Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology


Abstract
:1. Introduction
2. Results
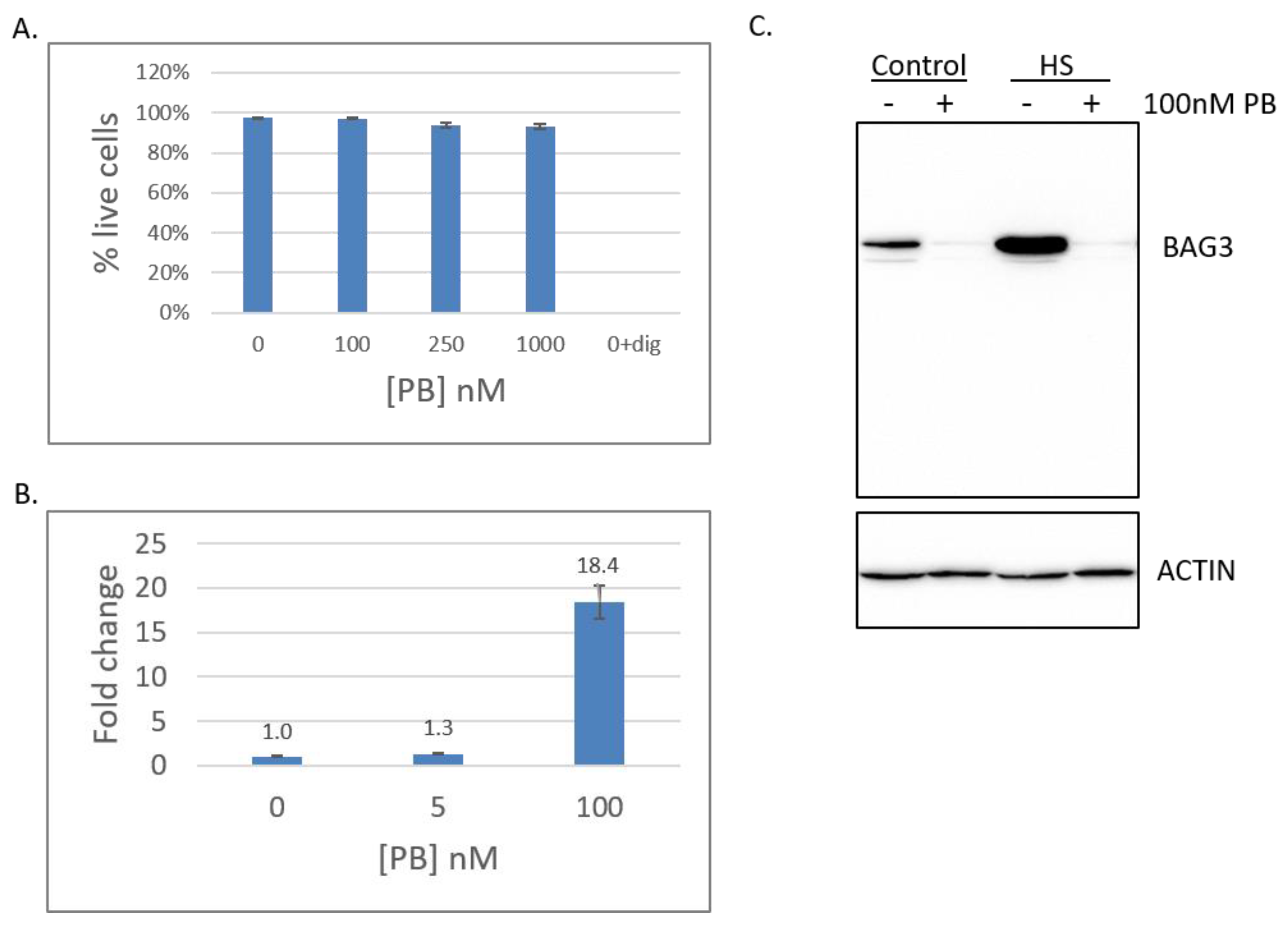
2.1. Pladienolide B Affects Splicing but not Cell Viability in HEK293T Cells
2.2. Transcriptomes of Cells Exposed to PB Reveal Massive Changes to Transcript Architecture
2.3. PB induced Defect in NMD
2.4. PB Induced Changes to Gene Expression
2.5. Sensitivity to Low PB Does not Predict Response to High PB
2.6. Low PB Enhances the Protein Folding Environment
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Harvest
4.3. Transcript Quantitation
4.4. RNA Sequencing
4.5. Transcriptome Analysis Pipeline
4.6. Trypan Blue Exclusion Assay
4.7. Western Blots
4.8. Nonsense-Mediated Decay Quantitation
4.9. PolyQ Aggregation Assay
4.10. Accession Numbers
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PB | Pladienolide B |
| HSR | Heat Shock Response |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | Fetal bovine serum |
| FDR | False Discovery Rate |
| NMD | Nonsense-mediated decay |
| UTR | Untranslated region |
| A3′SS | Alternative 3′ splice site |
| A5′SS | Alternative 5′ splice site |
| MXE | Mutually exclusive exons |
| RI | Retained intron |
| SE | Skipped exon |
| snRNP | Small nuclear ribonucleoprotein |
References
- Fu, X.-D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.-L.; Maquat, L.E. Organizing Principles of Mammalian Nonsense-Mediated mRNA Decay. Annu. Rev. Genet. 2013, 47, 139–165. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-F.; Imam, J.S.; Wilkinson, M.F. The Nonsense-Mediated Decay RNA Surveillance Pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef] [Green Version]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [Green Version]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Kataoka, N. Modulation of aberrant splicing in human RNA diseases by chemical compounds. Hum. Genet. 2017, 136, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Ogawa, S. Splicing factor mutations and cancer: Splicing factor mutations and cancer. Wiley RNA 2014, 5, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrionero, A.; Miñana, B.; Valcárcel, J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011, 25, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, E.H.S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.M.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome Sequencing Reveals Potential Mechanism of Cryptic 3′ Splice Site Selection in SF3B1-mutated Cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef] [Green Version]
- Dolatshad, H.; Pellagatti, A.; Yip, B.H.; Killick, S.; Kušec, R.; Taylor, S.; Mills, K.I.; Savage, K.I.; Smith, C.W.J.; Boultwood, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef] [Green Version]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; De La Grange, P.; Roman-Roman, S.; et al. SF3B1 Mutations Are Associated with Alternative Splicing in Uveal Melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1 K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.L.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, J.L.; Kielkopf, C.L. Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet. 2017, 33, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnal, S.; Vigevani, L.; Valcárcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- Webb, T.R.; Joyner, A.S.; Potter, P.M. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 2013, 18, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Mizui, Y.; Sakai, T.; Iwata, M.; Uenaka, T.; Okamoto, K.; Shimizu, H.; Yamori, T.; Yoshimatsu, K.; Asada, M. Pladienolides, New Substances from Culture of Streptomyces platensis Mer-11107 III. In Vitro and In Vivo Antitumor Activities. J. Antibiot. 2004, 57, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, A.; Kotake, Y.; Mizui, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide: SF3b as an antitumor target of pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Kumar, D.; Burkart, M.D.; Castro, J.E.; Villa, R.; La Clair, J.J.; Benner, C.; Sasik, R.; Jones, H.; Ghia, E.M.; et al. Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 2015, 100, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Convertini, P.; Shen, M.; Potter, P.M.; Palacios, G.; Lagisetti, C.; De La Grange, P.; Horbinski, C.; Fondufe-Mittendorf, Y.N.; Webb, T.R.; Stamm, S. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res. 2014, 42, 4947–4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furumai, R.; Uchida, K.; Komi, Y.; Yoneyama, M.; Ishigami, K.; Watanabe, H.; Kojima, S.; Yoshida, M. Spliceostatin A blocks angiogenesis by inhibiting global gene expression including VEGF. Cancer Sci. 2010, 101, 2483–2489. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Satoh, T.; Takasaki, I.; Kawamura, Y.; Yoshida, M.; Kaida, D. U2 snRNP Is Required for Expression of the 3′ End of Genes. PLoS ONE 2014, 9, e98015. [Google Scholar] [CrossRef]
- Vigevani, L.; Gohr, A.; Webb, T.; Irimia, M.; Valcárcel, J. Molecular basis of differential 3′ splice site sensitivity to anti-tumor drugs targeting U2 snRNP. Nat. Commun. 2017, 8, 2100. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Zheng, X.; Chen, H.; Guo, Y.; Jiang, H.; He, X.; Zhu, X.; Zheng, Y. Splicing function of mitotic regulators links R-loop–mediated DNA damage to tumor cell killing. J. Cell Biol. 2015, 209, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Fan, L.; Edmonson, M.N.; Shaw, T.I.; Boggs, K.; Easton, J.; Rusch, M.; Webb, T.R.; Zhang, J.; Potter, P.M.; et al. Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA 2018, 24, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, R.; Kaida, D.; Furuno, M.; Burroughs, A.M.; Noma, S.; Suzuki, H.; Kawamura, Y.; Hayashizaki, Y.; Mayeda, A.; Yoshida, M. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA 2016, 23, 47–57. [Google Scholar] [CrossRef]
- Jorge, J.; Petronilho, S.; Alves, R.; Coucelo, M.; Gonçalves, A.C.; Costa, J.M.N.; Sarmento-Ribeiro, A.B. Apoptosis induction and cell cycle arrest of pladienolide B in erythroleukemia cell lines. Investig. New Drugs 2020, 38, 369–377. [Google Scholar] [CrossRef]
- Guisbert, K.S.K.; Guisbert, E. SF3B1 is a stress-sensitive splicing factor that regulates both HSF1 concentration and activity. PLoS ONE 2017, 12, e0176382. [Google Scholar] [CrossRef] [Green Version]
- Guisbert, E.; Czyz, D.M.; Richter, K.; McMullen, P.D.; Morimoto, R.I. Identification of a Tissue-Selective Heat Shock Response Regulatory Network. PLoS Genet. 2013, 9, e1003466. [Google Scholar] [CrossRef] [Green Version]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Franceschelli, S.; Rosati, A.; Lerose, R.; De Nicola, S.; Turco, M.C.; Pascale, M. bag3 gene expression is regulated by heat shock factor 1. J. Cell. Physiol. 2008, 215, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Wang, E.T.-S.; Airoldi, E.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, T.D.; Cheng, K.C.-C.; Chen, Y.-C.; Bühler, E.; Martin, S.E.; Inglese, J.; Hogg, J.R. ICE1 promotes the link between splicing and nonsense-mediated mRNA decay. eLife 2018, 7, e33178. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Von Lindern, M.; Fornerod, M.; Van Baal, S.; Jaegle, M.; De Wit, T.; Buijs, A.; Grosveld, G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol. Cell. Biol. 1992, 12, 1687–1697. [Google Scholar] [CrossRef] [Green Version]
- Narain, Y.; Wyttenbach, A.; Rankin, J.; Furlong, R.A.; Rubinsztein, D.C. A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet. 1999, 36, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Wilkinson, M.F. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2012, 3, 807–828. [Google Scholar] [CrossRef]
- Chen, J.; Brunner, A.-D.; Weissman, J.S.; Cogan, J.Z.; Nuñez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; et al. Pervasive functional translation of noncanonical human open reading frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Increased Expression | Reduced Expression | |||
|---|---|---|---|---|
| Pathway | p-Value | Pathway | p-Value | |
| High PB | Nonsense-mediated decay | 9.66 × 10−15 | Monocarboxylic acid transport | 5.89 × 10−4 |
| SRP-dep cotrln protein targeting | 7.28 × 10−15 | Multicellular organismal signaling | 3.71 × 10−4 | |
| Cytoplasmic translation | 5.74 × 10−9 | Action potential | 3.13 × 10−4 | |
| Ribosomal large subunit assembly | 3.98 × 10−4 | |||
| Low PB | Regulation of transcription | 2.9 × 10−12 | Cellular amino acid catabolic process | 3.91 × 10−5 |
| Embryonic skeletal morphogenesis | 3.92 × 10−6 | Export across plasma membrane | 3.52 × 10−4 | |
| Nuclear division | 1.08 × 10−4 | Selective autophagy | 3.4 × 10−4 | |
| Neg regulation of axonogenesis | 1.26 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim Guisbert, K.S.; Mossiah, I.; Guisbert, E. Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology. Int. J. Mol. Sci. 2020, 21, 9641. https://doi.org/10.3390/ijms21249641
Kim Guisbert KS, Mossiah I, Guisbert E. Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology. International Journal of Molecular Sciences. 2020; 21(24):9641. https://doi.org/10.3390/ijms21249641
Chicago/Turabian StyleKim Guisbert, Karen S., Isiah Mossiah, and Eric Guisbert. 2020. "Titration of SF3B1 Activity Reveals Distinct Effects on the Transcriptome and Cell Physiology" International Journal of Molecular Sciences 21, no. 24: 9641. https://doi.org/10.3390/ijms21249641