Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range

 ,
,  ,
,  and
and
Abstract
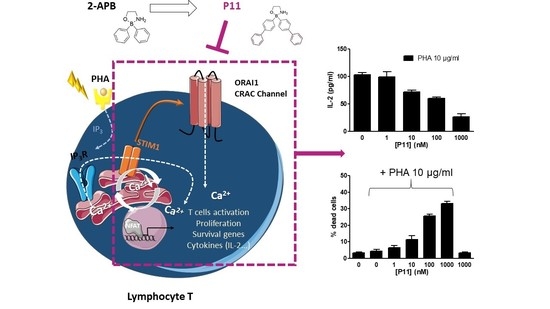
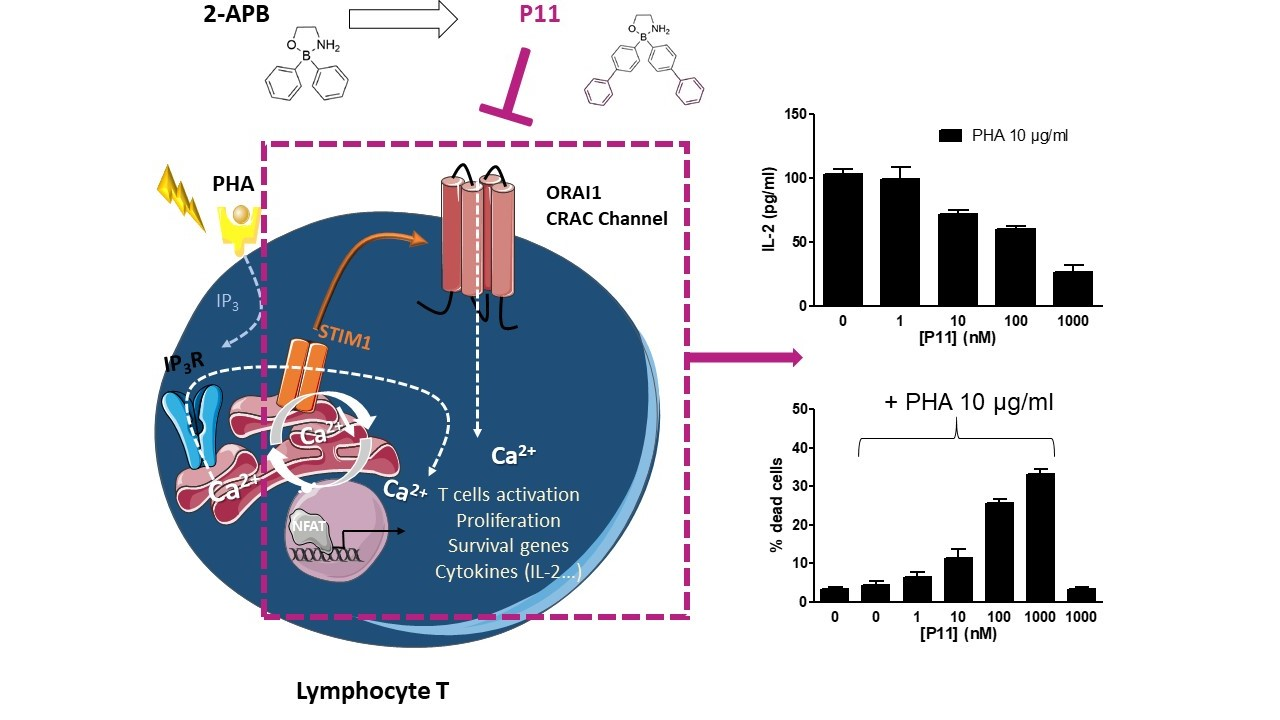
1. Introduction
2. Results
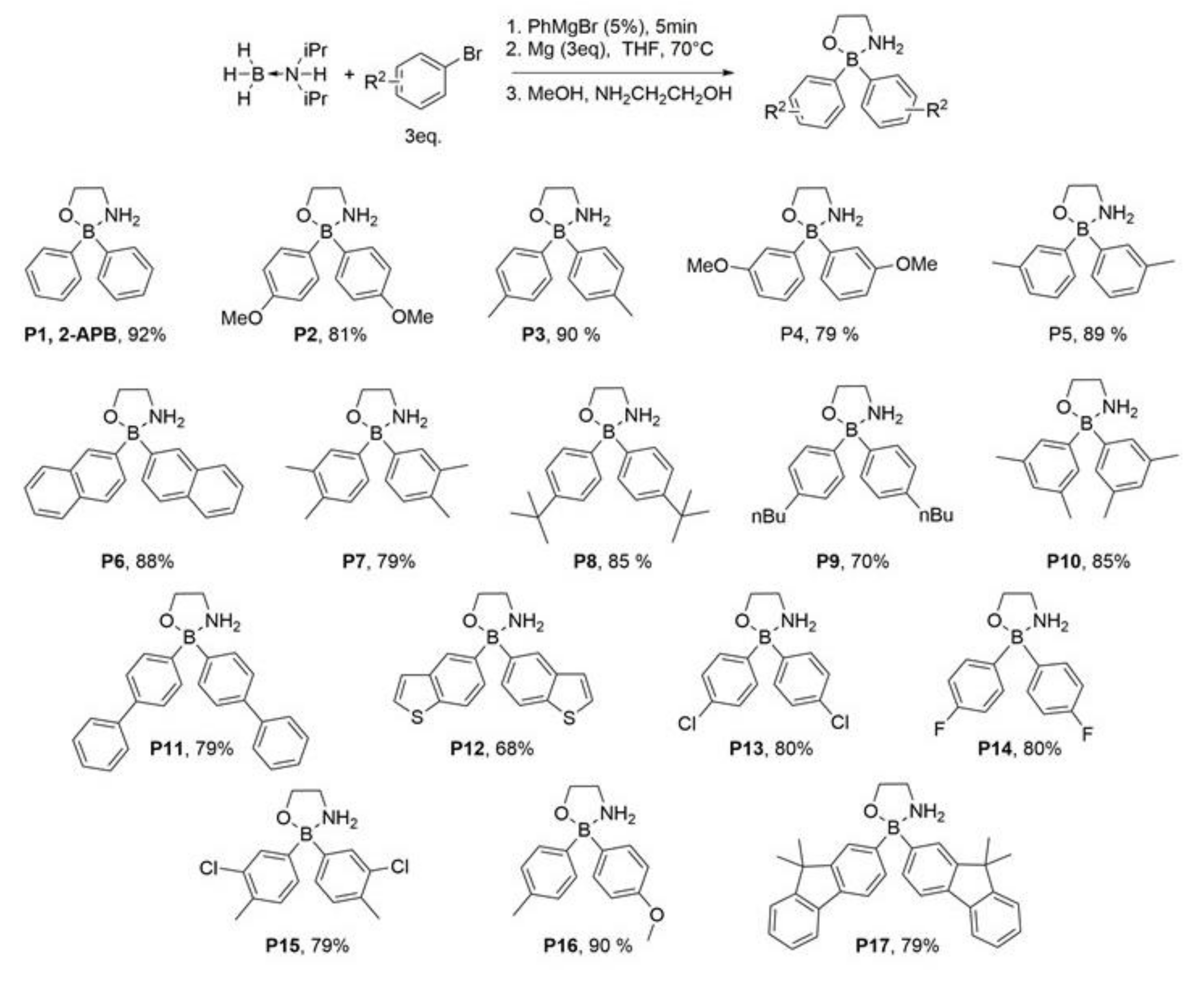
2.1. Addition of Methyl and/or Methoxyl Groups
2.2. Fluorine- and Chlorine-Containing Analogs
2.3. Compounds P8 and P9
2.4. Fused Ring Analogs
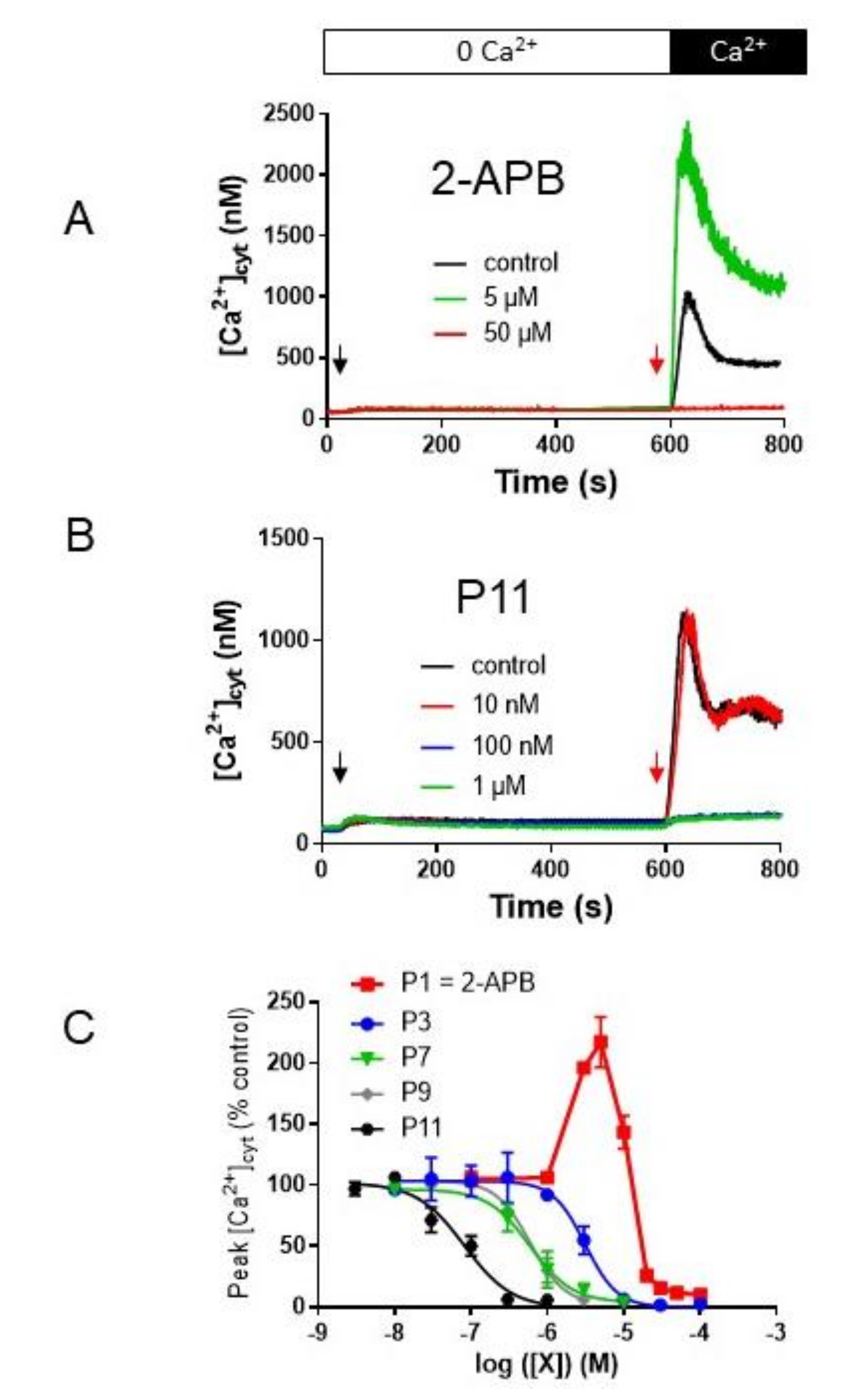
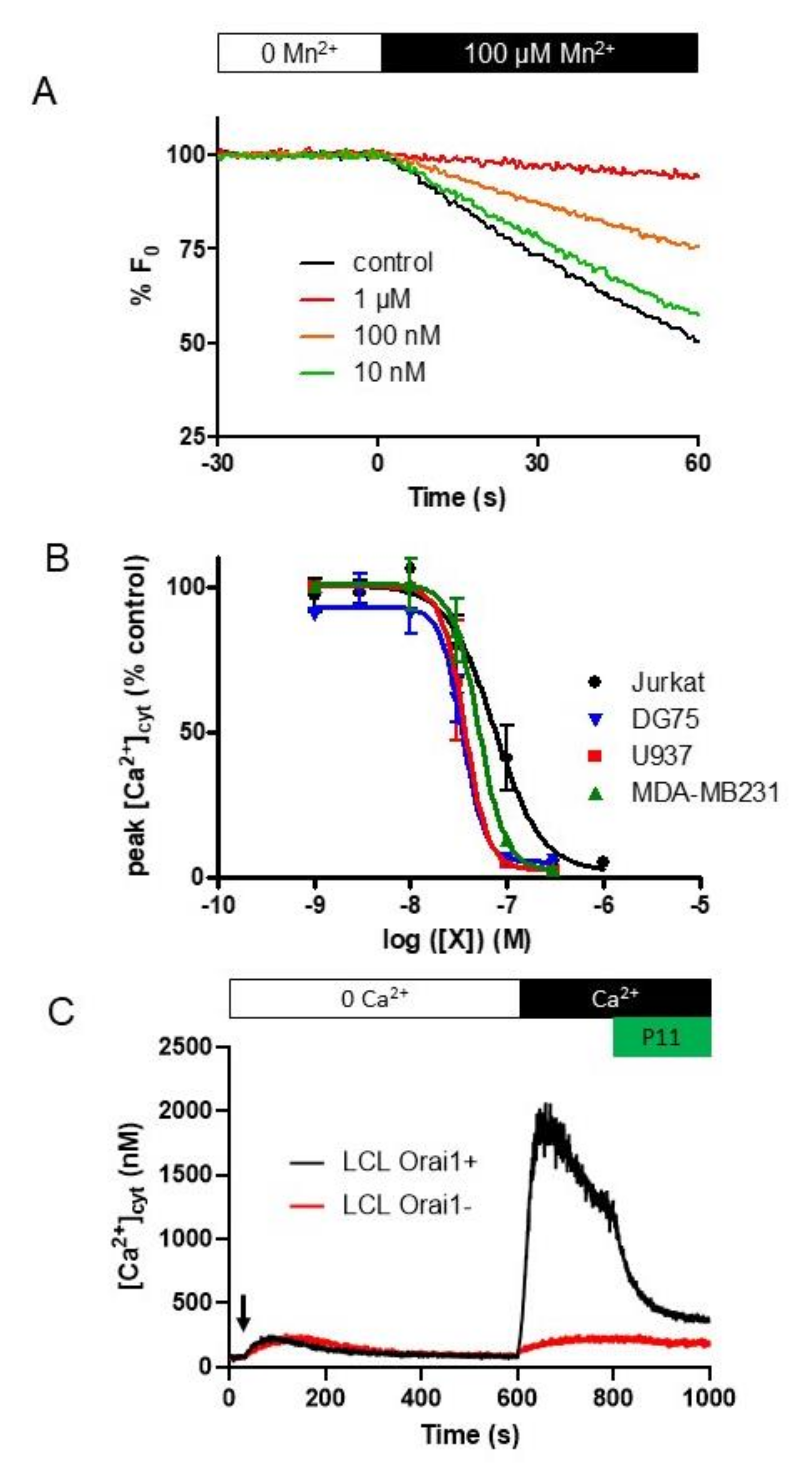
2.5. P11 Specifically Inhibits Store-Operated Calcium Entry (SOCE) but Does Not Affect the Ca2+-Extrusion Mechanisms
2.6. P11 Is Also Able to Block SOCE in Other Cell Types
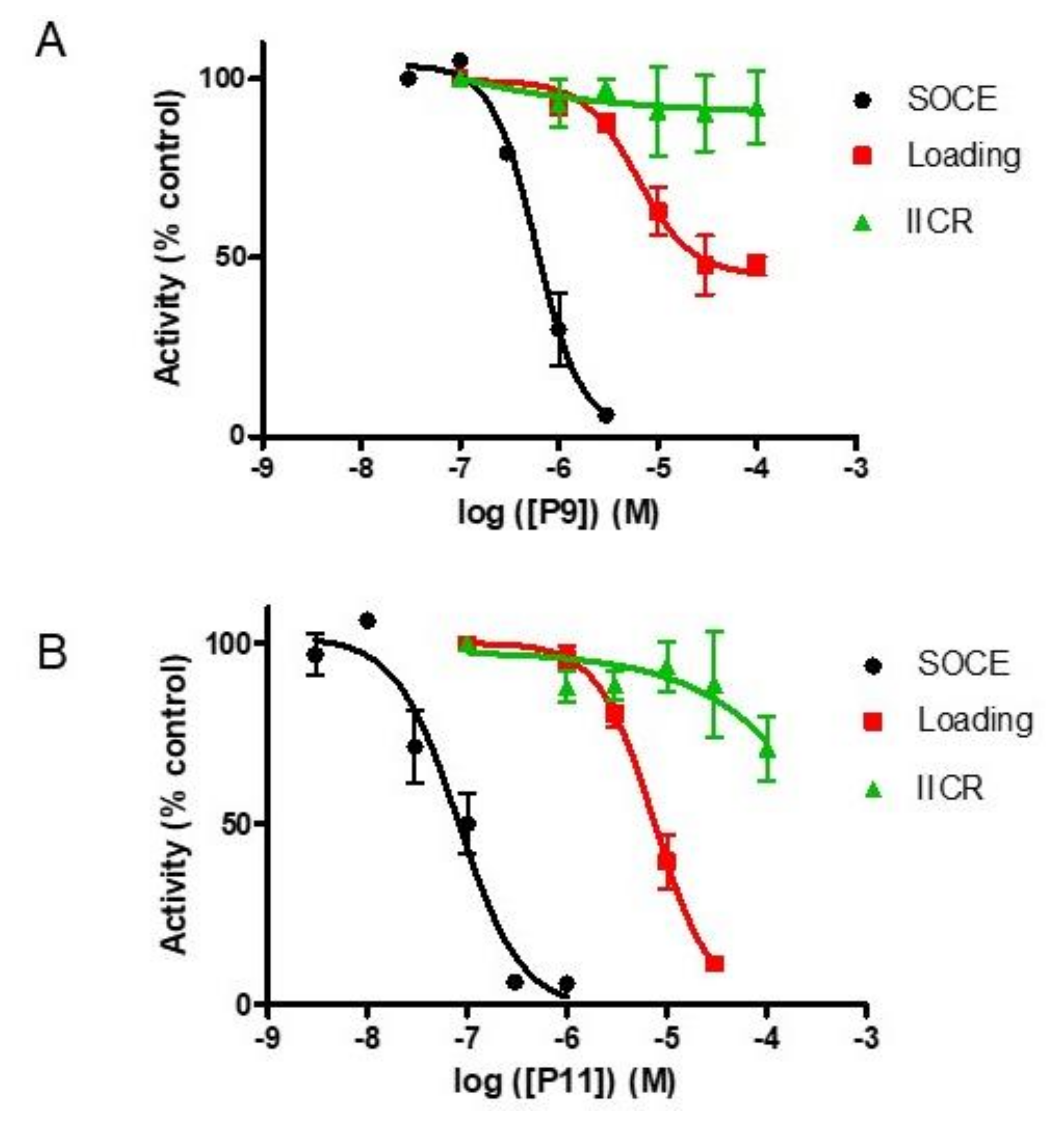
2.7. Selectivity of Compounds P11 and P9
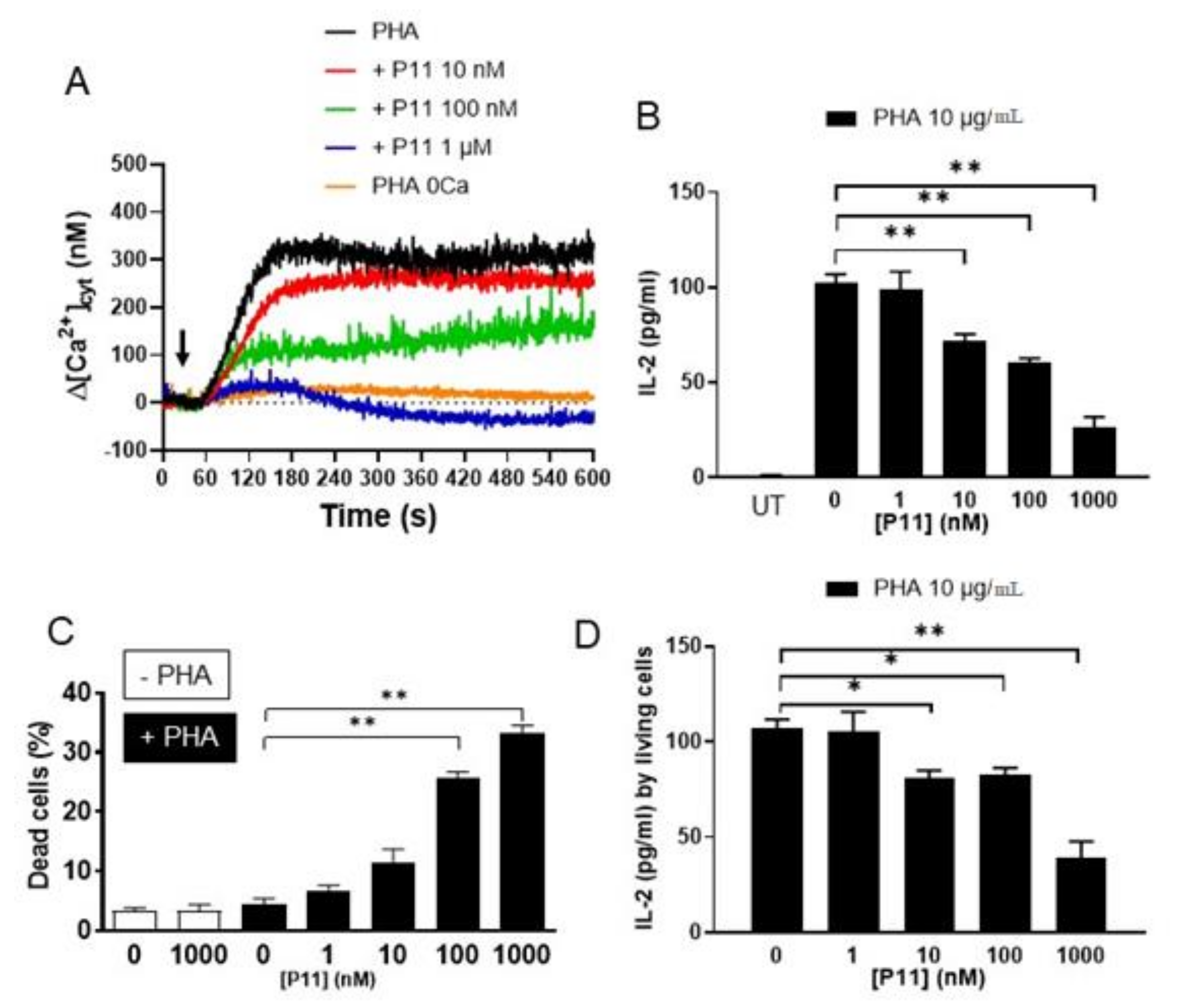
2.8. Compound P11 Impairs the Activation of Jurkat Cells
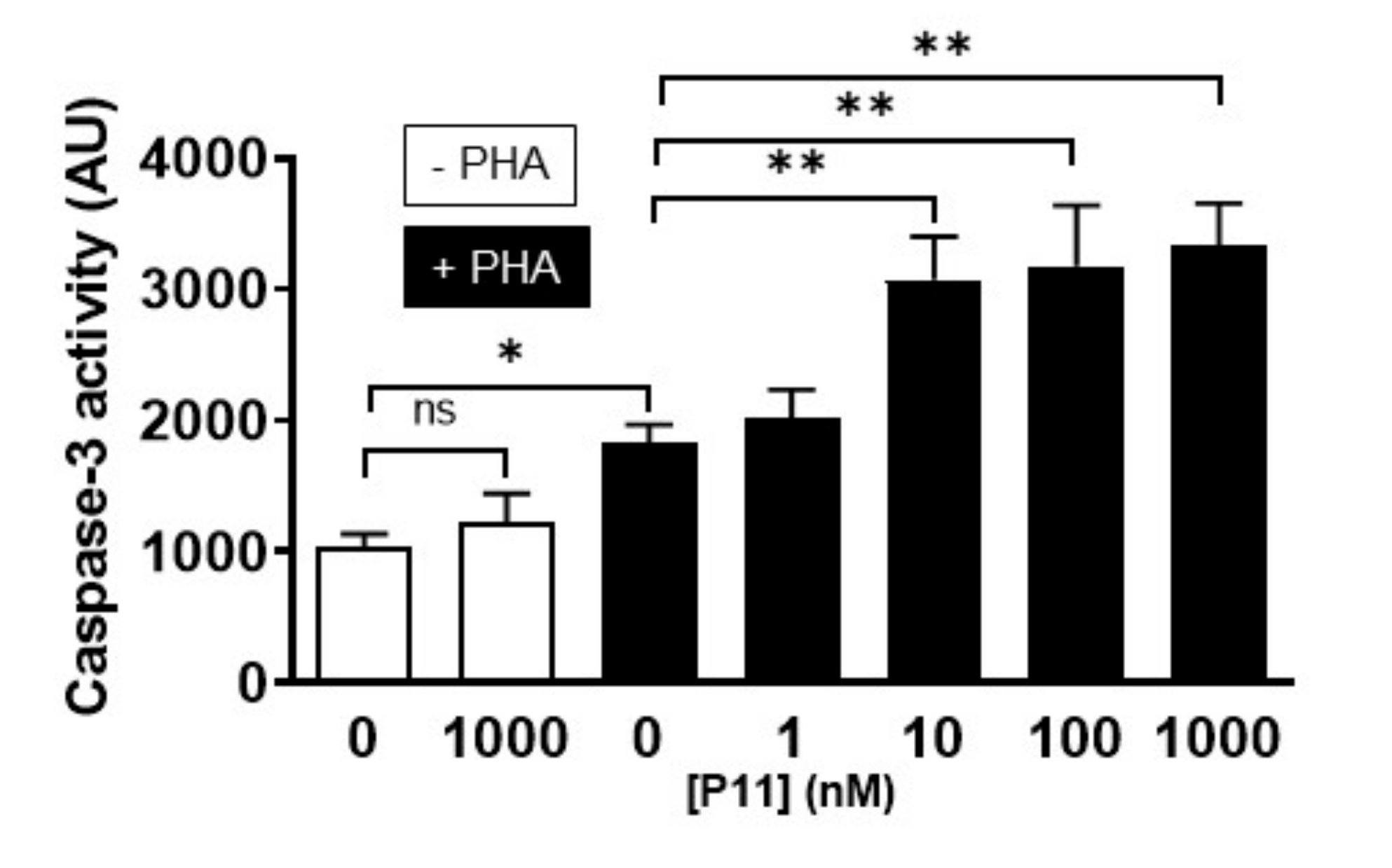
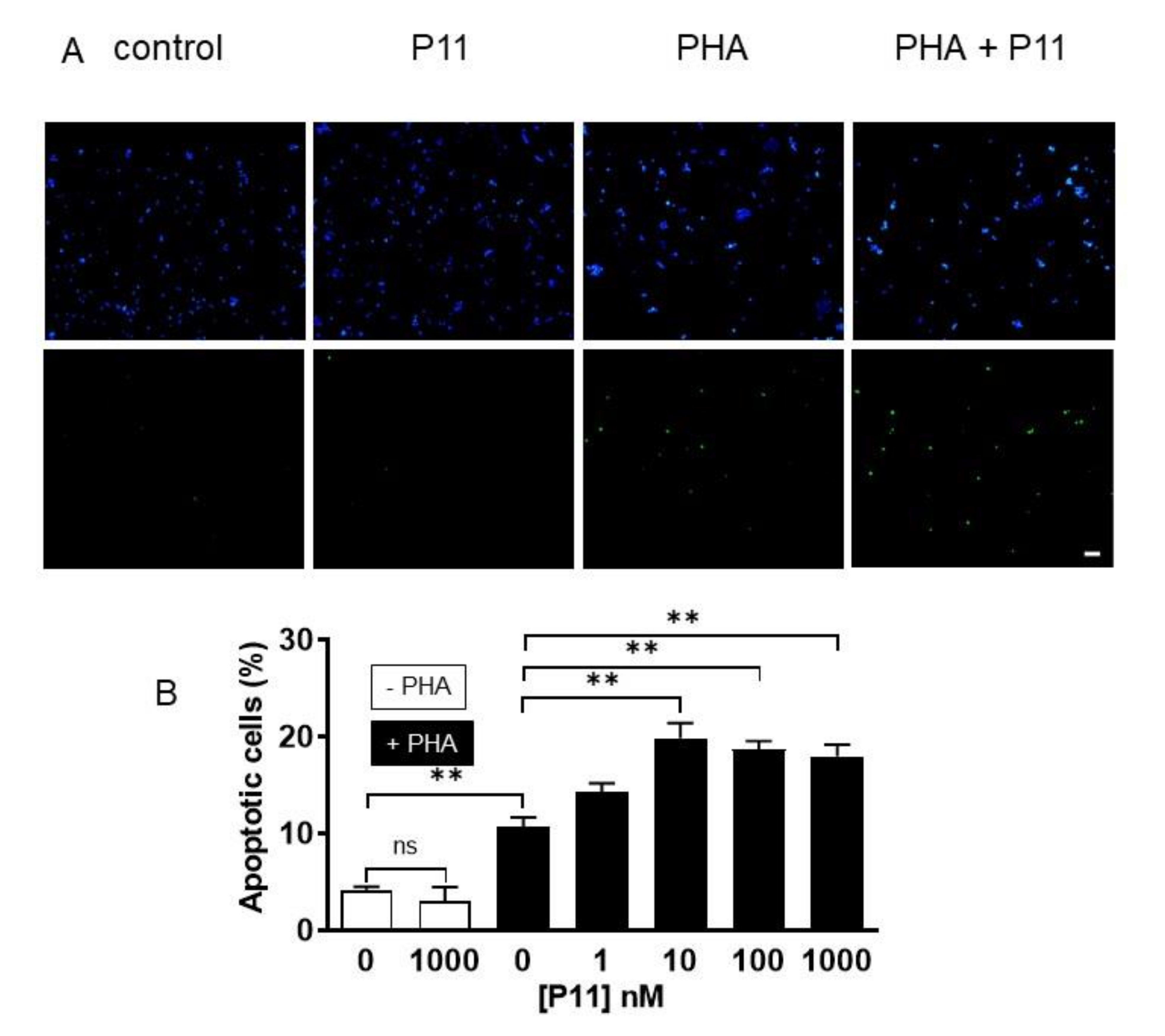
2.9. Compound P11 Induces Apoptosis of PHA-Stimulated Jurkat Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cytosolic Ca2+ Concentration and SOCE Measurement
4.3. Measurement of Mn2+ Influx by Indo-1 Quenching
4.4. Unidirectional 45Ca2+ Flux Experiments
4.5. Interleukin-2 (IL-2) Assay
4.6. Fluorometric Assay for Caspase-3 Activity
4.7. Apoptosis Detection by the Terminal Transferase dUTP Nick End Labelling (TUNEL) Method
4.8. Chemicals
4.9. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-APB | 2-aminoethyl diphenylborinate |
| [Ca2+]cyt | Cytosolic Ca2+ concentration |
| DAPI | di-aminodo Phenyl Indol |
| ELISA | Enzyme Linked Immunosorbent Assay |
| ER | Endoplasmic Reticulum |
| IL-2 | Interleukin-2 |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3R | IP3 receptor |
| Ki | Inhibition constant |
| SOCC | Store-Operated Calcium Channel |
| SOCE | Store-Operated Calcium Entry |
| TG | Thapsigargin |
| TUNEL | Terminal Uridine Nick-End Labeling |
| SOCC | Store-Operated Calcium Channel |
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.J.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nat. Cell Biol. 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Pharmacology of Store-operated Calcium Channels. Mol. Interv. 2010, 10, 209–218. [Google Scholar] [CrossRef]
- Dobrydneva, Y.; Abelt, C.J.; Dovel, B.; Thadigiri, C.M.; Williams, R.L.; Blackmore, P.F. 2-Aminoethoxydiphenyl Borate as a Prototype Drug for a Group of Structurally Related Calcium Channel Blockers in Human Platelets. Mol. Pharmacol. 2005, 69, 247–256. [Google Scholar] [CrossRef]
- Goto, J.-I.; Suzuki, A.Z.; Ozaki, S.; Matsumoto, N.; Nakamura, T.; Ebisui, E.; Fleig, A.; Penner, R.; Mikoshiba, K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca2+ entry via STIM proteins. Cell Calcium. 2010, 47, 1–10. [Google Scholar] [CrossRef]
- Djillani, A.; Nüße, O.; Dellis, O. Characterization of novel store-operated calcium entry effectors. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2014, 1843, 2341–2347. [Google Scholar] [CrossRef][Green Version]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 2001, 536, 3–19. [Google Scholar] [CrossRef]
- Djillani, A.; Doignon, I.; Luyten, T.; Lamkhioued, B.; Gangloff, S.C.; Parys, J.B.; Nüße, O.; Chomienne, C.; Dellis, O. Potentiation of the store-operated calcium entry (SOCE) induces phytohemagglutinin-activated Jurkat T cell apoptosis. Cell Calcium. 2015, 58, 171–185. [Google Scholar] [CrossRef]
- Bittremieux, M.; Gerasimenko, J.V.; Schuermans, M.; Luyten, T.; Stapleton, E.; Alzayady, K.J.; De Smedt, H.; Yule, D.I.; Mikoshiba, K.; Vangheluwe, P.; et al. DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium. 2017, 62, 60–70. [Google Scholar] [CrossRef]
- Schild, A.; Bhardwaj, R.; Wenger, N.; Tscherrig, D.; Kandasamy, P.; Dernič, J.; Baur, R.; Peinelt, C.; Hediger, M.A.; Lochner, M. Synthesis and Pharmacological Characterization of 2-Aminoethyl Diphenylborinate (2-APB) Derivatives for Inhibition of Store-Operated Calcium Entry (SOCE) in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5604. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-T.; Venkatachalam, K.; Parys, J.B.; Gill, D.L. Modification of Store-operated Channel Coupling and Inositol Trisphosphate Receptor Function by 2-Aminoethoxydiphenyl Borate in DT40 Lymphocytes. J. Biol. Chem. 2001, 277, 6915–6922. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Collins, T.J.; Mackenzie, L.; Roderick, H.L.; Berridge, M.J.; Peppiatt, C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002, 16, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, L.; Callewaert, G.; De Smedt, H.; Parys, J.B. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+pump and the non-specific Ca2+leak from the non-mitochondrial Ca2+stores in permeabilized A7r5 cells. Cell Calcium. 2001, 29, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Collins, T.J.; MacKenzie, L.; Conway, S.J.; Holmes, A.B.; Bootman, M.D.; Berridge, M.J.; Seo, J.T.; Roderick, H. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium. 2003, 34, 97–108. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W. Complex Actions of 2-Aminoethyldiphenyl Borate on Store-operated Calcium Entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef]
- Peinelt, C.; Lis, A.; Beck, A.; Fleig, A.; Penner, R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 2008, 586, 3061–3073. [Google Scholar] [CrossRef]
- Zhang, S.L.; Kozak, J.A.; Jiang, W.; Yeromin, A.V.; Chen, J.; Yu, Y.; Penna, A.; Shen, W.; Chi, V.; Cahalan, M.D. Store-dependent and -independent Modes Regulating Ca2+Release-activated Ca2+Channel Activity of Human Orai1 and Orai3. J. Biol. Chem. 2008, 283, 17662–17671. [Google Scholar] [CrossRef]
- Rosenwasser, L. Faculty Opinions recommendation of ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2010, 124, 1311–1318. [Google Scholar]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-Aminoethoxydiphenyl Borate, a Membrane-Penetrable Modulator of Ins(1,4,5)P3-Induced Ca2+ Release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef]
- Trebak, M.; Bird, G.S.; McKay, R.R.; Putney, J.W., Jr. Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J Biol Chem. 2002, 277, 21617–21623. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.C.; McDonald, T.V.; Gardner, P. Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br. J. Pharmacol. 1994, 113, 8618–8668. [Google Scholar]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Futur. Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nat. Cell Biol. 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Feske, S.; Picard, C.; Fischer, A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin. Immunol. 2010, 135, 169–182. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations inORAI1andSTIM1. Ann. New York Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Jani, P.K.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3:Selective requirement of Orai3 versus Orai1 in estrogen receptor positive versus estrogen receptor negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nat. Cell Biol. 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Dickson, E.J.; Duman, J.G.; Moody, M.W.; Chen, L.; Hille, B. Orai-STIM-mediated Ca2+ release from secretory granules revealed by a targeted Ca2+ and pH probe. Proc. Natl. Acad. Sci. USA 2012, 109, E3539–E3548. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Tanaka, K.; Smalley, J.L.; Bampton, E.T.W.; Pellecchia, M.; Dinsdale, D.; Willars, G.B.; Cohen, G.M. Endoplasmic Reticulum Membrane Reorganization Is Regulated by Ionic Homeostasis. PLoS ONE 2013, 8, e56603. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, M.; Yamanoue, K.; Mochizuki, Y.; Konishi, H.; Tadokoro, S.; Tanaka, M.; Suzuki, R.; Hirashima, N. Orai-2 is localized on secretory granules and regulates antigen-evoked Ca2+ mobilization and exocytosis in mast cells. Biochem. Biophys. Res. Commun. 2014, 451, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.; Lokteva, L.A.; Lewis, R.S. Functional Analysis of Orai1 Concatemers Supports a Hexameric Stoichiometry for the CRAC Channel. Biophys. J. 2016, 111, 1897–1907. [Google Scholar] [CrossRef]
- Cai, X.; Zhou, Y.; Nwokonko, R.M.; Loktionova, N.A.; Wang, X.; Xin, P.; Trebak, M.; Wang, Y.; Gill, D.L. The Orai1 Store-operated Calcium Channel Functions as a Hexamer. J. Biol. Chem. 2016, 291, 25764–25775. [Google Scholar] [CrossRef]
- Dellis, O.; Mercier, P.; Chomienne, C. The boron-oxygen core of borinate esters is responsible for the store-operated calcium entry potentiation ability. BMC Pharmacol. 2011, 11, 1. [Google Scholar] [CrossRef]
- Scrimgeour, N.; Litjens, T.; Ma, L.; Barritt, G.J.; Rychkov, G.Y. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J. Physiol. 2009, 587, 2903–2918. [Google Scholar] [CrossRef]
- Liao, Y.; Erxleben, C.; Abramowitz, J.; Flockerzi, V.; Zhu, M.X.; Armstrong, D.L.; Birnbaumer, L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2895–2900. [Google Scholar] [CrossRef]
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflügers Archiv. Eur. J. Physiol. 2018, 470, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Furuichi, T.; Maeda, N.; Mikoshiba, K. Expressed cerebellar-type inositol 1,4,5-trisphosphate receptor, P400, has calcium release activity in a fibroblast L cell line. Neuron 1990, 5, 11–18. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Doignon, I.; Fayol, O.; Dellis, O. Improvement of the rituximab–induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Oncotarget 2019, 10, 4466–4478. [Google Scholar] [CrossRef][Green Version]
- Dellis, O.; Gangloff, S.C.; Paulais, M.; Tondelier, D.; Rona, J.-P.; Brouillard, F.; Bouteau, F.; Guenounou, M.; Teulon, J. Inhibition of the Calcium Release-activated Calcium (CRAC) Current in Jurkat T Cells by the HIV-1 Envelope Protein gp160. J. Biol. Chem. 2002, 277, 6044–6050. [Google Scholar] [CrossRef]
- Brooks, S.; Storey, K. Bound and determined: A computer program for making buffers of defined ion concentrations. Anal. Biochem. 1992, 201, 119–126. [Google Scholar] [CrossRef]
- Luyten, T.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Measurement of intracellular Ca2+ release in permeabilized cells using 45Ca2+. Cold Spring Harb. Protoc. 2014, 2014, 289–294. [Google Scholar] [CrossRef]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-Trisphosphate and Its Receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar]
- Pinet, S.; Pucheault, M.; Richard, J.; Birepinte, M.; Charbonnier, J.B.; Liautard, V. Borinic Acids via Direct Arylation of Amine–Borane Complexes: An Air- and Water-Stable Boron Source. Synthesis 2016, 49, 736–744. [Google Scholar] [CrossRef][Green Version]
- Zhou, H.; Iwasaki, H.; Nakamura, T.; Nakamura, K.; Maruyama, T.; Hamano, S.-I.; Ozaki, S.; Mizutani, A.; Mikoshiba, K. 2-Aminoethyl diphenylborinate analogues: Selective inhibition for store-operated Ca2+ entry. Biochem. Biophys. Res. Commun. 2007, 352, 277–282. [Google Scholar] [CrossRef]
- Ohane, K.; Hayase, Y. Nematicide for Plant Containing Organoboron Compound. JP2002322007A, 8 November 2002. [Google Scholar]
- Mikoshiba, K.; Iwasaki, H.; Maruyama, T.; Hamano, S.-I. Intracellular Calcium Concentration Increase Inhibitors. WO2003033002A1, 24 April 2003. [Google Scholar]
- Ozaki, S.; Suzuki, A.Z.; Bauer, P.O.; Ebisui, E.; Mikoshiba, K. 2-Aminoethyl diphenylborinate (2-APB) analogues: Regulation of Ca2+ signaling. Biochem. Biophys. Res. Commun. 2013, 441, 286–290. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (nM) | Compound | Ki (µM) |
|---|---|---|---|
| P11 | 75 ± 21 | P13 | 1.8 ± 0.1 |
| P6 | 275 ± 17 | P5 | 2.1 ± 0.6 |
| P15 | 294 ± 74 | P16 | 3.1 ± 0.6 |
| P8 | 350 ± 40 | P3 | 3.5 ± 0.6 |
| P12 | 374 ± 96 | P2 | 3.5 ± 0.65 |
| Dibenzothienyl-APB [7] | 405 ± 23 | P14 | 5.4 ± 0.3 |
| P10 | 484 ± 116 | P4 | 75 ± 21 |
| P9 | 641 ± 103 | P17 | >> 100 |
| P7 | 751 ± 97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Guilcher, C.; Luyten, T.; Parys, J.B.; Pucheault, M.; Dellis, O. Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. Int. J. Mol. Sci. 2020, 21, 9777. https://doi.org/10.3390/ijms21249777
Le Guilcher C, Luyten T, Parys JB, Pucheault M, Dellis O. Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. International Journal of Molecular Sciences. 2020; 21(24):9777. https://doi.org/10.3390/ijms21249777
Chicago/Turabian StyleLe Guilcher, Camille, Tomas Luyten, Jan B. Parys, Mathieu Pucheault, and Olivier Dellis. 2020. "Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range" International Journal of Molecular Sciences 21, no. 24: 9777. https://doi.org/10.3390/ijms21249777
APA StyleLe Guilcher, C., Luyten, T., Parys, J. B., Pucheault, M., & Dellis, O. (2020). Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. International Journal of Molecular Sciences, 21(24), 9777. https://doi.org/10.3390/ijms21249777