Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity



Abstract
1. Introduction
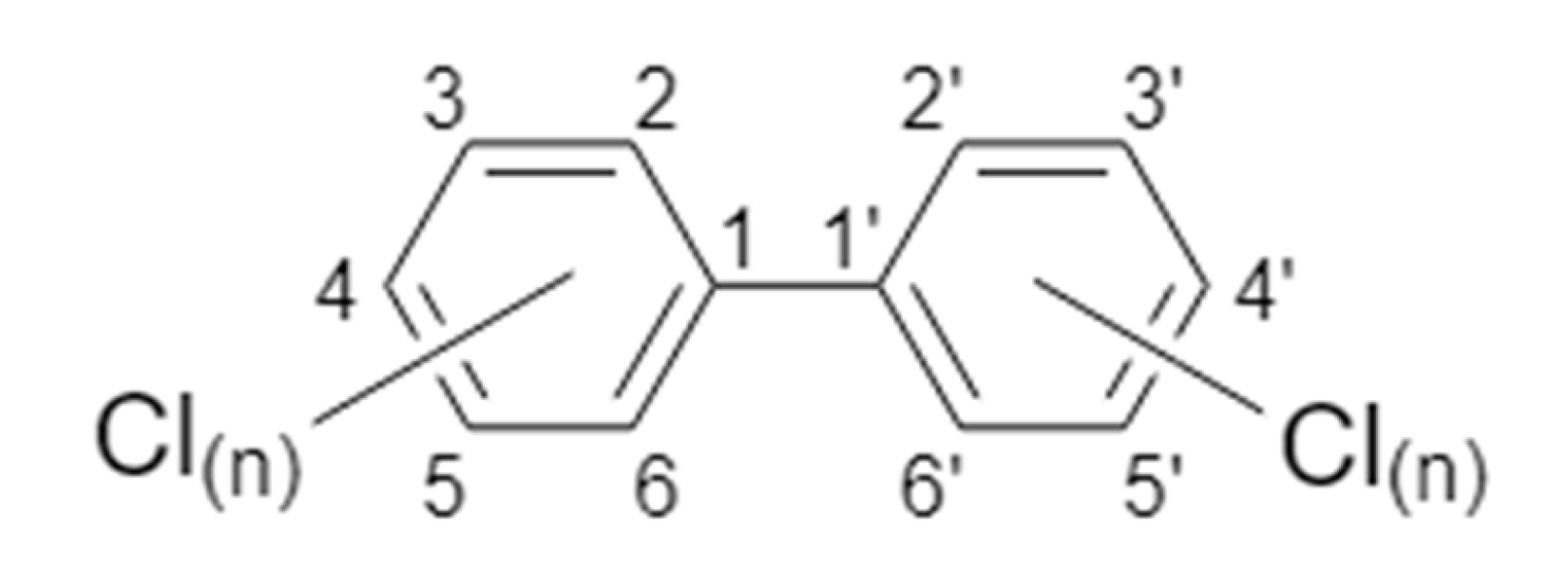
2. PCB Nomenclature and Classifications
3. Neurobehavioral Studies of PCB Developmental Neurotoxicity
3.1. Hyperactivity
3.2. Social Deficits
3.3. Cognitive Impairment and Executive Dysfunction
4. Neurodevelopmental Processes Altered by PCBs
4.1. PCB Effects on Neuronal Apoptosis
4.2. PCB Effects on Axonal and Dendritic Morphogenesis
5. Molecular Mechanisms of PCB Developmental Neurotoxicity
5.1. Arylhydrocarbon Receptor (AhR) as a Molecular Target in PCB Developmental Neurotoxicity
5.2. Thyroid Hormone-Mediated Mechanisms of PCB Developmental Neurotoxicity
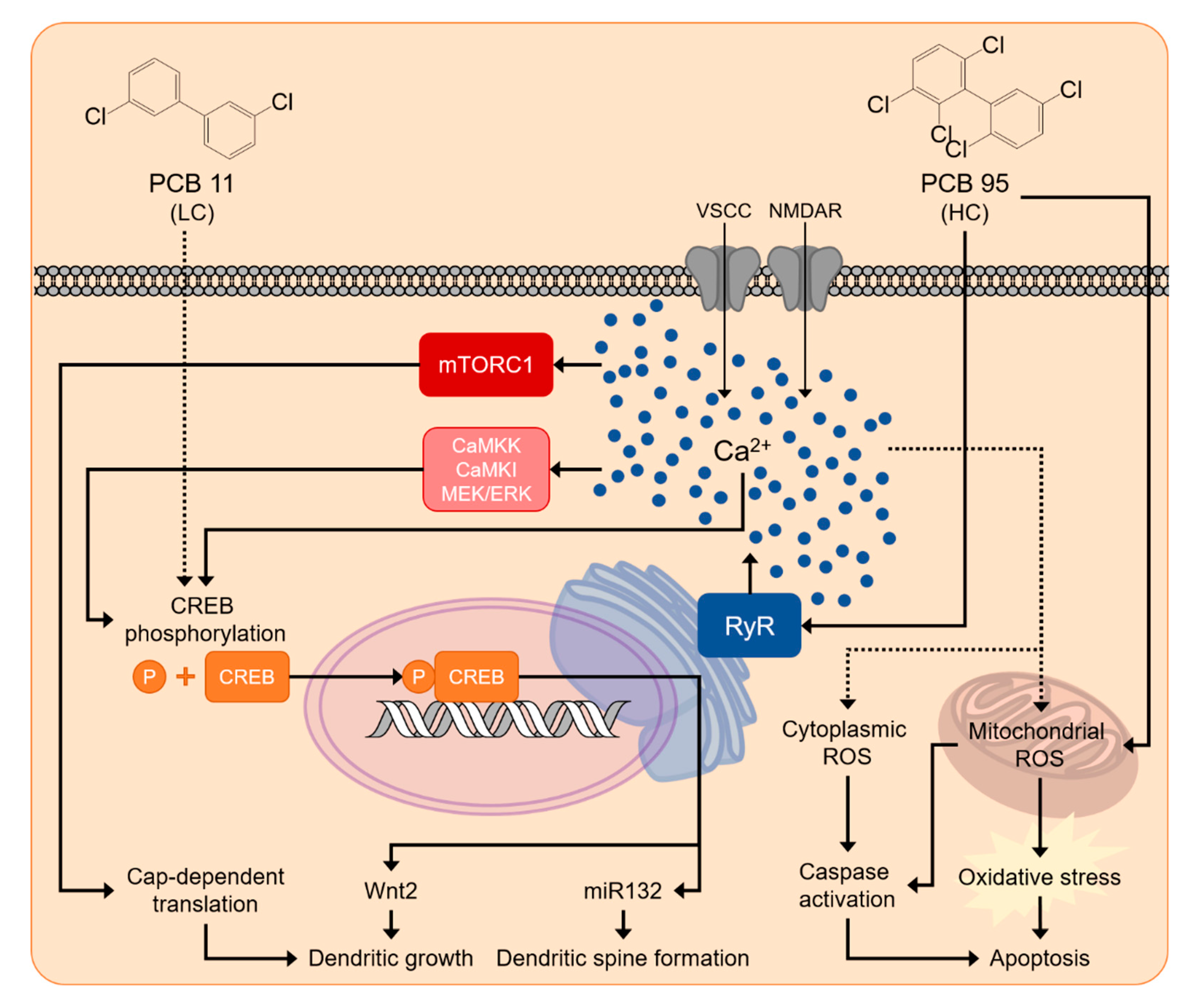
5.3. Calcium Signaling
5.4. Increased ROS as a Mechanism of PCB Developmental Neurotoxicity
6. Emerging Evidence that Non-Legacy LC-PCBs Interfere with Typical Neurodevelopment
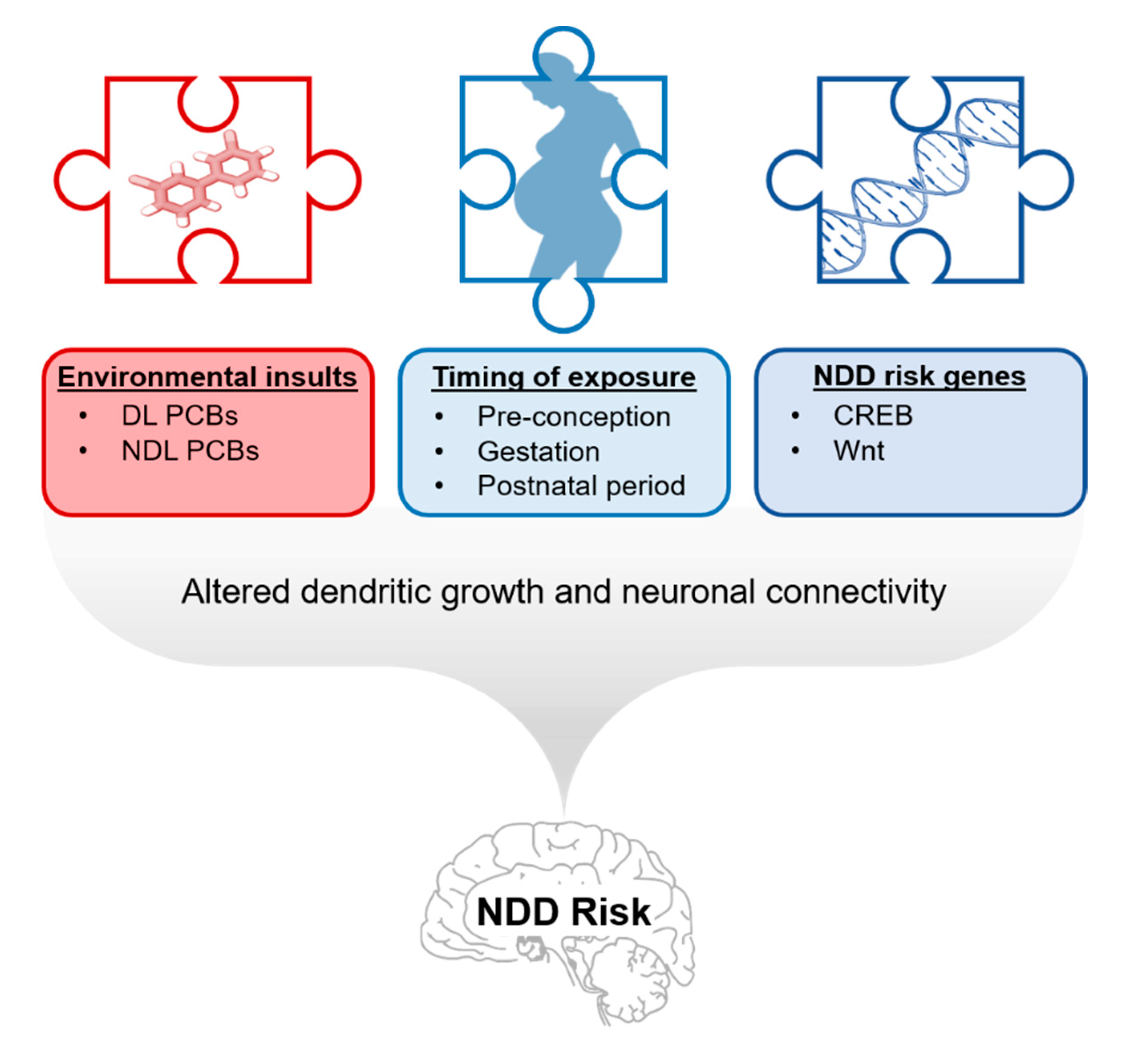
7. Relevance of PCB Developmental Neurotoxicity to NDDs
8. Conclusions, Data Gaps, and Directions for Future Study
Funding
Conflicts of Interest
Abbreviations
| ABR | Auditory brain stem response |
| ADHD | Attention-deficit hyperactivity disorder |
| AhR | Arylhydrocarbon receptor |
| ASD | Autism spectrum disorder |
| bHLH/PAS | Basic helix-loop-helix/Per-Arnt-Sim |
| CREB | cAMP response element-binding protein |
| DL | Dioxin-like |
| DNT | Developmental neurotoxicity |
| DRL | Differential reinforcement of low-rate |
| DSA | Delayed spatial alternation |
| EPM | Elevated plus maze |
| GD | Gestational day |
| 4-HNE | 4-Hydroxynonenal |
| GWAS | Genome-wide association study |
| LDB | Light-dark box |
| MWM | Morris water maze |
| NDD | Neurodevelopmental disorder |
| NDL | Non-dioxin-like |
| NOR | Novel object recognition |
| 3-NT | 3-Nitrotyrosine |
| PCB | Polychlorinated biphenyl |
| PKC | Protein kinase C |
| PND | Postnatal day |
| POPs | Persistent organic pollutants |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SAR | Structure activity relationship |
| SNP | Single nucleotide polymorphism |
| T4 | Thyroxine |
| TCDD | 2,3,7,8-Tetrachlorodibenzo-p-dioxin |
| TH | Thyroid hormone |
| THR | Thyroid hormone receptor |
| TUNEL | Terminal deoxynucelotidyl transferase nick-end labeling |
| U.S. | United States |
References
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Polychlorinated Biphenyls (PCBs). Available online: https://www.atsdr.cdc.gov/toxprofiles/tp17.pdf (accessed on 10 January 2020).
- Secretariat. Protecting Human Health and the Environment from Persistent Organic Pollutants. Available online: http://chm.pops.int/Home/tabid/2121/mctl/ViewDetails/EventModID/1126/EventID/468/xmid/6922/Default.aspx (accessed on 1 June 2019).
- Secretariat. The New POPs under the Stockholm Convention. Available online: http://chm.pops.int/TheConvention/ThePOPs/TheNewPOPs/tabid/2511/Default.aspx (accessed on 1 June 2019).
- Consonni, D.; Sindaco, R.; Bertazzi, P.A. Blood levels of dioxins, furans, dioxin-like PCBs, and TEQs in general populations: A review, 1989–2010. Environ. Int. 2012, 44, 151–162. [Google Scholar] [CrossRef]
- Hopf, N.B.; Ruder, A.M.; Succop, P. Background levels of polychlorinated biphenyls in the U.S. population. Sci. Total Environ. 2009, 407, 6109–6119. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.X.; Hornbuckle, K.C.; Thorne, P.S. Human Serum from Urban and Rural Adolescents and Their Mothers Shows Exposure to Polychlorinated Biphenyls Not Found in Commercial Mixtures. Environ. Sci. Technol. 2015, 49, 8105–8112. [Google Scholar] [CrossRef] [PubMed]
- Hornbuckle, K.; Robertson, L. Polychlorinated biphenyls (PCBs): Sources, exposures, toxicities. Environ. Sci. Technol. 2010, 44, 2749–2751. [Google Scholar] [CrossRef]
- Grimm, F.A.; Hu, D.; Kania-Korwel, I.; Lehmler, H.J.; Ludewig, G.; Hornbuckle, K.C.; Duffel, M.W.; Bergman, A.; Robertson, L.W. Metabolism and metabolites of polychlorinated biphenyls. Crit. Rev. Toxicol. 2015, 45, 245–272. [Google Scholar] [CrossRef]
- Basu, I.; Arnold, K.A.; Venier, M.; Hites, R.A. Partial pressures of PCB-11 in air from several Great Lakes sites. Environ. Sci. Technol. 2009, 43, 6488–6492. [Google Scholar] [CrossRef]
- Choi, S.D.; Baek, S.Y.; Chang, Y.S.; Wania, F.; Ikonomou, M.G.; Yoon, Y.J.; Park, B.K.; Hong, S. Passive air sampling of polychlorinated biphenyls and organochlorine pesticides at the Korean Arctic and Antarctic research stations: Implications for long-range transport and local pollution. Environ. Sci. Technol. 2008, 42, 7125–7131. [Google Scholar] [CrossRef]
- Hu, D.; Hornbuckle, K.C. Inadvertent polychlorinated biphenyls in commercial paint pigments. Environ. Sci. Technol. 2010, 44, 2822–2827. [Google Scholar] [CrossRef]
- Jahnke, J.C.; Hornbuckle, K.C. PCB Emissions from Paint Colorants. Environ. Sci. Technol. 2019, 53, 5187–5194. [Google Scholar] [CrossRef]
- Sethi, S.; Keil, K.P.; Chen, H.; Hayakawa, K.; Li, X.; Lin, Y.; Lehmler, H.J.; Puschner, B.; Lein, P.J. Detection of 3,3’-Dichlorobiphenyl in Human Maternal Plasma and Its Effects on Axonal and Dendritic Growth in Primary Rat Neurons. Toxicol. Sci. 2017, 158, 401–411. [Google Scholar] [CrossRef]
- Pessah, I.N.; Lein, P.J.; Seegal, R.F.; Sagiv, S.K. Neurotoxicity of polychlorinated biphenyls and related organohalogens. Acta Neuropathol. 2019, 138, 363–387. [Google Scholar] [CrossRef]
- Berghuis, S.A.; Bos, A.F.; Sauer, P.J.; Roze, E. Developmental neurotoxicity of persistent organic pollutants: An update on childhood outcome. Arch. Toxicol. 2015, 89, 687–709. [Google Scholar] [CrossRef]
- Schantz, S.L.; Widholm, J.J.; Rice, D.C. Effects of PCB exposure on neuropsychological function in children. Environ. Health Perspect. 2003, 111, 357–576. [Google Scholar] [CrossRef]
- Boucher, O.; Muckle, G.; Bastien, C.H. Prenatal exposure to polychlorinated biphenyls: A neuropsychologic analysis. Environ. Health Perspect. 2009, 117, 7–16. [Google Scholar] [CrossRef]
- Cheslack-Postava, K.; Rantakokko, P.V.; Hinkka-Yli-Salomaki, S.; Surcel, H.M.; McKeague, I.W.; Kiviranta, H.A.; Sourander, A.; Brown, A.S. Maternal serum persistent organic pollutants in the Finnish Prenatal Study of Autism: A pilot study. Neurotoxicol. Teratol. 2013, 38, 1–5. [Google Scholar] [CrossRef]
- Granillo, L.; Sethi, S.; Keil, K.P.; Lin, Y.; Ozonoff, S.; Iosif, A.M.; Puschner, B.; Schmidt, R.J. Polychlorinated biphenyls influence on autism spectrum disorder risk in the MARBLES cohort. Environ. Res. 2019, 171, 177–184. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.A.; Sjodin, A.; Yoshida, C.K.; Zerbo, O.; Kharrazi, M.; Windham, G.C. Polychlorinated Biphenyl and Organochlorine Pesticide Concentrations in Maternal Mid-Pregnancy Serum Samples: Association with Autism Spectrum Disorder and Intellectual Disability. Environ. Health Perspect. 2017, 125, 474–480. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Ye, B.S.; Leung, A.O.W.; Wong, M.H. The association of environmental toxicants and autism spectrum disorders in children. Environ. Pollut. 2017, 227, 234–242. [Google Scholar] [CrossRef]
- De Cock, M.; Maas, Y.G.; van de Bor, M. Does perinatal exposure to endocrine disruptors induce autism spectrum and attention deficit hyperactivity disorders? Review. Acta Paediatr. 2012, 101, 811–818. [Google Scholar] [CrossRef]
- Eubig, P.A.; Aguiar, A.; Schantz, S.L. Lead and PCBs as risk factors for attention deficit/hyperactivity disorder. Environ. Health Perspect. 2010, 118, 1654–1667. [Google Scholar] [CrossRef]
- Rosenquist, A.H.; Hoyer, B.B.; Julvez, J.; Sunyer, J.; Pedersen, H.S.; Lenters, V.; Jonsson, B.A.G.; Bonde, J.P.; Toft, G. Prenatal and Postnatal PCB-153 and p,p’-DDE Exposures and Behavior Scores at 5-9 Years of Age among Children in Greenland and Ukraine. Environ. Health Perspect. 2017, 125, 107002. [Google Scholar] [CrossRef]
- Sagiv, S.K.; Thurston, S.W.; Bellinger, D.C.; Tolbert, P.E.; Altshul, L.M.; Korrick, S.A. Prenatal organochlorine exposure and behaviors associated with attention deficit hyperactivity disorder in school-aged children. Am. J. Epidemiol. 2010, 171, 593–601. [Google Scholar] [CrossRef]
- Stewart, P.W.; Lonky, E.; Reihman, J.; Pagano, J.; Gump, B.B.; Darvill, T. The relationship between prenatal PCB exposure and intelligence (IQ) in 9-year-old children. Environ. Health Perspect. 2008, 116, 1416–1422. [Google Scholar] [CrossRef]
- Ikeno, T.; Miyashita, C.; Nakajima, S.; Kobayashi, S.; Yamazaki, K.; Saijo, Y.; Kita, T.; Sasaki, S.; Konishi, K.; Kajiwara, J.; et al. Effects of low-level prenatal exposure to dioxins on cognitive development in Japanese children at 42 months. Sci. Total Environ. 2018, 618, 1423–1430. [Google Scholar] [CrossRef]
- Kyriklaki, A.; Vafeiadi, M.; Kampouri, M.; Koutra, K.; Roumeliotaki, T.; Chalkiadaki, G.; Anousaki, D.; Rantakokko, P.; Kiviranta, H.; Fthenou, E.; et al. Prenatal exposure to persistent organic pollutants in association with offspring neuropsychological development at 4years of age: The Rhea mother-child cohort, Crete, Greece. Environ. Int. 2016, 97, 204–211. [Google Scholar] [CrossRef]
- Tatsuta, N.; Nakai, K.; Murata, K.; Suzuki, K.; Iwai-Shimada, M.; Kurokawa, N.; Hosokawa, T.; Satoh, H. Impacts of prenatal exposures to polychlorinated biphenyls, methylmercury, and lead on intellectual ability of 42-month-old children in Japan. Environ. Res. 2014, 133, 321–326. [Google Scholar] [CrossRef]
- Baibergenova, A.; Kudyakov, R.; Zdeb, M.; Carpenter, D.O. Low birth weight and residential proximity to PCB-contaminated waste sites. Environ. Health Perspect. 2003, 111, 1352–1357. [Google Scholar] [CrossRef]
- Govarts, E.; Nieuwenhuijsen, M.; Schoeters, G.; Ballester, F.; Bloemen, K.; de Boer, M.; Chevrier, C.; Eggesbo, M.; Guxens, M.; Kramer, U.; et al. Birth weight and prenatal exposure to polychlorinated biphenyls (PCBs) and dichlorodiphenyldichloroethylene (DDE): A meta-analysis within 12 European Birth Cohorts. Environ. Health Perspect. 2012, 120, 162–170. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Charles, M.J.; James, R.A.; Keller, J.A.; Willman, E.; Teplin, S. In utero polychlorinated biphenyl exposures in relation to fetal and early childhood growth. Epidemiology 2005, 16, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Patandin, S.; Koopman-Esseboom, C.; de Ridder, M.A.; Weisglas-Kuperus, N.; Sauer, P.J. Effects of environmental exposure to polychlorinated biphenyls and dioxins on birth size and growth in Dutch children. Pediatric Res. 1998, 44, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Stelma, J.M.; Lawrence, C.E. The relation of polychlorinated biphenyls to birth weight and gestational age in the offspring of occupationally exposed mothers. Am. J. Epidemiol. 1989, 129, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Govarts, E.; Iszatt, N.; Trnovec, T.; de Cock, M.; Eggesbo, M.; Palkovicova Murinova, L.; van de Bor, M.; Guxens, M.; Chevrier, C.; Koppen, G.; et al. Prenatal exposure to endocrine disrupting chemicals and risk of being born small for gestational age: Pooled analysis of seven European birth cohorts. Environ. Int. 2018, 115, 267–278. [Google Scholar] [CrossRef]
- Lauritzen, H.B.; Larose, T.L.; Oien, T.; Sandanger, T.M.; Odland, J.O.; van de Bor, M.; Jacobsen, G.W. Maternal serum levels of perfluoroalkyl substances and organochlorines and indices of fetal growth: A Scandinavian case-cohort study. Pediatric Res. 2017, 81, 33–42. [Google Scholar] [CrossRef]
- Longnecker, M.P.; Klebanoff, M.A.; Brock, J.W.; Guo, X. Maternal levels of polychlorinated biphenyls in relation to preterm and small-for-gestational-age birth. Epidemiology 2005, 16, 641–647. [Google Scholar] [CrossRef]
- Linsell, L.; Malouf, R.; Johnson, S.; Morris, J.; Kurinczuk, J.J.; Marlow, N. Prognostic Factors for Behavioral Problems and Psychiatric Disorders in Children Born Very Preterm or Very Low Birth Weight: A Systematic Review. J. Dev. Behav. Pediatr. 2016, 37, 88–102. [Google Scholar] [CrossRef]
- Linsell, L.; Malouf, R.; Morris, J.; Kurinczuk, J.J.; Marlow, N. Prognostic Factors for Poor Cognitive Development in Children Born Very Preterm or With Very Low Birth Weight: A Systematic Review. JAMA Pediatr. 2015, 169, 1162–1172. [Google Scholar] [CrossRef]
- Movsas, T.Z.; Pinto-Martin, J.A.; Whitaker, A.H.; Feldman, J.F.; Lorenz, J.M.; Korzeniewski, S.J.; Levy, S.E.; Paneth, N. Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. J. Pediatr. 2013, 163, 73–78. [Google Scholar] [CrossRef]
- Salmaso, N.; Jablonska, B.; Scafidi, J.; Vaccarino, F.M.; Gallo, V. Neurobiology of premature brain injury. Nat. Neurosci. 2014, 17, 341–346. [Google Scholar] [CrossRef]
- Gore, A.C.; Krishnan, K.; Reilly, M.P. Endocrine-disrupting chemicals: Effects on neuroendocrine systems and the neurobiology of social behavior. Horm. Behav. 2019, 111, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Sable, H.J.K.; Schantz, S.L. Executive Function following Developmental Exposure to Polychlorinated Biphenyls (PCBs): What Animal Models Have Told Us. In Animal Models of Cognitive Impairment; Levin, E.D., Buccafusco, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Winneke, G. Developmental aspects of environmental neurotoxicology: Lessons from lead and polychlorinated biphenyls. J. Neurol. Sci. 2011, 308, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, K.H.; Phimister, A.; Bachstetter, A.D.; Ward, T.R.; Stackman, R.W.; Mervis, R.F.; Wisniewski, A.B.; Klein, S.L.; Kodavanti, P.R.; et al. Developmental exposure to polychlorinated biphenyls interferes with experience-dependent dendritic plasticity and ryanodine receptor expression in weanling rats. Environ. Health Perspect. 2009, 117, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Ballschmiter, K.; Mennel, A.; Buyten, J. Long chain alkyl-polysiloxanes as non-polar stationary phases in capillary gas chromatography. Fresenius J. Anal. Chem. 1993, 346, 396–402. [Google Scholar] [CrossRef]
- Thomas, K.; Xue, J.; Williams, R.; Jones, P.; Whitaker, D. Polychlorinated Biphenyls (PCBs) in School Buildings: Sources, Environmental Levels, and Exposures; United States Environmental Protection Agency: Washington, DC, USA, 2012; p. 150. [Google Scholar]
- Carpenter, D.O. Polychlorinated biphenyls (PCBs): Routes of exposure and effects on human health. Rev. Environ. Health 2006, 21, 1–23. [Google Scholar] [CrossRef]
- Erickson, M.D.; Kaley, R.G., 2nd. Applications of polychlorinated biphenyls. Environ. Sci. Pollut. Res. Int. 2011, 18, 135–151. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.; Bosveld, A.T.; Brunstrom, B.; Cook, P.; Feeley, M.; Giesy, J.P.; Hanberg, A.; Hasegawa, R.; Kennedy, S.W.; et al. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998, 106, 775–792. [Google Scholar] [CrossRef]
- USEPA. Recommended Toxicity Equivalence Factors (TEFs) for Human Health Risk Assessments of 2,3,7,8-Tetrachlorodibenzo-p-dioxin and Dioxin-Like Compounds; U.S. Environmental Protection Agency Risk Assessment Forum: Washington, DC, USA, 2010.
- Beyer, A.; Biziuk, M. Environmental fate and global distribution of polychlorinated biphenyls. Rev. Environ. Contam. Toxicol. 2009, 201, 137–158. [Google Scholar] [CrossRef]
- Pessah, I.N.; Cherednichenko, G.; Lein, P.J. Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol. Ther. 2010, 125, 260–285. [Google Scholar] [CrossRef]
- Mitchell, M.M.; Woods, R.; Chi, L.H.; Schmidt, R.J.; Pessah, I.N.; Kostyniak, P.J.; LaSalle, J.M. Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder. Environ. Mol. Mutagenes. 2012, 53, 589–598. [Google Scholar] [CrossRef]
- Bock, K.W. Toward elucidation of dioxin-mediated chloracne and Ah receptor functions. Biochem. Pharmacol. 2016, 112, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mellor, C.L.; Steinmetz, F.P.; Cronin, M.T. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit. Rev. Toxicol. 2016, 46, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Rothhammer, V.; Quintana, F.J. Control of immune-mediated pathology via the aryl hydrocarbon receptor. J. Biol. Chem. 2017, 292, 12383–12389. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Loomis, D.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K.; et al. Carcinogenicity of polychlorinated biphenyls and polybrominated biphenyls. Lancet Oncol. 2013, 14, 287–288. [Google Scholar] [CrossRef]
- Nowack, N.; Wittsiepe, J.; Kasper-Sonnenberg, M.; Wilhelm, M.; Scholmerich, A. Influence of Low-Level Prenatal Exposure to PCDD/Fs and PCBs on Empathizing, Systemizing and Autistic Traits: Results from the Duisburg Birth Cohort Study. PLoS ONE 2015, 10, e0129906. [Google Scholar] [CrossRef] [PubMed]
- Hany, J.; Lilienthal, H.; Roth-Harer, A.; Ostendorp, G.; Heinzow, B.; Winneke, G. Behavioral effects following single and combined maternal exposure to PCB 77 (3,4,3’,4’-tetrachlorobiphenyl) and PCB 47 (2,4,2’,4’-tetrachlorobiphenyl) in rats. Neurotoxicol. Teratol. 1999, 21, 147–156. [Google Scholar] [CrossRef]
- Bernhoft, A.; Skaare, J.U. Levels of selected individual polychlorinated biphenyls in different tissues of harbour seals (phoca vitulina) from the Southern coast of Norway. Environ. Pollut. 1994, 86, 99–107. [Google Scholar] [CrossRef]
- Bushnell, P.J.; Rice, D.C. Behavioral assessments of learning and attention in rats exposed perinatally to 3,3’,4,4’,5-pentachlorobiphenyl (PCB 126). Neurotoxicol. Teratol. 1999, 21, 381–392. [Google Scholar] [CrossRef]
- Schantz, S.L.; Seo, B.W.; Moshtaghian, J.; Peterson, R.E.; Moore, R.W. Effects of gestational and lactational exposure to TCDD or coplanar PCBs on spatial learning. Neurotoxicol. Teratol. 1996, 18, 305–313. [Google Scholar] [CrossRef]
- Verner, M.A.; Plusquellec, P.; Desjardins, J.L.; Cartier, C.; Haddad, S.; Ayotte, P.; Dewailly, E.; Muckle, G. Prenatal and early-life polychlorinated biphenyl (PCB) levels and behavior in Inuit preschoolers. Environ. Int. 2015, 78, 90–94. [Google Scholar] [CrossRef]
- Klocke, C.; Sethi, S.; Lein, P.J. The developmental neurotoxicity of legacy vs. contemporary polychlorinated biphenyls (PCBs): Similarities and differences. Environ. Sci. Pollut. Res. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, B.; Stahlmann, R. Developmental toxicity of polychlorinated biphenyls (PCBs): A systematic review of experimental data. Arch. Toxicol. 2004, 78, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Morgan, R.K.; Feng, W.; Lin, Y.; Li, X.; Luna, C.; Koch, M.; Bansal, R.; Duffel, M.W.; Puschner, B.; et al. Comparative Analyses of the 12 Most Abundant PCB Congeners Detected in Human Maternal Serum for Activity at the Thyroid Hormone Receptor and Ryanodine Receptor. Environ. Sci. Technol. 2019, 53, 3948–3958. [Google Scholar] [CrossRef] [PubMed]
- Dewailly, E.; Mulvad, G.; Pedersen, H.S.; Ayotte, P.; Demers, A.; Weber, J.P.; Hansen, J.C. Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland. Environ. Health Perspect. 1999, 107, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Covaci, A.; Schepens, P. Levels and chiral signatures of persistent organochlorine pollutants in human tissues from Belgium. Environ. Res. 2003, 93, 167–176. [Google Scholar] [CrossRef]
- Covaci, A.; de Boer, J.; Ryan, J.J.; Voorspoels, S.; Schepens, P. Distribution of organobrominated and organochlorinated contaminants in Belgian human adipose tissue. Environ. Res. 2002, 88, 210–218. [Google Scholar] [CrossRef]
- Craig, F.; Margari, F.; Legrottaglie, A.R.; Palumbi, R.; de Giambattista, C.; Margari, L. A review of executive function deficits in autism spectrum disorder and attention-deficit/hyperactivity disorder. Neuropsychiatr. Dis. Treat. 2016, 12, 1191–1202. [Google Scholar] [CrossRef]
- Tian, Y.H.; Hwan Kim, S.; Lee, S.Y.; Jang, C.G. Lactational and postnatal exposure to polychlorinated biphenyls induces sex-specific anxiolytic behavior and cognitive deficit in mice offspring. Synapse 2011, 65, 1032–1041. [Google Scholar] [CrossRef]
- Nam, Y.; Shin, E.J.; Shin, S.W.; Lim, Y.K.; Jung, J.H.; Lee, J.H.; Ha, J.R.; Chae, J.S.; Ko, S.K.; Jeong, J.H.; et al. YY162 prevents ADHD-like behavioral side effects and cytotoxicity induced by Aroclor1254 via interactive signaling between antioxidant potential, BDNF/TrkB, DAT and NET. Food Chem. Toxicol. 2014, 65, 280–292. [Google Scholar] [CrossRef]
- Elnar, A.A.; Diesel, B.; Desor, F.; Feidt, C.; Bouayed, J.; Kiemer, A.K.; Soulimani, R. Neurodevelopmental and behavioral toxicity via lactational exposure to the sum of six indicator non-dioxin-like-polychlorinated biphenyls ( summation operator6 NDL-PCBs) in mice. Toxicology 2012, 299, 44–54. [Google Scholar] [CrossRef]
- Gillette, R.; Reilly, M.P.; Topper, V.Y.; Thompson, L.M.; Crews, D.; Gore, A.C. Anxiety-like behaviors in adulthood are altered in male but not female rats exposed to low dosages of polychlorinated biphenyls in utero. Horm. Behav. 2017, 87, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Cauli, O.; Leslie, H.; Felipo, V. Differential long-term effects of developmental exposure to polychlorinated biphenyls 52, 138 or 180 on motor activity and neurotransmission. Gender dependence and mechanisms involved. Neurochem. Int. 2011, 58, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Cauli, O.; Felipo, V. Developmental exposure to polychlorinated biphenyls 52, 138 or 180 affects differentially learning or motor coordination in adult rats. Mechanisms involved. Neuroscience 2010, 167, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.; Kannan, N.; Yamashita, N.; Derr-Yellin, E.C.; Ward, T.R.; Burgin, D.E.; Tilson, H.A.; Birnbaum, L.S. Differential effects of two lots of Aroclor 1254: Congener-specific analysis and neurochemical end points. Environ. Health Perspect. 2001, 109, 1153–1161. [Google Scholar] [CrossRef]
- Supekar, K.; Uddin, L.Q.; Khouzam, A.; Phillips, J.; Gaillard, W.D.; Kenworthy, L.E.; Yerys, B.E.; Vaidya, C.J.; Menon, V. Brain hyperconnectivity in children with autism and its links to social deficits. Cell Rep. 2013, 5, 738–747. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef]
- Mohrle, D.; Fernandez, M.; Penagarikano, O.; Frick, A.; Allman, B.; Schmid, S. What we can learn from a genetic rodent model about autism. Neurosci. Biobehav. Rev. 2020, 109, 29–53. [Google Scholar] [CrossRef]
- Yang, M.; Silverman, J.L.; Crawley, J.N. Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci. 2011. Chapter 8. [Google Scholar] [CrossRef]
- Crawley, J.N. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007, 17, 448–459. [Google Scholar] [CrossRef]
- Reilly, M.P.; Weeks, C.D.; Crews, D.; Gore, A.C. Application of a novel social choice paradigm to assess effects of prenatal endocrine-disrupting chemical exposure in rats (Rattus norvegicus). J. Comp. Psychol. 2018, 132, 253–267. [Google Scholar] [CrossRef]
- Hofer, M.A.; Shair, H.N.; Brunelli, S.A. Ultrasonic vocalizations in rat and mouse pups. Curr. Protoc. Neurosci. 2002. Chapter 8. [Google Scholar] [CrossRef] [PubMed]
- Winslow, J.T. Mouse social recognition and preference. Curr. Protoc. Neurosci. 2003. Chapter 8. [Google Scholar] [CrossRef] [PubMed]
- Karkaba, A.; Soualeh, N.; Soulimani, R.; Bouayed, J. Perinatal effects of exposure to PCBs on social preferences in young adult and middle-aged offspring mice. Horm. Behav. 2017, 96, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.R.; Thompson, L.M.; Rodriguez, K.; Gore, A.C. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 1. Sexually dimorphic effects on social and anxiety-like behaviors. Horm. Behav. 2016, 78, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.P.; Weeks, C.D.; Topper, V.Y.; Thompson, L.M.; Crews, D.; Gore, A.C. The effects of prenatal PCBs on adult social behavior in rats. Horm. Behav. 2015, 73, 47–55. [Google Scholar] [CrossRef]
- Topper, V.Y.; Reilly, M.P.; Wagner, L.M.; Thompson, L.M.; Gillette, R.; Crews, D.; Gore, A.C. Social and neuromolecular phenotypes are programmed by prenatal exposures to endocrine-disrupting chemicals. Mol. Cell Endocrinol. 2019, 479, 133–146. [Google Scholar] [CrossRef]
- Jolous-Jamshidi, B.; Cromwell, H.C.; McFarland, A.M.; Meserve, L.A. Perinatal exposure to polychlorinated biphenyls alters social behaviors in rats. Toxicol. Lett. 2010, 199, 136–143. [Google Scholar] [CrossRef]
- Bushnell, P.J. Advanced behavioral testing in rodents: Assessment of cognitive function in animals. Curr. Protoc. Toxicol. 2011. Chapter 11. [Google Scholar] [CrossRef]
- Winstanley, C.A.; Eagle, D.M.; Robbins, T.W. Behavioral models of impulsivity in relation to ADHD: Translation between clinical and preclinical studies. Clin. Psychol. Rev. 2006, 26, 379–395. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci. 2014, 69, 8.31.1–8.31.17. [Google Scholar] [CrossRef]
- Elnar, A.A.; Allouche, A.; Desor, F.; Yen, F.T.; Soulimani, R.; Oster, T. Lactational exposure of mice to low levels of non-dioxin-like polychlorinated biphenyls increases susceptibility to neuronal stress at a mature age. Neurotoxicology 2016, 53, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.E.; Miller, M.M.; Nelms Sprowles, J.L.; Levine, L.R.; Sable, H.J. A comparison of presynaptic and postsynaptic dopaminergic agonists on inhibitory control performance in rats perinatally exposed to PCBs. Neurotoxicol. Teratol. 2015, 50, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Monaikul, S.; Eubig, P.; Floresco, S.; Schantz, S. Strategy set-shifting and response inhibition in adult rats exposed to an environmental polychlorinated biphenyl mixture during adolescence. Neurotoxicol. Teratol. 2017, 63, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kostyniak, P.J.; Hansen, L.G.; Widholm, J.J.; Fitzpatrick, R.D.; Olson, J.R.; Helferich, J.L.; Kim, K.H.; Sable, H.J.; Seegal, R.F.; Pessah, I.N.; et al. Formulation and characterization of an experimental PCB mixture designed to mimic human exposure from contaminated fish. Toxicol. Sci. 2005, 88, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.L.; Moshtaghian, J.; Ness, D.K. Spatial learning deficits in adult rats exposed to ortho-substituted PCB congeners during gestation and lactation. Fundam. Appl. Toxicol. 1995, 26, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Seegal, R.F. Epidemiological and laboratory evidence of PCB-induced neurotoxicity. Crit. Rev. Toxicol. 1996, 26, 709–737. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.E. In vitro systems as simulations of in vivo conditions: The study of cognition and synaptic plasticity in neurotoxicology. Ann. N. Y. Acad. Sci. 2000, 919, 119–132. [Google Scholar] [CrossRef]
- Stamou, M.; Streifel, K.M.; Goines, P.E.; Lein, P.J. Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 2013, 36, 3–16. [Google Scholar] [CrossRef]
- Copf, T. Impairments in dendrite morphogenesis as etiology for neurodevelopmental disorders and implications for therapeutic treatments. Neurosci. Biobehav. Rev. 2016, 68, 946–978. [Google Scholar] [CrossRef]
- Rakic, P.; Bourgeois, J.P.; Goldman-Rakic, P.S. Synaptic development of the cerebral cortex: Implications for learning, memory, and mental illness. Prog. Brain Res. 1994, 102, 227–243. [Google Scholar] [CrossRef]
- Bourgeron, T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009, 19, 231–234. [Google Scholar] [CrossRef]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 2005, 64, 537–544. [Google Scholar] [CrossRef]
- Dikranian, K.; Ishimaru, M.J.; Tenkova, T.; Labruyere, J.; Qin, Y.Q.; Ikonomidou, C.; Olney, J.W. Apoptosis in the in vivo mammalian forebrain. Neurobiol. Dis. 2001, 8, 359–379. [Google Scholar] [CrossRef]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury (Review). Int. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [CrossRef]
- Lein, P.J.; Supasai, S.; Guignet, M. Apoptosis as a mechanism of developmental neurotoxicity. In Handbook of Developmental Neurotoxicology; Slikker, W., Jr., Paule, M.G., Wang, C., Eds.; Elsevier, Inc.: Amsterdam, The Netherlands, 2018; pp. 91–112. [Google Scholar]
- White, L.D.; Barone, S., Jr. Qualitative and quantitative estimates of apoptosis from birth to senescence in the rat brain. Cell Death Differ. 2001, 8, 345–356. [Google Scholar] [CrossRef]
- Barone, S., Jr.; Das, K.P.; Lassiter, T.L.; White, L.D. Vulnerable processes of nervous system development: A review of markers and methods. Neurotoxicology 2000, 21, 15–36. [Google Scholar]
- Sastry, P.S.; Rao, K.S. Apoptosis and the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Lein, P.J. Polychlorinated biphenyls increase apoptosis in the developing rat brain. Curr. Neurobiol. 2010, 1, 70–76. [Google Scholar] [PubMed]
- Sanchez-Alonso, J.A.; Lopez-Aparicio, P.; Recio, M.N.; Perez-Albarsanz, M.A. Polychlorinated biphenyl mixtures (Aroclors) induce apoptosis via Bcl-2, Bax and caspase-3 proteins in neuronal cell cultures. Toxicol. Lett. 2004, 153, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.S.; Fitzpatrick, R.; Pessah, I.; Kostyniak, P.; Lein, P.J. Polychlorinated biphenyls induce caspase-dependent cell death in cultured embryonic rat hippocampal but not cortical neurons via activation of the ryanodine receptor. Toxicol. Appl. Pharmacol. 2003, 190, 72–86. [Google Scholar] [CrossRef]
- Hwang, S.G.; Lee, H.C.; Lee, D.W.; Kim, Y.S.; Joo, W.H.; Cho, Y.K.; Moon, J.Y. Induction of apoptotic cell death by a p53-independent pathway in neuronal SK-N-MC cells after treatment with 2,2’,5,5’-tetrachlorobiphenyl. Toxicology 2001, 165, 179–188. [Google Scholar] [CrossRef]
- Sanchez-Alonso, J.A.; Lopez-Aparicio, P.; Recio, M.N.; Perez-Albarsanz, M.A. Apoptosis-mediated neurotoxic potential of a planar (PCB 77) and a nonplanar (PCB 153) polychlorinated biphenyl congeners in neuronal cell cultures. Toxicol. Lett. 2003, 144, 337–349. [Google Scholar] [CrossRef]
- Kennedy, M.B. Signal-processing machines at the postsynaptic density. Science 2000, 290, 750–754. [Google Scholar] [CrossRef]
- Matus, A.; Shepherd, G.M. The millennium of the dendrite? Neuron 2000, 27, 431–434. [Google Scholar] [CrossRef][Green Version]
- Purves, D. Functional and structural changes in mammalian sympathetic neurones following interruption of their axons. J. Physiol. 1975, 252, 429–463. [Google Scholar] [CrossRef]
- Purves, D. Body and Brain: A Trophic Theory of Neural Connections; Harvard University Press: Cambridge, MA, USA, 1988. [Google Scholar]
- Miller, J.P.; Jacobs, G.A. Relationships between neuronal structure and function. J. Exp. Biol. 1984, 112, 129–145. [Google Scholar]
- Sejnowski, T.J. The year of the dendrite. Science 1997, 275, 178–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leuner, B.; Shors, T.J. New spines, new memories. Mol. Neurobiol. 2004, 29, 117–130. [Google Scholar] [CrossRef]
- Engle, E.C. Human genetic disorders of axon guidance. Cold Spring Harb. Perspect. Biol. 2010, 2, a001784. [Google Scholar] [CrossRef] [PubMed]
- Garey, L. When cortical development goes wrong: Schizophrenia as a neurodevelopmental disease of microcircuits. J. Anat. 2010, 217, 324–333. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef]
- Robichaux, M.A.; Cowan, C.W. Signaling mechanisms of axon guidance and early synaptogenesis. Curr. Top. Behav. Neurosci. 2014, 16, 19–48. [Google Scholar] [CrossRef]
- Cremer, H.; Chazal, G.; Goridis, C.; Represa, A. NCAM is essential for axonal growth and fasciculation in the hippocampus. Mol. Cell Neurosci. 1997, 8, 323–335. [Google Scholar] [CrossRef]
- Maier, D.L.; Mani, S.; Donovan, S.L.; Soppet, D.; Tessarollo, L.; McCasland, J.S.; Meiri, K.F. Disrupted cortical map and absence of cortical barrels in growth-associated protein (GAP)-43 knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 9397–9402. [Google Scholar] [CrossRef]
- Berger-Sweeney, J.; Hohmann, C.F. Behavioral consequences of abnormal cortical development: Insights into developmental disabilities. Behav. Brain Res. 1997, 86, 121–142. [Google Scholar] [CrossRef]
- Hoffman, M.P.; Kidder, B.L.; Steinberg, Z.L.; Lakhani, S.; Ho, S.; Kleinman, H.K.; Larsen, M. Gene expression profiles of mouse submandibular gland development: FGFR1 regulates branching morphogenesis in vitro through BMP- and FGF-dependent mechanisms. Development 2002, 129, 5767–5778. [Google Scholar] [CrossRef]
- Jones, L.; Fischer, I.; Levitt, P. Nonuniform alteration of dendritic development in the cerebral cortex following prenatal cocaine exposure. Cereb Cortex 1996, 6, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.B.; Stanwood, G.D.; Reinoso, B.S.; Washington, R.A.; Wang, H.Y.; Friedman, E.; Levitt, P. In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J. Neurosci. 2000, 20, 4606–4614. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, D.L.; Meserve, L.A.; Bingman, V.P. Reduced growth of intra- and infra-pyramidal mossy fibers is produced by continuous exposure to polychlorinated biphenyl. Toxicology 1999, 138, 11–17. [Google Scholar] [CrossRef]
- Lein, P.J.; Yang, D.; Bachstetter, A.D.; Tilson, H.A.; Harry, G.J.; Mervis, R.F.; Kodavanti, P.R. Ontogenetic alterations in molecular and structural correlates of dendritic growth after developmental exposure to polychlorinated biphenyls. Environ. Health Perspect. 2007, 115, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Roegge, C.S.; Morris, J.R.; Villareal, S.; Wang, V.C.; Powers, B.E.; Klintsova, A.Y.; Greenough, W.T.; Pessah, I.N.; Schantz, S.L. Purkinje cell and cerebellar effects following developmental exposure to PCBs and/or MeHg. Neurotoxicol. Teratol. 2006, 28, 74–85. [Google Scholar] [CrossRef]
- Wayman, G.A.; Yang, D.; Bose, D.D.; Lesiak, A.; Ledoux, V.; Bruun, D.; Pessah, I.N.; Lein, P.J. PCB-95 promotes dendritic growth via ryanodine receptor-dependent mechanisms. Environ. Health Perspect. 2012, 120, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Keil, K.P.; Miller, G.W.; Chen, H.; Sethi, S.; Schmuck, M.R.; Dhakal, K.; Kim, J.W.; Lein, P.J. PCB 95 promotes dendritic growth in primary rat hippocampal neurons via mTOR-dependent mechanisms. Arch. Toxicol. 2018, 92, 3163–3173. [Google Scholar] [CrossRef]
- Wayman, G.A.; Bose, D.D.; Yang, D.; Lesiak, A.; Bruun, D.; Impey, S.; Ledoux, V.; Pessah, I.N.; Lein, P.J. PCB-95 modulates the calcium-dependent signaling pathway responsible for activity-dependent dendritic growth. Environ. Health Perspect. 2012, 120, 1003–1009. [Google Scholar] [CrossRef]
- Yang, D.; Kania-Korwel, I.; Ghogha, A.; Chen, H.; Stamou, M.; Bose, D.D.; Pessah, I.N.; Lehmler, H.J.; Lein, P.J. PCB 136 atropselectively alters morphometric and functional parameters of neuronal connectivity in cultured rat hippocampal neurons via ryanodine receptor-dependent mechanisms. Toxicol. Sci. 2014, 138, 379–392. [Google Scholar] [CrossRef]
- Keil, K.P.; Sethi, S.; Lein, P.J. Sex-Dependent Effects of 2,2’,3,5’,6-Pentachlorobiphenyl on Dendritic Arborization of Primary Mouse Neurons. Toxicol. Sci. 2019, 168, 95–109. [Google Scholar] [CrossRef]
- Lesiak, A.; Zhu, M.; Chen, H.; Appleyard, S.M.; Impey, S.; Lein, P.J.; Wayman, G.A. The environmental neurotoxicant PCB 95 promotes synaptogenesis via ryanodine receptor-dependent miR132 upregulation. J. Neurosci. 2014, 34, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Kuroda, J.; Nagata, I.; Kuroda, Y. Disrupting effects of hydroxy-polychlorinated biphenyl (PCB) congeners on neuronal development of cerebellar Purkinje cells: A possible causal factor for developmental brain disorders? Chemosphere 2007, 67, S412–S420. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Kuroda, J.; Nagata, I.; Kuroda, Y. Hydroxylated metabolites of polychlorinated biphenyls inhibit thyroid-hormone-dependent extension of cerebellar Purkinje cell dendrites. Brain Res. Dev. Brain Res. 2005, 154, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.; Matsuyoshi, C.; Benner, S.; Hosokawa, M.; Endo, T.; Ling, W.; Kohda, M.; Yokoyama, K.; Nakajima, K.; et al. Developmental origin of abnormal dendritic growth in the mouse brain induced by in utero disruption of aryl hydrocarbon receptor signaling. Neurotoxicol. Teratol. 2015, 52, 42–50. [Google Scholar] [CrossRef]
- Crofton, K.M.; Zoeller, R.T. Mode of action: Neurotoxicity induced by thyroid hormone disruption during development--hearing loss resulting from exposure to PHAHs. Crit. Rev. Toxicol. 2005, 35, 757–769. [Google Scholar] [CrossRef]
- Zoeller, R.T. Environmental chemicals impacting the thyroid: Targets and consequences. Thyroid 2007, 17, 811–817. [Google Scholar] [CrossRef]
- Mariussen, E.; Fonnum, F. Neurochemical targets and behavioral effects of organohalogen compounds: An update. Crit. Rev. Toxicol. 2006, 36, 253–289. [Google Scholar] [CrossRef]
- Fonnum, F.; Mariussen, E.; Reistad, T. Molecular mechanisms involved in the toxic effects of polychlorinated biphenyls (PCBs) and brominated flame retardants (BFRs). J. Toxicol. Environ. Health A 2006, 69, 21–35. [Google Scholar] [CrossRef]
- Seegal, R.F.; Brosch, K.O.; Okoniewski, R.J. Effects of in utero and lactational exposure of the laboratory rat to 2,4,2’,4’- and 3,4,3’,4’-tetrachlorobiphenyl on dopamine function. Toxicol. Appl. Pharmacol. 1997, 146, 95–103. [Google Scholar] [CrossRef]
- Seegal, R.F.; Bush, B.; Brosch, K.O. Decreases in dopamine concentrations in adult, non-human primate brain persist following removal from polychlorinated biphenyls. Toxicology 1994, 86, 71–87. [Google Scholar] [CrossRef]
- Seegal, R.F.; Bush, B.; Shain, W. Lightly chlorinated ortho-substituted PCB congeners decrease dopamine in nonhuman primate brain and in tissue culture. Toxicol. Appl. Pharmacol. 1990, 106, 136–144. [Google Scholar] [CrossRef]
- Antunes Fernandes, E.C.; Hendriks, H.S.; van Kleef, R.G.; Reniers, A.; Andersson, P.L.; van den Berg, M.; Westerink, R.H. Activation and potentiation of human GABAA receptors by non-dioxin-like PCBs depends on chlorination pattern. Toxicol. Sci. 2010, 118, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Antunes Fernandes, E.C.; Hendriks, H.S.; van Kleef, R.G.; van den Berg, M.; Westerink, R.H. Potentiation of the human GABA(A) receptor as a novel mode of action of lower-chlorinated non-dioxin-like PCBs. Environ. Sci. Technol. 2010, 44, 2864–2869. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, H.S.; Antunes Fernandes, E.C.; Bergman, A.; van den Berg, M.; Westerink, R.H. PCB-47, PBDE-47, and 6-OH-PBDE-47 differentially modulate human GABAA and alpha4beta2 nicotinic acetylcholine receptors. Toxicol. Sci. 2010, 118, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.; Shin, D.S.; Tilson, H.A.; Harry, G.J. Comparative effects of two polychlorinated biphenyl congeners on calcium homeostasis in rat cerebellar granule cells. Toxicol. Appl. Pharmacol. 1993, 123, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.; Ward, T.R.; McKinney, J.D.; Tilson, H.A. Inhibition of microsomal and mitochondrial Ca2+-sequestration in rat cerebellum by polychlorinated biphenyl mixtures and congeners. Structure-activity relationships. Arch. Toxicol. 1996, 70, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Tilson, H.A.; Kodavanti, P.R. The neurotoxicity of polychlorinated biphenyls. Neurotoxicology 1998, 19, 517–525. [Google Scholar]
- Kimura, E.; Tohyama, C. Embryonic and Postnatal Expression of Aryl Hydrocarbon Receptor mRNA in Mouse Brain. Front. Neuroanat. 2017, 11, 4. [Google Scholar] [CrossRef]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. 2005, 83, 340–348. [Google Scholar] [CrossRef]
- Latchney, S.E.; Hein, A.M.; O’Banion, M.K.; DiCicco-Bloom, E.; Opanashuk, L.A. Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 2013, 125, 430–445. [Google Scholar] [CrossRef]
- Dever, D.P.; Adham, Z.O.; Thompson, B.; Genestine, M.; Cherry, J.; Olschowka, J.A.; DiCicco-Bloom, E.; Opanashuk, L.A. Aryl hydrocarbon receptor deletion in cerebellar granule neuron precursors impairs neurogenesis. Dev. Neurobiol. 2016, 76, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.I.; Endo, T.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Excessive activation of AhR signaling disrupts neuronal migration in the hippocampal CA1 region in the developing mouse. J. Toxicol. Sci. 2017, 42, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Lioy, D.T.; Henry, E.C.; Gasiewicz, T.A.; Strathmann, F.G.; Mayer-Proschel, M.; Opanashuk, L.A. Neural precursor cell proliferation is disrupted through activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev. 2011, 20, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.I.; Endo, T.; Ling, W.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Impaired dendritic growth and positioning of cortical pyramidal neurons by activation of aryl hydrocarbon receptor signaling in the developing mouse. PLoS ONE 2017, 12, e0183497. [Google Scholar] [CrossRef]
- Kimura, E.; Tohyama, C. Vocalization as a novel endpoint of atypical attachment behavior in 2,3,7,8-tetrachlorodibenzo-p-dioxin-exposed infant mice. Arch. Toxicol. 2018, 92, 1741–1749. [Google Scholar] [CrossRef]
- Chepelev, N.L.; Moffat, I.D.; Bowers, W.J.; Yauk, C.L. Neurotoxicity may be an overlooked consequence of benzoapyrene exposure that is relevant to human health risk assessment. Mutat. Res. Rev. Mutat. Res. 2015, 764, 64–89. [Google Scholar] [CrossRef]
- Poland, A.; Palen, D.; Glover, E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994, 46, 915–921. [Google Scholar]
- Nebert, D.W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog. Lipid Res. 2017, 67, 38–57. [Google Scholar] [CrossRef]
- Klinefelter, K.; Hooven, M.K.; Bates, C.; Colter, B.T.; Dailey, A.; Infante, S.K.; Kania-Korwel, I.; Lehmler, H.J.; Lopez-Juarez, A.; Ludwig, C.P.; et al. Genetic differences in the aryl hydrocarbon receptor and CYP1A2 affect sensitivity to developmental polychlorinated biphenyl exposure in mice: Relevance to studies of human neurological disorders. Mamm. Genome 2018, 29, 112–127. [Google Scholar] [CrossRef]
- Curran, C.P.; Nebert, D.W.; Genter, M.B.; Patel, K.V.; Schaefer, T.L.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V. In utero and lactational exposure to PCBs in mice: Adult offspring show altered learning and memory depending on Cyp1a2 and Ahr genotypes. Environ. Health Perspect. 2011, 119, 1286–1293. [Google Scholar] [CrossRef]
- Curran, C.P.; Altenhofen, E.; Ashworth, A.; Brown, A.; Kamau-Cheggeh, C.; Curran, M.; Evans, A.; Floyd, R.; Fowler, J.; Garber, H.; et al. Ahrd Cyp1a2(-/-) mice show increased susceptibility to PCB-induced developmental neurotoxicity. Neurotoxicology 2012, 33, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, J.H.; Schwartz, H.L. Molecular basis of thyroid hormone-dependent brain development. Endocr. Rev. 1997, 18, 462–475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rovet, J.F. The role of thyroid hormones for brain development and cognitive function. Endocr. Dev. 2014, 26, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R. Neurodevelopmental and neurophysiological actions of thyroid hormone. J. Neuroendocrinol. 2008, 20, 784–794. [Google Scholar] [CrossRef]
- Salazar, P.; Cisternas, P.; Martinez, M.; Inestrosa, N.C. Hypothyroidism and Cognitive Disorders during Development and Adulthood: Implications in the Central Nervous System. Mol. Neurobiol. 2019, 56, 2952–2963. [Google Scholar] [CrossRef]
- Kalikiri, M.K.; Mamidala, M.P.; Rao, A.N.; Rajesh, V. Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients. Autism Res. 2017, 10, 1919–1928. [Google Scholar] [CrossRef]
- Lyall, K.; Anderson, M.; Kharrazi, M.; Windham, G.C. Neonatal thyroid hormone levels in association with autism spectrum disorder. Autism Res. 2017, 10, 585–592. [Google Scholar] [CrossRef]
- Drover, S.S.M.; Villanger, G.D.; Aase, H.; Skogheim, T.S.; Longnecker, M.P.; Zoeller, R.T.; Reichborn-Kjennerud, T.; Knudsen, G.P.; Zeiner, P.; Engel, S.M. Maternal Thyroid Function During Pregnancy or Neonatal Thyroid Function and Attention Deficit Hyperactivity Disorder: A Systematic Review. Epidemiology 2019, 30, 130–144. [Google Scholar] [CrossRef]
- Modesto, T.; Tiemeier, H.; Peeters, R.P.; Jaddoe, V.W.; Hofman, A.; Verhulst, F.C.; Ghassabian, A. Maternal Mild Thyroid Hormone Insufficiency in Early Pregnancy and Attention-Deficit/Hyperactivity Disorder Symptoms in Children. JAMA Pediatr. 2015, 169, 838–845. [Google Scholar] [CrossRef]
- Itoh, S.; Baba, T.; Yuasa, M.; Miyashita, C.; Kobayashi, S.; Araki, A.; Sasaki, S.; Kajiwara, J.; Hori, T.; Todaka, T.; et al. Association of maternal serum concentration of hydroxylated polychlorinated biphenyls with maternal and neonatal thyroid hormones: The Hokkaido birth cohort study. Environ. Res. 2018, 167, 583–590. [Google Scholar] [CrossRef]
- Li, Z.M.; Hernandez-Moreno, D.; Main, K.M.; Skakkebaek, N.E.; Kiviranta, H.; Toppari, J.; Feldt-Rasmussen, U.; Shen, H.; Schramm, K.W.; De Angelis, M. Association of In Utero Persistent Organic Pollutant Exposure with Placental Thyroid Hormones. Endocrinology 2018, 159, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Crofton, K.M.; Kodavanti, P.R.; Derr-Yellin, E.C.; Casey, A.C.; Kehn, L.S. PCBs, thyroid hormones, and ototoxicity in rats: Cross-fostering experiments demonstrate the impact of postnatal lactation exposure. Toxicol. Sci. 2000, 57, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Goldey, E.S.; Crofton, K.M. Thyroxine replacement attenuates hypothyroxinemia, hearing loss, and motor deficits following developmental exposure to Aroclor 1254 in rats. Toxicol. Sci. 1998, 45, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Sharlin, D.S.; Bansal, R.; Zoeller, R.T. Polychlorinated biphenyls exert selective effects on cellular composition of white matter in a manner inconsistent with thyroid hormone insufficiency. Endocrinology 2006, 147, 846–858. [Google Scholar] [CrossRef]
- Powers, B.E.; Poon, E.; Sable, H.J.; Schantz, S.L. Developmental exposure to PCBs, MeHg, or both: Long-term effects on auditory function. Environ. Health Perspect. 2009, 117, 1101–1107. [Google Scholar] [CrossRef]
- Zahalka, E.A.; Ellis, D.H.; Goldey, E.S.; Stanton, M.E.; Lau, C. Perinatal exposure to polychlorinated biphenyls Aroclor 1016 or 1254 did not alter brain catecholamines nor delayed alternation performance in Long-Evans rats. Brain Res. Bull. 2001, 55, 487–500. [Google Scholar] [CrossRef]
- Ness, D.K.; Schantz, S.L.; Moshtaghian, J.; Hansen, L.G. Effects of perinatal exposure to specific PCB congeners on thyroid hormone concentrations and thyroid histology in the rat. Toxicol. Lett. 1993, 68, 311–323. [Google Scholar] [CrossRef]
- Craft, E.S.; DeVito, M.J.; Crofton, K.M. Comparative responsiveness of hypothyroxinemia and hepatic enzyme induction in Long-Evans rats versus C57BL/6J mice exposed to TCDD-like and phenobarbital-like polychlorinated biphenyl congeners. Toxicol. Sci. 2002, 68, 372–380. [Google Scholar] [CrossRef][Green Version]
- Rice, D.C.; Hayward, S. Lack of effect of 3,3’4,4’,5-pentachlorobiphenyl (PCB 126) throughout gestation and lactation on multiple fixed interval-fixed ratio and DRL performance in rats. Neurotoxicol. Teratol. 1998, 20, 645–650. [Google Scholar] [CrossRef]
- Rice, D.C.; Hayward, S. Effects of exposure to 3,3’,4,4’,5-pentachlorobiphenyl (PCB 126) throughout gestation and lactation on behavior (concurrent random interval-random interval and progressive ratio performance) in rats. Neurotoxicol. Teratol. 1999, 21, 679–687. [Google Scholar] [CrossRef]
- Kapfhammer, J.P. Cellular and molecular control of dendritic growth and development of cerebellar Purkinje cells. Prog. Histochem. Cytochem. 2004, 39, 131–182. [Google Scholar] [CrossRef] [PubMed]
- Kenet, T.; Froemke, R.C.; Schreiner, C.E.; Pessah, I.N.; Merzenich, M.M. Perinatal exposure to a noncoplanar polychlorinated biphenyl alters tonotopy, receptive fields, and plasticity in rat primary auditory cortex. Proc. Natl. Acad. Sci. USA 2007, 104, 7646–7651. [Google Scholar] [CrossRef] [PubMed]
- Gauger, K.J.; Kato, Y.; Haraguchi, K.; Lehmler, H.J.; Robertson, L.W.; Bansal, R.; Zoeller, R.T. Polychlorinated biphenyls (PCBs) exert thyroid hormone-like effects in the fetal rat brain but do not bind to thyroid hormone receptors. Environ. Health Perspect. 2004, 112, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Miyazaki, W.; Takeshita, A.; Kuroda, Y.; Koibuchi, N. Polychlorinated biphenyls suppress thyroid hormone-induced transactivation. Biochem. Biophys. Res. Commun. 2002, 299, 384–388. [Google Scholar] [CrossRef]
- Aamodt, S.M.; Constantine-Paton, M. The role of neural activity in synaptic development and its implications for adult brain function. Adv. Neurol. 1999, 79, 133–144. [Google Scholar]
- Cline, H.T. Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 2001, 11, 118–126. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J. Neurobiol. 1998, 37, 110–130. [Google Scholar] [CrossRef]
- Levitt, P. Structural and functional maturation of the developing primate brain. J. Pediatr. 2003, 143, S35–S45. [Google Scholar] [CrossRef]
- Moody, W.J.; Bosma, M.M. Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol. Rev. 2005, 85, 883–941. [Google Scholar] [CrossRef]
- Borodinsky, L.N.; Root, C.M.; Cronin, J.A.; Sann, S.B.; Gu, X.; Spitzer, N.C. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 2004, 429, 523–530. [Google Scholar] [CrossRef]
- Spitzer, N.C. Activity-dependent neurotransmitter respecification. Nat. Rev. Neurosci. 2012, 13, 94–106. [Google Scholar] [CrossRef]
- Lohmann, C. Calcium signaling and the development of specific neuronal connections. Prog. Brain Res. 2009, 175, 443–452. [Google Scholar] [CrossRef]
- Lohmann, C.; Wong, R.O. Regulation of dendritic growth and plasticity by local and global calcium dynamics. Cell Calcium 2005, 37, 403–409. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Konur, S.; Ghosh, A. Calcium signaling and the control of dendritic development. Neuron 2005, 46, 401–405. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium microdomains: Organization and function. Cell Calcium 2006, 40, 405–412. [Google Scholar] [CrossRef]
- Krey, J.F.; Dolmetsch, R.E. Molecular mechanisms of autism: A possible role for Ca2+ signaling. Curr. Opin. Neurobiol. 2007, 17, 112–119. [Google Scholar] [CrossRef]
- Pessah, I.N.; Lein, P.J. Evidence for environmental susceptibility in autism: What we need to know about gene x environment interactions. In Autism: Current Theories and Evidence; Zimmerman, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 409–428. [Google Scholar]
- Mundy, W.R.; Shafer, T.J.; Tilson, H.A.; Kodavanti, P.R. Extracellular calcium is required for the polychlorinated biphenyl-induced increase of intracellular free calcium levels in cerebellar granule cell culture. Toxicology 1999, 136, 27–39. [Google Scholar] [CrossRef]
- Inglefield, J.R.; Shafer, T.J. Polychlorinated biphenyl-stimulation of Ca(2+) oscillations in developing neocortical cells: A role for excitatory transmitters and L-type voltage-sensitive Ca(2+) channels. J. Pharmacol. Exp. Ther. 2000, 295, 105–113. [Google Scholar]
- Wong, P.W.; Brackney, W.R.; Pessah, I.N. Ortho-substituted polychlorinated biphenyls alter microsomal calcium transport by direct interaction with ryanodine receptors of mammalian brain. J. Biol. Chem. 1997, 272, 15145–15153. [Google Scholar] [CrossRef]
- Wong, P.W.; Pessah, I.N. Ortho-substituted polychlorinated biphenyls alter calcium regulation by a ryanodine receptor-mediated mechanism: Structural specificity toward skeletal- and cardiac-type microsomal calcium release channels. Mol. Pharmacol. 1996, 49, 740–751. [Google Scholar] [PubMed]
- Wong, P.W.; Pessah, I.N. Noncoplanar PCB 95 alters microsomal calcium transport by an immunophilin FKBP12-dependent mechanism. Mol. Pharmacol. 1997, 51, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Inglefield, J.R.; Mundy, W.R.; Shafer, T.J. Inositol 1,4,5-triphosphate receptor-sensitive Ca(2+) release, store-operated Ca(2+) entry, and cAMP responsive element binding protein phosphorylation in developing cortical cells following exposure to polychlorinated biphenyls. J. Pharmacol. Exp. Ther. 2001, 297, 762–773. [Google Scholar]
- Samso, M.; Feng, W.; Pessah, I.N.; Allen, P.D. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 2009, 7, e85. [Google Scholar] [CrossRef]
- Holland, E.B.; Feng, W.; Zheng, J.; Dong, Y.; Li, X.; Lehmler, H.J.; Pessah, I.N. An Extended Structure-Activity Relationship of Nondioxin-Like PCBs Evaluates and Supports Modeling Predictions and Identifies Picomolar Potency of PCB 202 Towards Ryanodine Receptors. Toxicol. Sci. 2017, 155, 170–181. [Google Scholar] [CrossRef]
- Feng, W.; Zheng, J.; Robin, G.; Dong, Y.; Ichikawa, M.; Inoue, Y.; Mori, T.; Nakano, T.; Pessah, I.N. Enantioselectivity of 2,2’,3,5’,6-Pentachlorobiphenyl (PCB 95) Atropisomers toward Ryanodine Receptors (RyRs) and Their Influences on Hippocampal Neuronal Networks. Environ. Sci. Technol. 2017, 51, 14406–14416. [Google Scholar] [CrossRef]
- Fritsch, E.B.; Pessah, I.N. Structure-activity relationship of non-coplanar polychlorinated biphenyls toward skeletal muscle ryanodine receptors in rainbow trout (Oncorhynchus mykiss). Aquat. Toxicol. 2013, 140–141, 204–212. [Google Scholar] [CrossRef]
- Fritsch, E.B.; Stegeman, J.J.; Goldstone, J.V.; Nacci, D.E.; Champlin, D.; Jayaraman, S.; Connon, R.E.; Pessah, I.N. Expression and function of ryanodine receptor related pathways in PCB tolerant Atlantic killifish (Fundulus heteroclitus) from New Bedford Harbor, MA, USA. Aquat. Toxicol. 2015, 159, 156–166. [Google Scholar] [CrossRef]
- Holland, E.B.; Goldstone, J.V.; Pessah, I.N.; Whitehead, A.; Reid, N.M.; Karchner, S.I.; Hahn, M.E.; Nacci, D.E.; Clark, B.W.; Stegeman, J.J. Ryanodine receptor and FK506 binding protein 1 in the Atlantic killifish (Fundulus heteroclitus): A phylogenetic and population-based comparison. Aquat. Toxicol. 2017, 192, 105–115. [Google Scholar] [CrossRef]
- Niknam, Y.; Feng, W.; Cherednichenko, G.; Dong, Y.; Joshi, S.N.; Vyas, S.M.; Lehmler, H.J.; Pessah, I.N. Structure-activity relationship of selected meta- and para-hydroxylated non-dioxin like polychlorinated biphenyls: From single RyR1 channels to muscle dysfunction. Toxicol. Sci. 2013, 136, 500–513. [Google Scholar] [CrossRef]
- Pessah, I.N.; Hansen, L.G.; Albertson, T.E.; Garner, C.E.; Ta, T.A.; Do, Z.; Kim, K.H.; Wong, P.W. Structure-activity relationship for noncoplanar polychlorinated biphenyl congeners toward the ryanodine receptor-Ca2+ channel complex type 1 (RyR1). Chem. Res. Toxicol. 2006, 19, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Adasme, T.; Haeger, P.; Paula-Lima, A.C.; Espinoza, I.; Casas-Alarcon, M.M.; Carrasco, M.A.; Hidalgo, C. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc. Natl. Acad. Sci. USA 2011, 108, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Chiesi, M.; Schwaller, R.; Calviello, G. Inhibition of rapid Ca-release from isolated skeletal and cardiac sarcoplasmic reticulum (SR) membranes. Biochem. Biophys. Res. Commun. 1988, 154, 1–8. [Google Scholar] [CrossRef]
- Mack, W.M.; Zimanyi, I.; Pessah, I.N. Discrimination of multiple binding sites for antagonists of the calcium release channel complex of skeletal and cardiac sarcoplasmic reticulum. J. Pharmacol. Exp. Ther. 1992, 262, 1028–1037. [Google Scholar]
- Pessah, I.N.; Lehmler, H.J.; Robertson, L.W.; Perez, C.F.; Cabrales, E.; Bose, D.D.; Feng, W. Enantiomeric specificity of (-)-2,2’,3,3’,6,6’-hexachlorobiphenyl toward ryanodine receptor types 1 and 2. Chem. Res. Toxicol. 2009, 22, 201–207. [Google Scholar] [CrossRef]
- Royland, J.E.; Kodavanti, P.R. Gene expression profiles following exposure to a developmental neurotoxicant, Aroclor 1254: Pathway analysis for possible mode(s) of action. Toxicol. Appl. Pharmacol. 2008, 231, 179–196. [Google Scholar] [CrossRef]
- Royland, J.E.; Wu, J.; Zawia, N.H.; Kodavanti, P.R. Gene expression profiles in the cerebellum and hippocampus following exposure to a neurotoxicant, Aroclor 1254: Developmental effects. Toxicol. Appl. Pharmacol. 2008, 231, 165–178. [Google Scholar] [CrossRef]
- Roegge, C.S.; Schantz, S.L. Motor function following developmental exposure to PCBS and/or MEHG. Neurotoxicol. Teratol. 2006, 28, 260–277. [Google Scholar] [CrossRef]
- Schratt, G.M.; Nigh, E.A.; Chen, W.G.; Hu, L.; Greenberg, M.E. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 2004, 24, 7366–7377. [Google Scholar] [CrossRef] [PubMed]
- Tsokas, P.; Grace, E.A.; Chan, P.; Ma, T.; Sealfon, S.C.; Iyengar, R.; Landau, E.M.; Blitzer, R.D. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 2005, 25, 5833–5843. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Impey, S.; Marks, D.; Saneyoshi, T.; Grant, W.F.; Derkach, V.; Soderling, T.R. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 2006, 50, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Do, Y.; Lee, D.K. Effects of polychlorinated biphenyls on the development of neuronal cells in growth period; structure-activity relationship. Exp. Neurobiol. 2012, 21, 30–36. [Google Scholar] [CrossRef]
- Mariussen, E.; Myhre, O.; Reistad, T.; Fonnum, F. The polychlorinated biphenyl mixture aroclor 1254 induces death of rat cerebellar granule cells: The involvement of the N-methyl-D-aspartate receptor and reactive oxygen species. Toxicol. Appl. Pharmacol. 2002, 179, 137–144. [Google Scholar] [CrossRef]
- Dreiem, A.; Rykken, S.; Lehmler, H.J.; Robertson, L.W.; Fonnum, F. Hydroxylated polychlorinated biphenyls increase reactive oxygen species formation and induce cell death in cultured cerebellar granule cells. Toxicol. Appl. Pharmacol. 2009, 240, 306–313. [Google Scholar] [CrossRef]
- Elnar, A.A.; Desor, F.; Legay, S.; Nemos, C.; Yen, F.T.; Oster, T.; Bohn, T.; Soulimani, R. No evidence for oxidative stress in the cerebellar tissues or cells of juvenile male mice exposed via lactation to the 6 non-dioxin-like PCBs at levels below the regulatory safe limits for humans. Toxicol. Lett. 2016, 245, 7–14. [Google Scholar] [CrossRef]
- Carmody, R.J.; Cotter, T.G. Signalling apoptosis: A radical approach. Redox Rep. 2001, 6, 77–90. [Google Scholar] [CrossRef]
- Ravagnan, L.; Roumier, T.; Kroemer, G. Mitochondria, the killer organelles and their weapons. J. Cell. Physiol. 2002, 192, 131–137. [Google Scholar] [CrossRef]
- Robertson, J.D.; Chandra, J.; Gogvadze, V.; Orrenius, S. Biological reactive intermediates and mechanisms of cell death. Adv. Exp. Med. Biol. 2001, 500, 1–10. [Google Scholar] [CrossRef]
- Pessah, I.N. Ryanodine receptor acts as a sensor for redox stress. Pest. Manag. Sci. 2001, 57, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Liu, G.; Xia, R.; Abramson, J.J.; Pessah, I.N. Site-selective modification of hyperreactive cysteines of ryanodine receptor complex by quinones. Mol. Pharmacol. 1999, 55, 821–831. [Google Scholar] [PubMed]
- Okabe, E.; Tsujimoto, Y.; Kobayashi, Y. Calmodulin and cyclic ADP-ribose interaction in Ca2+ signaling related to cardiac sarcoplasmic reticulum: Superoxide anion radical-triggered Ca2+ release. Antioxid. Redox Signal. 2000, 2, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Ford, G.D. Redox regulation of signal transduction in cardiac and smooth muscle. J. Mol. Cell. Cardiol. 1999, 31, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Stangler, T.; Abramson, J.J. Skeletal muscle ryanodine receptor is a redox sensor with a well defined redox potential that is sensitive to channel modulators. J. Biol. Chem. 2000, 275, 36556–36561. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Liu, G.; Allen, P.D.; Pessah, I.N. Transmembrane redox sensor of ryanodine receptor complex. J. Biol. Chem. 2000, 275, 35902–35907. [Google Scholar] [CrossRef]
- Marek, R.F.; Thorne, P.S.; Wang, K.; Dewall, J.; Hornbuckle, K.C. PCBs and OH-PCBs in serum from children and mothers in urban and rural U.S. communities. Environ. Sci. Technol. 2013, 47, 3353–3361. [Google Scholar] [CrossRef]
- Sethi, S.; Chen, X.; Kass, P.H.; Puschner, B. Polychlorinated biphenyl and polybrominated diphenyl ether profiles in serum from cattle, sheep, and goats across California. Chemosphere 2017, 181, 63–73. [Google Scholar] [CrossRef]
- Chen, X.; Lin, Y.; Dang, K.; Puschner, B. Quantification of Polychlorinated Biphenyls and Polybrominated Diphenyl Ethers in Commercial Cows’ Milk from California by Gas Chromatography-Triple Quadruple Mass Spectrometry. PLoS ONE 2017, 12, e0170129. [Google Scholar] [CrossRef]
- Sethi, S.; Keil, K.P.; Lein, P.J. 3,3’-Dichlorobiphenyl (PCB 11) promotes dendritic arborization in primary rat cortical neurons via a CREB-dependent mechanism. Arch. Toxicol. 2018, 92, 3337–3345. [Google Scholar] [CrossRef]
- Delorme, R.; Ey, E.; Toro, R.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Progress toward treatments for synaptic defects in autism. Nat. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Bourgeron, T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat. Neurosci. 2006, 9, 1221–1225. [Google Scholar] [CrossRef]
- Martinez-Cerdeno, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Shu, N.; Cao, Q.; Wang, Y.; He, Y. Imaging functional and structural brain connectomics in attention-deficit/hyperactivity disorder. Mol. Neurobiol. 2014, 50, 1111–1123. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; King, P.G.; Sykes, L.K.; Mehta, J.A.; Geier, M.R. Shared Brain Connectivity Issues, Symptoms, and Comorbidities in Autism Spectrum Disorder, Attention Deficit/Hyperactivity Disorder, and Tourette Syndrome. Brain Connect. 2015, 5, 321–335. [Google Scholar] [CrossRef]
- Ecker, C.; Bookheimer, S.Y.; Murphy, D.G. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015, 14, 1121–1134. [Google Scholar] [CrossRef]
- Jung, M.; Tu, Y.; Lang, C.A.; Ortiz, A.; Park, J.; Jorgenson, K.; Kong, X.J.; Kong, J. Decreased structural connectivity and resting-state brain activity in the lateral occipital cortex is associated with social communication deficits in boys with autism spectrum disorder. NeuroImage 2019, 190, 205–212. [Google Scholar] [CrossRef]
- Rudie, J.D.; Dapretto, M. Convergent evidence of brain overconnectivity in children with autism? Cell Rep. 2013, 5, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Keown, C.L.; Shih, P.; Nair, A.; Peterson, N.; Mulvey, M.E.; Muller, R.A. Local functional overconnectivity in posterior brain regions is associated with symptom severity in autism spectrum disorders. Cell Rep. 2013, 5, 567–572. [Google Scholar] [CrossRef]
- Moseley, R.L.; Ypma, R.J.; Holt, R.J.; Floris, D.; Chura, L.R.; Spencer, M.D.; Baron-Cohen, S.; Suckling, J.; Bullmore, E.; Rubinov, M. Whole-brain functional hypoconnectivity as an endophenotype of autism in adolescents. Neuroimage Clin. 2015, 9, 140–152. [Google Scholar] [CrossRef]
- Lein, P.J. Overview of the role of environmental factors in neurodevelopmental disorders. In Environmental Factors in Neurodevelopmental and Neurodegenerative Disorders; Costa, L.G., Aschner, M., Eds.; Elsevier: Oxford, UK, 2015; pp. 3–20. [Google Scholar]
- Barrientos, G.C.; Feng, W.; Truong, K.; Matthaei, K.I.; Yang, T.; Allen, P.D.; Lopez, J.R.; Pessah, I.N. Gene dose influences cellular and calcium channel dysregulation in heterozygous and homozygous T4826I-RYR1 malignant hyperthermia-susceptible muscle. J. Biol. Chem. 2012, 287, 2863–2876. [Google Scholar] [CrossRef] [PubMed]
- Yuen, B.; Boncompagni, S.; Feng, W.; Yang, T.; Lopez, J.R.; Matthaei, K.I.; Goth, S.R.; Protasi, F.; Franzini-Armstrong, C.; Allen, P.D.; et al. Mice expressing T4826I-RYR1 are viable but exhibit sex- and genotype-dependent susceptibility to malignant hyperthermia and muscle damage. FASEB J. 2012, 26, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.A.; Pessah, I.N. Ryanodine receptor type 1 (RyR1) possessing malignant hyperthermia mutation R615C exhibits heightened sensitivity to dysregulation by non-coplanar 2,2’,3,5’,6-pentachlorobiphenyl (PCB 95). Neurotoxicology 2007, 28, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Tochigi, M.; Kato, C.; Ohashi, J.; Koishi, S.; Kawakubo, Y.; Yamamoto, K.; Matsumoto, H.; Hashimoto, O.; Kim, S.Y.; Watanabe, K.; et al. No association between the ryanodine receptor 3 gene and autism in a Japanese population. Psychiatry Clin. Neurosci. 2008, 62, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Cantor, R.M. Allowing for sex differences increases power in a GWAS of multiplex Autism families. Mol. Psychiatry 2012, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Pasca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB Signaling Is Involved in Rett Syndrome Pathogenesis. J. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef]
- D’Andrea, I.; Fardella, V.; Fardella, S.; Pallante, F.; Ghigo, A.; Iacobucci, R.; Maffei, A.; Hirsch, E.; Lembo, G.; Carnevale, D. Lack of kinase-independent activity of PI3Kgamma in locus coeruleus induces ADHD symptoms through increased CREB signaling. EMBO Mol. Med. 2015, 7, 904–917. [Google Scholar] [CrossRef]
- Ngounou Wetie, A.G.; Wormwood, K.L.; Charette, L.; Ryan, J.P.; Woods, A.G.; Darie, C.C. Comparative two-dimensional polyacrylamide gel electrophoresis of the salivary proteome of children with autism spectrum disorder. J. Cell. Mol. Med. 2015, 19, 2664–2678. [Google Scholar] [CrossRef]
- Todd, P.K.; Mack, K.J. Phosphorylation, CREB, and Mental Retardation. Pediatric Res. 2001, 50, 672. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Kasper, L.H.; Bedford, D.C.; Lerach, S.; Teubner, B.J.; Brindle, P.K. Mutation of the CH1 Domain in the Histone Acetyltransferase CREBBP Results in Autism-Relevant Behaviors in Mice. PLoS ONE 2016, 11, e0146366. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elneel, K.; Liu, T.; Gazzaniga, F.S.; Nishimura, Y.; Wall, D.P.; Geschwind, D.H.; Lao, K.; Kosik, K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 2008, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Sarachana, T.; Zhou, R.; Chen, G.; Manji, H.K.; Hu, V.W. Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med. 2010, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Talebizadeh, Z.; Butler, M.G.; Theodoro, M.F. Feasibility and relevance of examining lymphoblastoid cell lines to study role of microRNAs in autism. Autism. Res. 2008, 1, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol. Autism. 2012, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Belinson, H.; Nakatani, J.; Babineau, B.A.; Birnbaum, R.Y.; Ellegood, J.; Bershteyn, M.; McEvilly, R.J.; Long, J.M.; Willert, K.; Klein, O.D.; et al. Prenatal beta-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry 2016, 21, 1417–1433. [Google Scholar] [CrossRef]
- Bocchi, R.; Egervari, K.; Carol-Perdiguer, L.; Viale, B.; Quairiaux, C.; De Roo, M.; Boitard, M.; Oskouie, S.; Salmon, P.; Kiss, J.Z. Perturbed Wnt signaling leads to neuronal migration delay, altered interhemispheric connections and impaired social behavior. Nat. Commun. 2017, 8, 1158. [Google Scholar] [CrossRef]
- Bal-Price, A.; Crofton, K.M.; Sachana, M.; Shafer, T.J.; Behl, M.; Forsby, A.; Hargreaves, A.; Landesmann, B.; Lein, P.J.; Louisse, J.; et al. Putative adverse outcome pathways relevant to neurotoxicity. Crit. Rev. Toxicol. 2015, 45, 83–91. [Google Scholar] [CrossRef]
- Heiger-Bernays, W.J.; Tomsho, K.S.; Basra, K.; Petropoulos, Z.E.; Crawford, K.; Martinez, A.; Hornbuckle, K.C.; Scammell, M.K. Human health risks due to airborne polychlorinated biphenyls is highest in New Bedford Harbor communities living closest to the harbor. Sci. Total Environ. 2019. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model | Exposure | Dose(s) | Route of Exposure | Exposure Window | Findings | Ref. |
|---|---|---|---|---|---|---|
| Mouse (ICR) | A1254 | 6 mg/kg/d, 18 mg/kg/d | Gavage | Lactation (PND 7–21), Postnatal (PND 22–42) | ↑ Locomotor activity in females | [74] |
| Mouse (ICR) | A1254 | 18 mg/kg/d | Injection (i.p.) | Prenatal (GD 6-PND 0), Lactation (PND 0–21) | ↑ Locomotor activity | [75] |
| Mouse (Swiss albino) | NDL PCB mixture (PCBs 28, 52, 101, 138, 153, 180) | 1 ng/kg/d, 10 ng/kg/d, 100 ng/kg/d | Gavage | Lactation (PND 0–21) | ↓ Locomotor activity in 1 and 10 ng/kg males | [76] |
| Rat (Sprague-Dawley) | A1221 | 0.5 mg/kg, 1 mg/kg | Injection (i.p.) | Prenatal (GD 16, 18) | ↑ Distance traveled in LDB in male offspring | [77] |
| Rat (Wistar) | PCB 52, PCB 138, PCB 180 | 1 mg/kg/d | Dietary (jelly) | Prenatal (GD 7-PND 0), Lactation (PND 0–21) | ↓ Locomotor activity in PCB 138 and PCB 180 groups in males; ↓ Locomotor activity in PCB 138 group in females | [78] |
| Rat (Wistar) | PCB 52, PCB 138, PCB 180 | 1 mg/kg/d | Dietary (jelly) | Prenatal (GD 7-PND 0), Lactation (PND 0–21) | ↓ Time spent on rotarod in PCB 52 group for both sexes | [79] |
| Model | Exposure | Dose(s) | Route of Exposure | Exposure Window | Findings | Ref. |
|---|---|---|---|---|---|---|
| Mouse (CD1) | NDL PCB mixture (PCBs 28, 52, 101, 138, 153, 180) | 10 ng/kg/d, 1 µg/kg/d | Dietary (chow) | Prenatal (GD 6-PND 0), Lactation (PND 0–21) | ↑ Sociability in early adulthood in both sexes; ↑ Social novelty in both sexes (1 µg/kg/d); ↓ Social interaction in middle-aged males (1 µg/kg/d) | [89] |
| Rat (Sprague-Dawley) | A1221 | 1 mg/kg | Injection (i.p.) | Prenatal (GD 16, 18, 20), Postnatal 1 (PND 24, 26, 28) | ↑ USV calls in female (prenatal and postnatal); ↑ Affiliative wrestling behavior in female (prenatal and postnatal); ↓ Time spent near novel animal of opposite sex in males exposed postnatally | [90] |
| Rat (Sprague-Dawley) | A1221 | 0.5 mg/kg, 1 mg/kg | Injection (i.p.) | Prenatal (GD 16, 18) | ↓ Social interaction with novel conspecific in 0.5 mg/kg males | [91] |
| Rat (Sprague-Dawley) | A1221 | 0.5 mg/kg/d, 1 mg/kg/d | Injection (i.p.) | Prenatal (GD 16, 18) | ↑ USV calls in males in sociosexual context; No effect on sociosexual behavior in females; ↓ Sociosexual interaction (nose-nose sniffs) in males | [92] |
| Rat (Sprague-Dawley) | Mixture (PCBs 47, 77) | 12.5 mg/kg/d, 25 mg/kg/d | Dietary (chow) | Prenatal (GD 0-PND 0), Lactation (PND 0–21) | ↓ Social recognition in 25 mg/kg males; ↓ Social investigation in males (both doses) | [93] |
| Model | Exposure | Dose(s) | Route of Exposure | Exposure Window | Findings | Ref. |
|---|---|---|---|---|---|---|
| Mouse (ICR) | A1254 | 6 mg/kg/d, 8 mg/kg/d | Gavage | Lactation (PND 7–21), Juvenile (PND 22–42) | ↓ NOR performance in females; No effect on alternation behavior in either sex | [74] |
| Mouse (ICR) | A1254 | 18 mg/kg/d | Injection (i.p.) | Prenatal (GD 6-PND 0), Lactation (PND 0–21), Postnatal (PND 21–35) | ↓ Object-based attention in NOR task; ↓ Latency to enter open arms of EPM; ↑ Time spent in open arms of EPM | [75] |
| Mouse (Swiss albino) | NDL PCB mixture (PCBs 28, 52, 101, 138, 153, 180) | 1 ng/kg/d, 10 ng/kg/d, 100 ng/kg/d | Gavage | Lactation (PND 0–21) | ↑ Escape latency in water escape task in males (1 and 100 ng/kg), ↑ Anxiety-like behavior in EPM and LDB tasks (1 and 10 ng/kg), No effect on performance in MWM task | [76] |
| Mouse (Swiss albino) | NDL PCB mixture (PCBs 28, 52, 101, 138, 153, 180) | 10 ng/kg/d | Gavage | Lactation (PND 0–21) | No effect on short-term memory in spontaneous alternation task; No effect on learning acquisition in MWM task | [97] |
| Rat (Sprague-Dawley) | A1221 | 1 mg/kg/d | Injection (i.p.) | Prenatal (GD 16, 18, 20), Postnatal 1 (PND 24, 26, 28) | ↑ Entries into and time spent in open arms of EPM in females during both periods | [90] |
| Rat (Long-Evans) | Fox River | 3 mg/kg/d, 6 mg/kg/d | Dietary (cookie) | Pre-conception (28 d), Prenatal (GD 0-PND 0), Lactation (PND 0–21) | ↓ DRL performance in 3 mg/kg females; ↓ Inhibitory control in both sexes (both doses) | [98] |
| Rat (Long-Evans) | Fox River | 3 mg/kg/d, 6 mg/kg/d | Dietary (cookie) | Postnatal 1 (PND 27–50) | ↑ Response latency in cue discrimination phase of set-shifting task in males (3 mg/kg/d); ↓ Errors to criterion in position reversal in males (both doses); ↓ Perseverative errors in position reversal in males (both doses); No effect on DRL performance | [99] |
| Rat (Wistar) | PCB 52, PCB 138, PCB 180 | 1 mg/kg/d | Dietary (jelly) | Prenatal (GD 7-PND 0), Lactation (PND 0–21) | ↓ Learning in Y maze visual discrimination task for PCB 138 and 180 in both sexes | [79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klocke, C.; Lein, P.J. Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 1013. https://doi.org/10.3390/ijms21031013
Klocke C, Lein PJ. Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. International Journal of Molecular Sciences. 2020; 21(3):1013. https://doi.org/10.3390/ijms21031013
Chicago/Turabian StyleKlocke, Carolyn, and Pamela J. Lein. 2020. "Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity" International Journal of Molecular Sciences 21, no. 3: 1013. https://doi.org/10.3390/ijms21031013
APA StyleKlocke, C., & Lein, P. J. (2020). Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. International Journal of Molecular Sciences, 21(3), 1013. https://doi.org/10.3390/ijms21031013