies not so much in catabolism (yielding DHAP which can be catabolized in glycolysis or used as building block for glycerophospholipid formation) but rather in effective detoxification of an undesired metabolite. We found increased gene activity of the dhaK gene in GL4 which we take for evidence that the DHAKLM system is indeed involved in further metabolism of DHA which stems from F6P cleavage.
- (b)
- FSAA A129S not only opens a bypass at the block between F6P and F1,6BP (see Figure 4 and Figure S3). Through its action, G3P is formed which can be further metabolized in the lower glycolytic trunk to PEP and ultimately pyruvate. As well, G3P together with F6P serves as substrate for transketolase (TKT) from the PPP. TKT transfers two-carbon dihydroxyethyl units from F6P to G3P forming E4P and X5P; transaldolase then performs a DHA transfer reaction from F6P onto E4P yielding G3P and S7P [70,71]. FSA could also add DHA on E4P to form S7P and be a sink for DHA [25]; thus, not all DHA is to be expected to go to glycerol (in strain GL7).
- (c)
- The block of PFK in glycolysis leads to accumulation of phosphorylated sugars (G6P, F6P) from PTS substrates and causes the so-called sugar phosphate stress. This elicits activation of SgrS [59,72,73]. SgrS sRNA interacts with ptsG mRNA to reduce translation of new PtsG (enzyme IIBCGlc) molecules [74] whereas already existing PtsG molecules are still active as protein turnover is slow [75]; SgrS sRNA also activates synthesis of a sugar phosphatase (YigL) which dephosphorylates G6P to help with glucose homeostasis [75]. This could lead to less G6P and, in turn, to less F6P in the cells. Moreover, the gene product of sgrS, when expressed ectopically, is a small polypeptide SgrT which inhibits PtsG activity in vivo [76,77]. Thus, although FSAA A129S removes F6P, the uptake rate of glucose might be lower in GL3 than in LJ110 and contribute to the slower growth on glucose.
- (d)
- The missing glucose 6-phosphate dehydrogenase function (Δzwf) in strain GL3 and the resulting lack of a subsequent 6-phosphogluconate dehydrogenase step pose another problem as the NADPH supply by these two enzymes is not restored by introduction of FSAA A129S in the bypass pathway. Cells of GL3/pJF119fsaAA129S have thus to rely either on transhydrogenase or isocitrate dehydrogenase as NADPH sources [45] for anabolism. This could also contribute to the observed slower growth rates.
- (e)
- Why does the triple mutant GL3 grow slower on C sources other than glucose? Fructose is in part transported via the enzyme II for D-mannose to yield F6P [14] and thus could end up at the same blockade as with glucose. Xylose and other C sources which are initially routed through the PPP, also deliver F6P which could then accumulate and/or be interchanged with G6P. Both would not contribute to the EMP. In comparison to GL3, in GL3/pJF119fsaAA129S and strain GL7 growth on xylose and other C sources was only partially restored while being clearly slower than in the wild type, LJ110 (data not shown). This warrants further investigations.
4. Materials and Methods
4.1. Chemicals, Carbon Sources, and Enzymes
4.2. Bacterial Strains, Plasmid and Strain Constructions
4.3. Growth Conditions
4.4. Quantitative Real-Time PCR (qPCR)
4.5. Preparation of Cell-Free Extracts (cfe) and Enzyme Activity Assays
4.6. HPLC Analysis of Sugars, DHA, and Glycerol in Supernatants
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Amp | ampicillin |
| C | carbon |
| cfe | cell-free extracts |
| Cm | chloramphenicol |
| DHA | dihydroxyacetone |
| DHAKLM | dihydroxyacetone kinase |
| DHAP | dihydroxyacetone phosphate |
| ED | Entner–Doudoroff pathway |
| EIIGlc | glucose transporter |
| EMP | Embden–Meyerhof–Parnas pathway |
| F1,6BP | fructose 1,6-bisphosphate |
| F6P | fructose 6-phosphate |
| FRT | FLP recognition target |
| FSA | fructose 6-phosphate aldolase |
| G3P | glyceraldehyde 3-phosphate |
| G6P | glucose 6-phosphate |
| GLDA | glycerol dehydrogenase |
| HMP | hexose monophosphate shunt |
| IPTG | isopropyl β-D-1-thiogalactopyranoside |
| Km | kanamycin |
| MM | minimal medium |
| PFK | phosphofructokinase |
| PGI | phosphoglucoisomerase |
| PPP | pentose phosphate pathway |
| PTS | PEP-dependent sugar:phosphotransferase system |
| Spc | spectinomycin |
| TKT | transketolase |
| wt | wild type |
| ZWF | glucose 6-phosphate dehydrogenase |
References
- Jahreis, K.; Pimentel-Schmitt, E.F.; Brückner, R.; Titgemeyer, F. Ins and outs of glucose transport systems in eubacteria. FEMS Microbiol. Rev. 2008, 32, 891–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, J.; Ake, F.M.D.; Derkaoui, M.; Zebre, A.C.; Cao, T.N.; Bouraoui, A.; Kentache, T.; Mokhtari, A.; Milohanic, E.; Joyet, P. The bacterial phosphoenolpyruvate: Carbohydrate phosphotransferase system: Regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microb. Mol. Biol. Rev. 2014, 8, 231–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Even, A.; Flamholz, A.; Noor, E.; Milo, R. Rethinking glycolysis: On the biochemical logic of metabolic pathways. Nat. Chem. Biol. 2012, 8, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Babul, J. Phosphofructokinases from Escherichia coli. J. Biol. Chem. 1978, 253, 4350–4355. [Google Scholar] [PubMed]
- Fenton, A.W.; Reinhart, G.D. Disentangling the web of allosteric communication in a homotetramer: Heterotropic inhibition in phosphofructokinase from Escherichia coli. Biochemistry 2009, 8, 12323–12328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraenkel, D.G. Glycolysis. In Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; Neidhardt, F.C.M., Curtiss, R., III, Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., Reznikoff, W.S., Riley, M., Schaechter, M., Umbarger, H.E., Eds.; ASM Press: Washington, DC, USA, 1996; pp. 189–198. [Google Scholar]
- Romeo, T.; Snoep, J.L. Glycolysis and flux control. In EcoSal Plus 2013; ASMScience.org: Washington, DC, USA, 2013. [Google Scholar] [CrossRef]
- Robinson, J.P.; Fraenkel, D.G. Allosteric and non-allosteric E. coli phosphofructokinases: Effects on growth. Biochem. Biophys. Res. Commun. 1978, 81, 858–863. [Google Scholar] [CrossRef]
- Fraenkel, D.G.; Kotlarz, D.; Buc, H. Two fructose 6-phosphate kinase activities in Escherichia coli. J. Biol. Chem. 1973, 248, 4865–4866. [Google Scholar]
- Daldal, F. Molecular cloning of the gene for phosphofructokinase-2 of Escherichia coli and the nature of a mutation, pfkB1, causing a high level of the enzyme. J. Mol. Biol. 1983, 168, 285–305. [Google Scholar] [CrossRef]
- Lovingshimer, M.R.; Siegele, D.; Reinhart, G.D. Construction of an inducible, pfkA and pfkB deficient strain of Escherichia coli for the expression and purification of phosphofructokinase from bacterial sources. Protein Expr. Purif. 2006, 46, 475–482. [Google Scholar] [CrossRef]
- Siedler, S.; Bringer, S.; Blank, L.M.; Bott, M. Engineering yield and rate of reductive biotransformation in Escherichia coli by partial cyclization of the pentose phosphate pathway and PTS-independent glucose transport. Appl. Microbiol. Biotechnol. 2012, 93, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; San, K.-Y.; Bennett, G.N. Improvement of NADPH bioavailability in Escherichia coli through the use of phosphofructokinase deficient strains. Appl. Microbiol. Biotechnol. 2013, 97, 6883–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, H.L. Routes for fructose utilization by Escherichia coli. J. Mol. Microbiol. Biotechnol. 2001, 3, 355–359. [Google Scholar] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, C.A.C.; Bennett, R.K.; Papoutsakis, E.T.; Antoniewicz, M.R. Deletion of four genes in Escherichia coli enables preferential consumption of xylose and secretion of glucose. Metab. Eng. 2019, 52, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Gleizer, S.; Ben-Nissan, R.; Bar-On, Y.M.; Antonovsky, N.; Noor, E.; Zohar, N.; Jona, G.; Krieger, E.; Shamshoum, M.; Bar-Even, A.; et al. Conversion of Escherichia coli to generate all biomass carbon from CO2. Cell 2019, 179, 1255–1263. [Google Scholar] [CrossRef] [Green Version]
- Satanowski, A.; Dronsella, B.; Noor, E.; Vögeli, B.; He, H.; Wichmann, P.; Erb, T.J.; Lindner, S.N.; Bar-Even, A. Awakening a latent carbon fixation cycle in Escherichia coli. Nat. Commun. 2020, 11, 5812. [Google Scholar] [CrossRef]
- Brockman, I.M.; Prather, K.L.J. Dynamic knockdown of E. coli central metabolism for redirecting fluxes of primary metabolites. Metab. Eng. 2015, 28, 104–113. [Google Scholar] [CrossRef]
- Schürmann, M.; Sprenger, G.A. Fructose-6-phosphate aldolase is a novel class I aldolase from Escherichia coli and is related to a novel group of bacterial transaldolases. J. Biol. Chem. 2001, 276, 11055–11061. [Google Scholar] [CrossRef] [Green Version]
- Pieper, R.; Zhang, Q.; Clark, D.J.; Parmar, P.P.; Alami, H.; Suh, M.-J.; Kuntumalla, S.; Braisted, J.C.; Huang, S.-T.; Tzipori, S. Proteomic view of interactions of Shiga toxin-producing Escherichia coli with the intestinal environment in gnotobiotic piglets. PLoS ONE 2013, 8, e66462. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samland, A.K.; Sprenger, G.A. Synthetic potential of dihydroxyacetone-utilizing aldolases. In Industrial Biocatalysis; Grunwald, P., Ed.; Pan Stanford Publishing Pte. Ltd.: Singapore, 2015; pp. 783–816. [Google Scholar]
- Schürmann, M. Biochemische Charakterisierung und Struktur-Funktionsbeziehungen bakterieller Transaldolasenund Fruktose-6-Phosphat Aldolasen. Ph.D. Thesis, University of Düsseldorf, Düsseldorf, Germany, 2001. [Google Scholar]
- Schürmann, M.; Schürmann, M.; Sprenger, G.A. Fructose 6-phosphate aldolase and 1-deoxy-D-xylulose 5-phosphate synthase from Escherichia coli as tools in enzymatic synthesis of 1-deoxysugars. J. Mol. Catal. B Enzym. 2002, 19, 247–252. [Google Scholar] [CrossRef]
- Garrabou, X.; Castillo, J.A.; Guérard-Hélaine, C.; Parella, T.; Joglar, J.; Lemaire, M.; Clapés, P. Asymmetric self-and cross-aldol reactions of glycolaldehyde catalyzed by D-fructose-6-phosphate aldolase. Angew. Chem. 2009, 121, 5629–5633. [Google Scholar] [CrossRef]
- Castillo, J.A.; Guérard-Hélaine, C.; Gutiérrez, M.; Garrabou, X.; Sancelme, M.; Schürmann, M.; Inoue, T.; Hélaine, V.; Charmantray, F.; Gefflaut, T.; et al. A mutant of D-fructose-6-phosphate aldolase (Ala129Ser) with improved affinity towards dihydroxyacetone for the synthesis of polyhydroxylated compounds. Adv. Synth. Catal. 2010, 352, 1039–1046. [Google Scholar] [CrossRef]
- Peiro, C.; Millard, P.; de Simone, A.; Cahoreau, E.; Peyriga, L.; Enjalbert, B.; Heux, S. Chemical and metabolic controls on dihydroxyacetone metabolism lead to suboptimal growth of Escherichia coli. Appl. Environm. Microbiol. 2019, 85, e00768-19. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.N.; Aslan, S.; Müller, A.; Hoffart, E.; Behrens, P.; Edlich-Muth, C.; Blombach, B.; Bar-Even, A. A synthetic glycerol assimilation pathway demonstrates biochemical constraints of cellular metabolism. FEBS J. 2020, 287, 160–172. [Google Scholar] [CrossRef]
- Lachaux, C.; Frazao, C.J.R.; Kraußer, F.; Morin, N.; Walther, T.; Francois, J.M. A new synthetic pathway for the bioproduction of glycolic acid from lignocellulosic sugars aimed at maximal carbon conservation. Front. Bioeng. Biotechnol. 2019, 7, 359. [Google Scholar] [CrossRef]
- King, J.R.; Woolston, B.M.; Stephanopoulos, G. Designing a new entry point into isoprenoid metabolism by exploiting fructose-6-phosphate aldolase side reactivity of Escherichia coli. ACS Synth. Biol. 2017, 6, 1416–1426. [Google Scholar] [CrossRef]
- Riemer, S.A.; Rex, R.; Schomburg, D. A metabolite-centric view on flux distributions in genome-scale metabolic models. BMC Syst. Biol. 2013, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Andreozzi, S.; Chakrabarti, A.; Soh, K.C.; Burgard, A.; Yang, T.H.; van Dien, S.; Miskovic, L.; Hatzimanikatis, V. Identification of metabolic engineering targets for the enhancement of 1,4-butanediol production in recombinant E. coli using large-scale kinetic models. Metab. Eng. 2016, 35, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Haedicke, O.; Klamt, S. EColiCore2: A reference network model of the central metabolism of Escherichia coli and relationships to its genome-scale parent model. Sci. Rep. 2017, 7, 39647. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, G.A.; Schürmann, M.; Schürmann, M.; Johnen, S.; Sprenger, G.; Sahm, H.; Inoue, T.; Schörken, U. C-C-bonding microbial enzymes: Thiamine diphosphate-dependent enzymes and class I aldolases. In Asymmetric Synthesis with Chemical and Biological Methods; Enders, E., Jaeger, K.-E., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 312–326. [Google Scholar]
- Sprenger, G.A. Glycerol as carbon source for production of added-value compounds. In Engineering of Microorganisms for the Production of Chemicals and Biofuels from Renewable Resources; Gosset, G., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 93–123. [Google Scholar] [CrossRef]
- Semkiv, M.V.; Ruchala, J.; Dmytruk, K.V.; Sibirny, A.A. 100 years later, what is new in glycerol bioproduction? Trends Biotechnol. 2020, 38, 907–916. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Zhuge, J.; Fang, H.; Prior, B.A. Glycerol production by microbial fermentation: A review. Biotechnol. Adv. 2001, 19, 201–223. [Google Scholar] [CrossRef]
- Lim, C.; Lin, A.L.; Zhao, H. Metabolic strategies for microbial glycerol overproduction. J. Chem. Technol. Biotechnol. 2018, 93, 624–628. [Google Scholar] [CrossRef]
- Hartlep, M.; Hussmann, W.; Prayitn, N.; Meynial-Salles, I.; Zeng, A.-P. Study of two-stage processes for the microbial production of 1,3-propanediol from glucose. Appl. Microbiol. Biotechnol. 2002, 60, 60–66. [Google Scholar]
- Nakamura, C.E.; Whited, G.M. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 2003, 14, 454–459. [Google Scholar] [CrossRef]
- Meynial-Salles, I.; Forchhammer, N.; Croux, C.; Girbal, L.; Soucaille, P. Evolution of a Saccharomyces cerevisiae metabolic pathway in Escherichia coli. Metab. Eng. 2007, 9, 152–159. [Google Scholar] [CrossRef]
- Balagurunathan, B.; Jain, V.K.; Tear, C.J.Y.; Lim, C.Y.; Zhao, H. In silico design of anaerobic growth-coupled product formation in Escherichia coli: Experimental validation using a simple polyol, glycerol. Bioprocess Biosyst. Eng. 2017, 40, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, T.; Larisch, C.; Lengeler, J.W.; Jahreis, K. Glucose transporter mutants of Escherichia coli K-12 with changes in substrate recognition of IICBGlc and induction behavior of the ptsG gene. J. Bacteriol. 2000, 182, 4443–4452. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Baba, T.; Mori, H.; Shimizu, K. Effect of zwf gene knockout on the metabolism of Escherichia coli grown on glucose or acetate. Metab. Eng. 2004, 6, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Vinopal, R.T.; Fraenkel, D.G. Phenotypic suppression of phosphofructokinase mutations in Escherichia coli by constitutive expression of the glyoxylate shunt. J. Bacteriol. 1974, 118, 1090–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Tanaka, S.; Lerner, S.A.; Lin, E.C.C. Replacement of a phosphoenolpyruvate-dependent phosphotransferase by a nicotinamide adenine dinucleotide-linked dehydrogenase for the utilization of mannitol. J. Bacteriol. 1967, 93, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Fürste, J.P.; Pansegrau, W.; Frank, R.; Blöcker, H.; Scholz, P.; Bagdasarian, M.; Lanka, E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 1986, 48, 119–131. [Google Scholar] [CrossRef]
- Inoue, T. Microbial Aldolases as C-C Bonding Enzymes: Investigations of Structural-functional Characteristics and Application for Stereoselective Reactions. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2006. [Google Scholar]
- Trachtmann, N.; Alvarez Fong, K.F.; Guitart Font, E.; Sprenger, G.A. Construction of chromosomally encoded lacZ and gfp reporter strains of Escherichia coli for the study of global regulation of metabolism. Eng. Life Sci. 2016, 16, 675–681. [Google Scholar] [CrossRef]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef]
- Erni, B.; Siebold, C.; Christen, S.; Srinivas, A.; Oberholzer, A.; Baumann, U. Small substrate, big surprise: Fold, function and phylogeny of dihydroxyacetone kinases. Cell. Mol. Life Sci. 2006, 63, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.Z.; Lin, E.C.C. An inducible phosphoenolpyruvate: Dihydroxyacetone phosphotransferase system in Escherichia coli. J. Gen. Microbiol. 1984, 130, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Gutknecht, R.; Beutler, R.; Garcia-Alles, L.F.; Baumann, U.; Erni, B. The dihydroxyacetone kinase of Escherichia coli utilizes a phosphoprotein instead of ATP as phosphoryl donor. EMBO J. 2001, 20, 2480–2486. [Google Scholar] [CrossRef] [Green Version]
- Bächler, C.; Schneider, P.; Bähler, P.; Lustig, A.; Erni, B. Escherichia coli dihydroxyacetone kinase controls gene expression by binding to transcription factor DhaR. EMBO J. 2005, 24, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Vanderpool, C.K.; Gottesman, S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol. Microbiol. 2004, 54, 1076–1089. [Google Scholar] [CrossRef]
- Morita, T.; El-Kazzaz, W.; Tanaka, Y.; Inada, T.; Aiba, H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J. Biol. Chem. 2003, 278, 15608–15614. [Google Scholar] [CrossRef] [Green Version]
- Cleton-Jansen, A.-M.; Goosen, N.; Fayet, O.; Van de Putte, P. Cloning, mapping, and sequencing of the gene encoding Escherichia coli quinoprotein glucose dehydrogenase. J. Bacteriol. 1990, 172, 6308–6315. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, K.; Arents, J.C.; Bader, J.; Yamada, M.; Adachi, O.; Postma, P.W. Escherichia coli is unable to produce pyrroloquinoline quinone (PQQ). Microbiology 1997, 143, 3149–3156. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.C.C. Dissimilatory pathways for sugars, polyols, and carboxylates. In Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; Neidhardt, F.C.M., Curtiss, R., III, Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., Reznikoff, W.S., Riley, M., Schaechter, M., Umbarger, H.E., Eds.; ASM Press: Washington, DC, USA, 1996; pp. 307–342. [Google Scholar]
- Fong, S.S.; Nanchen, A.; Palsson, B.Ø.; Sauer, U. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J. Biol. Chem. 2006, 281, 8024–8033. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Matsuoka, Y. Regulation of glycolytic flux and overflow metabolism depending on the source of energy generation for energy demand. Biotechnol. Adv. 2019, 37, 284–305. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Rubion, S.A.; Xu, Y.-F.; Amador-Noguez, D.; Fan, J.; Shlomi, T.; Rabinowitz, J.D. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat. Chem. Biol. 2016, 12, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Benov, L.; Beema, A.F. Superoxide-dependence of the short chain sugars-induced mutagenesis. Free Radic. Biol. Med. 2003, 34, 429–433. [Google Scholar] [CrossRef]
- Wittgenstein, E.; Berry, H.K. Staining of skin with dihydroxyacetone. Science 1960, 132, 894–895. [Google Scholar] [CrossRef]
- Shi, R.; McDonald, L.; Cui, Q.; Matte, A.; Cygler, M.; Ekiel, I. Structural and mechanistic insight into covalent substrate binding by Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 1302–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenger, G.A. Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch. Microbiol. 1995, 164, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Tittmann, K. Sweet siblings with different faces: The mechanisms of FBP and F6P aldolase, transaldolase, transketolase and phosphoketolase revisited in light of recent structural data. Bioorg. Chem. 2014, 57, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Bobrovskyy, M.; Vanderpool, C.K. The small RNA SgrS: Roles in metabolism and pathogenesis of enteric bacteria. Front. Cell. Infect. Microbiol. 2014, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.R.; Cobe, B.L.; Richards, G.R. Stringent response regulators contribute to recovery from glucose phosphate stress in Escherichia coli. Appl. Environ. Microbiol. 2017, 83, e01636-17. [Google Scholar] [CrossRef] [Green Version]
- Maki, K.; Morita, T.; Otaka, H.; Aiba, H. A minimal base-pairing region of a bacterial small RNA SgrS required for translational repression of ptsG mRNA. Mol. Microbiol. 2010, 76, 782–792. [Google Scholar] [CrossRef]
- Papenfort, K.; Sun, Y.; Miyakoshi, M.; Vanderpool, C.K.; Vogel, J. Small mRNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 2013, 153, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Wadler, C.S.; Vanderpool, C.K. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc. Natl. Acad. Sci. USA 2007, 104, 20454–20459. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.R.; Park, S.; Fei, J.; Vanderpool, C.K. The small protein SgrT controls transport activity of the glucose-specific phosphotransferase system. J. Bacteriol. 2017, 199, e00869-16. [Google Scholar] [CrossRef] [Green Version]
- Valle, F.; Munoz, E.; Ponce, E.; Flores, N.; Bolivar, F. Basic and applied aspects of metabolic diversity: The phosphoenolpyruvate node. J. Ind. Microbiol. 1996, 17, 458–462. [Google Scholar] [CrossRef]
- Kochanowski, K.; Volkmer, B.; Gerosa, L.; van Rijsewijk, B.R.; Schmidt, A.; Heinemann, M. Functioning of a metabolic flux sensor in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 1130–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.-I.; Lin, E.C.C. Purification and properties of glycerol kinase from Escherichia coli. J. Biol. Chem. 1967, 242, 1030–1035. [Google Scholar] [PubMed]
- Subedi, K.P.; Kim, I.; Kim, J.; Min, B.; Park, C. Role of GldA in dihydroxyacetone and methylglyoxal metabolism of Escherichia coli K12. FEMS Microbiol. Lett. 2008, 279, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Origin/Reference |
|---|---|---|
| DH5α | F–, Φ80d, lacZΔM15, Δ(lacZYA-argF) U169, recA1, endA1, hsdR17 (rK–, mK+), phoA, supE44, λ–, thi-1, gyrA96, relA1 | [49] |
| LJ110 | W3110 fnr+, wild type | [44] |
| GL1 | LJ110, Δzwf::FRT | This study a |
| GL2 | LJ110, Δzwf::FRT ΔpfkB::FRT | This study a |
| GL3 | LJ110, Δzwf::FRT ΔpfkB::FRT ΔpfkA::FRT | This study a |
| GL4 | LJ110, Δzwf::FRT ΔpfkB::FRT ΔpfkA::FRT ΔlacZ::Ptac-fsaAA129S | This study b |
| GL5 | LJ110, Δzwf::FRT ΔpfkB::FRT ΔpfkA::FRT ΔdhaKLM ΔlacZ::Ptac-fsaAA129S | This study b |
| GL6 | LJ110, Δzwf::FRT ΔpfkB::FRT ΔpfkA::FRT ΔdhaKLM ΔlacZ::Ptac-fsaAA129S ΔglpK | This study b |
| GL7 | LJ110, Δzwf::FRT ΔpfkB::FRT ΔpfkA::FRT ΔdhaKLM ΔlacZ::Ptac-fsaAA129S ΔglpK Δrbsk::Ptac-gldA | This study b |
| Strain: | LJ110 | GL3 | GL4 | |
|---|---|---|---|---|
| LB-agar | Normal (1) | Normal (1) | Normal (1) | |
| MM-agar + 0.5% C-source (w/v) | No C-source | - | - | - |
| D-Fructose | Normal (1) | Small (2) | Small (2) * | |
| D-Glucose | Normal (1) | - | Small (2) | |
| Glycerol | Normal (1) | Small (1) | Small (1) | |
| Lactate (pH 7.0) | Small (1) | Small (1) | Small (1) | |
| Mannitol | Normal (1) | - | - | |
| Succinate (pH 7.0) | Small (1) | Small (1) | Small (1) | |
| Sorbitol | Normal (1) | - | Small (2) * | |
| D-Xylose | Small (1) | - | Small (2) | |
| Maltose | Normal (1) | - | - | |
| D-Galactose | Small (1) | - | - | |
| Gluconate (pH 7.0) | Normal (1) | Small (2) | Small (1) | |
| L-Arabinose | Normal (1) | Small (2) | Small (2) | |
| Lactose | Normal (1) | - | - ** | |
| Plasmid | Relevant Characteristics | Source/Reference |
|---|---|---|
| pJF119EH | Ptac, lacIq, RBS, AmpR | [51] |
| pJF119fsaA | fsaA wildtype gene, cloned into pJF119EH | [52] |
| pJF119fsaAA129S | fsaAA129S gene, cloned into pJF119EH | [52] |
| pJF119ΔEPtac-gldA | PtacgldA, lacIq, ΔEcoRI, ΔNdeI, optimized RBS, AmpR | Stefan Riemer (2010, unpublished) |
| pKD46 | repA101(ts), araC, ParaB-ϒ-β-exo (λred recombinase), AmpR | [47] |
| pCO1-cat | FRT-cat-FRT, AmpR, CmR | [53] |
| pCP20 | FLP+, λ cl857+, λ pR repts, AmpR, CmR | [54] |
| pCas | repA101 (Ts), ParaB-γ-β-exo (λred recombinase), Pcas-cas9, lacIq, Ptrc-sgRNA-pMB1, KmR | AddGene [48] |
| pTarget with sgRNAs | With (T) or without (F) donor DNA | AddGene [48] |
| pTargetF-dhaKLM | pMB1, sgRNA-dhaKLM, SpcR | This study |
| pTargetF-Cm-glpK | pMB1, sgRNA-glpK, CmR | This study |
| pTargetT-Cm-ΔlacZ::Ptac-fsaAA129S | pMB1, sgRNA-lacZ, ΔlacZ::Ptac-fsaAA129S, CmR | This study |
| pTargetF-Cm-rbsK | pMB1, sgRNA-rbsK, CmR | This study |
| pJNTN-m-L | Ptac, lacIq, KmR | Natalie Trachtmann (unpublished) |
| pJNTN-m-L-pfkA | pfkA gene cloned into pJNTN-m-L | Natalie Trachtmann (unpublished) |
| C Source | Strain | Time (h) until Max. OD600 | Max. OD600 | C Source (mM) Consumed | DHA | Glycerol | µ (h−1) | Gt (min) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Max. Conc. (mM) | Res * | Max. Conc. (mM) | Res * | |||||||
| 28 mM glucose | LJ110 | 24 | 4.562 ± 0.003 | 33.8 | 0.0 ± 0.0 | No | 0.0 ± 0.0 | No | 0.59 ± 0.01 | 71 ± 1 |
| GL3 | n.g. | n.g. | n.a | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| GL4 | 96 | 4.399 ± 0.675 | 32.2 | 0.5 ± 0.0 | No | 0.0 ± 0.0 | No | 0.05 ± 0.00 | 808 ± 11 | |
| GL5 | 216 | 2.410 ± 0.489 | 30.2 | 3.1 ± 0.3 | No | 0.0 ± 0.0 | No | 0.02 ± 0.00 | 2293 ± 85 | |
| GL6 | 288 | 1.497 ± 0.033 | 30.6 | 8.8 ± 0.3 | Yes | 1.9 ± 0.2 | Yes | 0.03 ± 0.01 | 1655 ± 103 | |
| GL7 | 240 | 1.395 ± 0.165 | 30.2 | 3.5 ± 0.3 | Yes | 21.8 ± 0.0 | Yes | 0.03 ± 0.00 | 1526 ± 49 | |
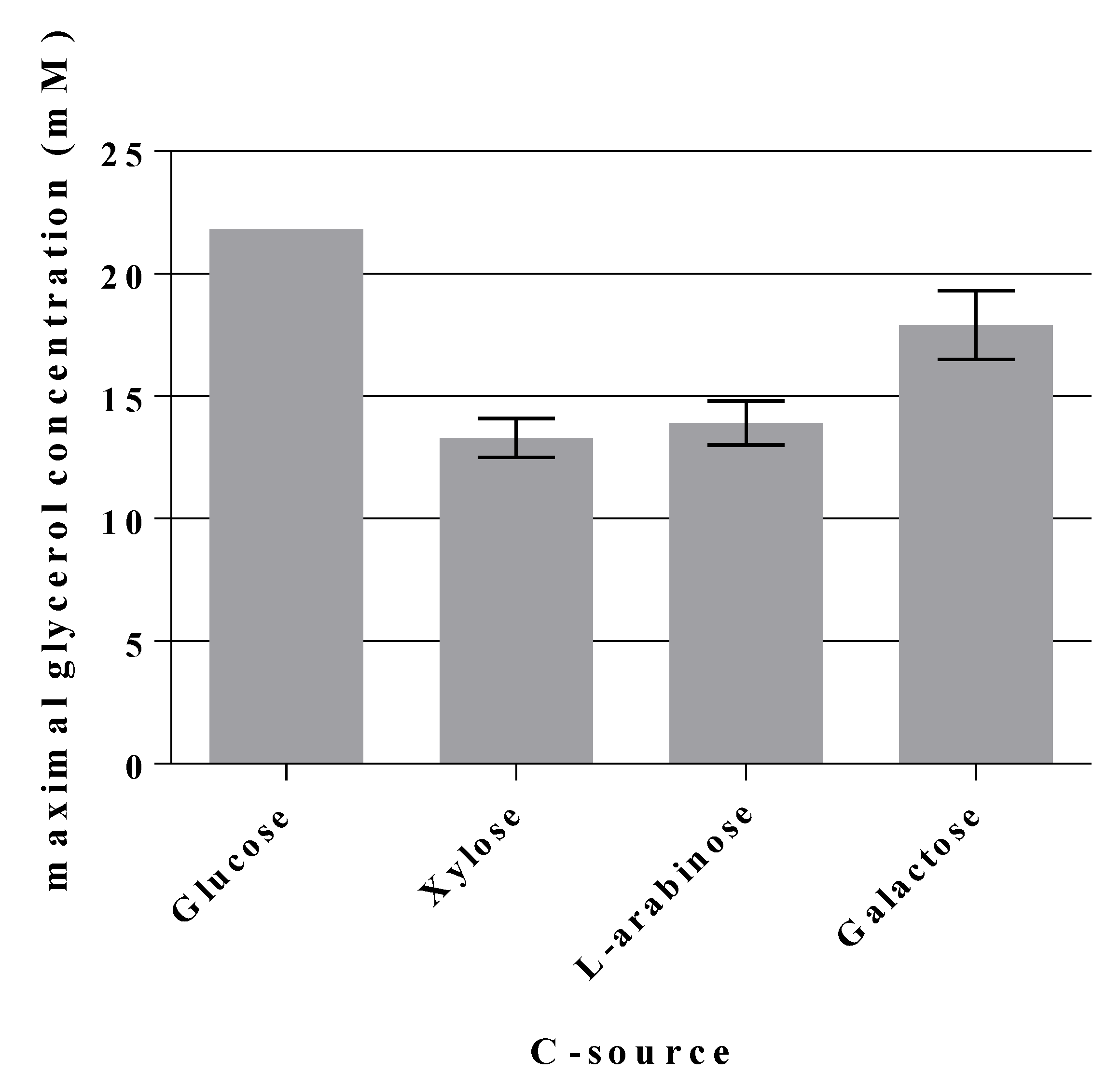
| 33 mM xylose | LJ110 | 24 | 4.492 ± 0.011 | 32.7 | 0.0 ± 0.0 | No | 0.0 ± 0.0 | No | 0.55 ± 0.01 | 76 ± 1 |
| GL7 | 48 | 4.431 ± 0.187 | 32.9 | 3.2 ± 0.1 | Yes | 13.3 ± 0.8 | Yes | 0.12 ± 0.01 | 361 ± 8 | |
| 33 mM L-arabinose | LJ110 | 24 | 1.991 ± 0.107 | 34.5 | 0.0 ± 0.0 | No | 0.0 ± 0.0 | No | 0.53 ± 0.01 | 79 ± 4 |
| GL7 | 48 | 2.024 ± 0.061 | 35.0 | 4.1 ± 0.7 | Yes | 13.9 ± 0.9 | Yes | 0.12 ± 0.01 | 360 ± 21 | |
| 28 mM galactose | LJ110 | 28 | 4.133 ± 0.160 | 28.0 | 0.0 ± 0.0 | No | 0.0 ± 0.0 | No | 0.19 ± 0.01 | 222 ± 21 |
| GL7 | 120 | 1.264 ± 0.001 | 28.2 | 3.5 ± 1.3 | Yes | 17.9 ± 1.4 | Yes | 0.04 ± 0.01 | 1266 ± 143 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guitart Font, E.; Sprenger, G.A. Opening a Novel Biosynthetic Pathway to Dihydroxyacetone and Glycerol in Escherichia coli Mutants through Expression of a Gene Variant (fsaAA129S) for Fructose 6-Phosphate Aldolase. Int. J. Mol. Sci. 2020, 21, 9625. https://doi.org/10.3390/ijms21249625
Guitart Font E, Sprenger GA. Opening a Novel Biosynthetic Pathway to Dihydroxyacetone and Glycerol in Escherichia coli Mutants through Expression of a Gene Variant (fsaAA129S) for Fructose 6-Phosphate Aldolase. International Journal of Molecular Sciences. 2020; 21(24):9625. https://doi.org/10.3390/ijms21249625
Chicago/Turabian StyleGuitart Font, Emma, and Georg A. Sprenger. 2020. "Opening a Novel Biosynthetic Pathway to Dihydroxyacetone and Glycerol in Escherichia coli Mutants through Expression of a Gene Variant (fsaAA129S) for Fructose 6-Phosphate Aldolase" International Journal of Molecular Sciences 21, no. 24: 9625. https://doi.org/10.3390/ijms21249625
APA StyleGuitart Font, E., & Sprenger, G. A. (2020). Opening a Novel Biosynthetic Pathway to Dihydroxyacetone and Glycerol in Escherichia coli Mutants through Expression of a Gene Variant (fsaAA129S) for Fructose 6-Phosphate Aldolase. International Journal of Molecular Sciences, 21(24), 9625. https://doi.org/10.3390/ijms21249625