Leptin, Adiponectin, and Sam68 in Bone Metastasis from Breast Cancer


IRCCS Istituto Ortopedico Galeazzi, Via R. Galeazzi 4, 20161 Milano, Italy
Int. J. Mol. Sci. 2020, 21(3), 1051; https://doi.org/10.3390/ijms21031051
Submission received: 16 January 2020
/
Revised: 3 February 2020
/
Accepted: 4 February 2020
/
Published: 5 February 2020
(This article belongs to the Special Issue Adiposity and Adipokines: Roles in the Local Spread of Cancer and the Metastasis Development)
Abstract
:The most serious aspect of neoplastic disease is the spread of cancer cells to secondary sites. Skeletal metastases can escape detection long after treatment of the primary tumour and follow-up. Bone tissue is a breeding ground for many types of cancer cells, especially those derived from the breast, prostate, and lung. Despite advances in diagnosis and therapeutic strategies, bone metastases still have a profound impact on quality of life and survival and are often responsible for the fatal outcome of the disease. Bone and the bone marrow environment contain a wide variety of cells. No longer considered a passive filler, bone marrow adipocytes have emerged as critical contributors to cancer progression. Released by adipocytes, adipokines are soluble factors with hormone-like functions and are currently believed to affect tumour development. Src-associated in mitosis of 68 kDa (Sam68), originally discovered as a protein physically associated with and phosphorylated by c-Src during mitosis, is now recognised as an important RNA-binding protein linked to tumour onset and progression of disease. Sam68 also regulates splicing events and recent evidence reports that dysregulation of these events is a key step in neoplastic transformation and tumour progression. The present review reports recent findings on adipokines and Sam68 and their role in breast cancer progression and metastasis.
1. Introduction
Bone metastases are a leading cause of death in patients with advanced breast and prostate cancers. Mineral content, matrix composition, extreme rigidity, highly hypoxic environment, acidic pH, and a high concentration of extracellular calcium all make the bone an attractive site for colonization by cancer cells. Furthermore, residing within the bone matrix is a vast array of cytokines, growth factors, and matrix proteins that can contribute to the development of bone metastases. Host cells in the tumour microenvironment drive metastatic behaviour and influence tumour growth and progression. In addition, the bone marrow environment contains a variety of cells, including mesenchymal stem cells (MSC), which give rise to several cell lineages such as osteoblasts and adipocytes.
Once considered passive fillers of the bone marrow niche or cells that occupy the space after trabecular bone loss, adipocytes are now known to be endocrine targets and to possess endocrine-like functions: besides responding to growth hormones, insulin, and thyroid hormone, they release a plethora of adipokines among which leptin and adiponectin [1]. The present review focuses on these two factors and their possible roles in bone marrow colonization by breast carcinoma cells. Furthermore, I discuss the Src-associated in mitosis of 68 kDa (Sam68), a signal transduction and activation of RNA (STAR) protein and eclectic component of signal transduction of leptin, which is strongly involved in breast cancer progression. The clinical consequences of bone metastases from breast cancer, as for other types of cancer, are often associated with reduced quality and life expectancy of patients due to skeletal-related events (SRE), which include pathological bone fractures, spinal cord compression, severe pain, and hypercalcaemia [2,3,4], with an increase in healthcare costs. Shedding light on the mechanisms that link adipokines and breast cancer progression and metastasis could help to identify new therapeutic opportunities.
2. Bone Marrow Adipose Tissue (BMAT), the Third Fat Depot
Now recognized as the body’s largest endocrine organ, adipose tissue secretes a variety of different factors: adipokines, cytokines, and exosomal microRNAs [5]. The two main subtypes of adipose tissue are white and brown adipose tissue (WAT and BAT, respectively). WAT, the more abundant of the two, is localized subcutaneously and viscerally. Its main function is to release energy from lipid stores as needed. In contrast, BAT is found in variable amounts in more distinct areas in adult humans. BAT works in adaptive thermogenesis through the uncoupling of ATP production and substrate oxidation [6].
A less known third type of adipose tissue is BMAT, which has garnered recent interest owing to its probable role in cancer progression. This discovery is particularly relevant for breast and prostate carcinoma-derived metastasis and for multiple myeloma arising in the bone. BMAT constitutes a major component of the bone marrow microenvironment in which the number of adipocytes increases with age: 40–70% of appendicular skeleton of lean adults (after age 25 years) is occupied by adipocytes [7,8], with continued gradual accumulation of BMAT throughout life [9].
Two types of adipocytes make up BMAT: constitutive bone marrow adipocytes (cBMA) residing in distal areas of bones and regulated bone marrow adipocytes (rBMA) found in more metabolically active bone sites (Figure 1).
The two adipocyte populations differ also in lipid content: rBMA stores prevalently saturated fats whereas cBMA stores unsaturated fats [10]. rBMA undergo constant changes during aging and promptly react to changes in nutritional, hormonal, and temperature states.
Furthermore, BMAT serves as an energy reservoir and a producer of cytokines and adipokines. Its unique characteristics make it different from WAT or BAT. BMAs are derived from multipotent mesenchymal progenitors that express Osterix, a transcription factor essential for osteoblastogenesis. In further contrast to WAT, BMAT expands under conditions of energy restriction and does not seem to be affected by energy surplus, at least not in humans. BMAs can influence the balance between adipogenesis and osteoblastogenesis: through the secretion of extracellular vesicles containing adipogenic mRNA transcript, BMAs weaken osteoblast lineage commitment [11]. BMAs can thus act as negative regulators of osteoblastogenesis by reducing the number of osteoblasts and decrease bone formation and resistance as a consequence. BMAs also inhibit haematopoiesis. Indeed, the haematopoietic activity in bone marrow is inversely correlated with adiposity [12]. The influence of BMAs in the context of cancer development and progression is still poorly understood, however.
The contribution of BMAs to tumour growth and metastasis has been amply discussed (Figure 2). Adipocytes exert a critical role in disease progression in the tumour microenvironment by providing cancer cells with fatty acids, pro-inflammatory cytokines, and proteases [13,14,15]. Also, BMAs promote tumour cell oxidative stress [16] and metabolic reprogramming toward a glycolytic phenotype [17]. BMAT is crucial in the development of blood disease, in which BMAs directly interact with blood tumour cells in the bone marrow. In addition, BMAT has been identified as a key driver in the progression of multiple myeloma [1,18,19].
3. Adipocytes as Source of Factors in Tumour Stroma: Adiponectin and Leptin
Adipokines are soluble factors with hormone-like functions secreted by adipocytes. The most extensively studied are leptin, adiponectin, inflammatory factors such as interleukin (IL)-6, and tumour necrosis factor (TNF)-α, autotaxin, and hepatocyte growth factor (HGF). In the stroma of breast cancer, adipocytes, along with fibroblasts, macrophages, and cancer cells produce adipokines such as leptin, adiponectin, autotoxin, and IL-6. Breast cancer cells express adipokines’ receptors; adipokines can induce initiation, progression, and metastasis of breast cancer cells with different effects.
Adipokines activate several signalling networks in target cells, including the JAK/STAT, AKT, and ERK1/2 signalling pathways, which are frequently activated in tumour tissues [20,21]. These pathways are associated with cell migration, proliferation, angiogenesis, fibrosis, and apoptosis. Moreover, IL-6 or leptin can increase procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) expression in breast cancer cells through the activation of the JAK/STAT3 and the AKT signalling pathway. PLOD2 contributes to the formation of triple helical pro-collagen molecules by triggering the hydroxylation of collagen lysine residues [22,23]. PLOD2 deregulation has been described in sarcoma, breast cancer, glioma, lung cancer, and cervical cancer [23,24,25,26,27].
3.1. Adiponectin
Adiponectin (ACRP30), a peptide hormone of 30kDa, plays a major role in glucose metabolism and energy homeostasis [28]. After its secretion by adipocytes as a monomeric protein, adiponectin oligomerizes to form low-molecular-weight, high-molecular-weight, and multimeric complexes [29]. Adiponectin binds to seven pass receptors, AdipoR1 and AdipoR2, and acts centrally to enhance energy homeostasis by increasing energy expenditure through the activation of hypothalamic leptin and insulin signalling pathways. It also improves glucose homeostasis by attenuating insulin resistance. Adiponectin influences adipose tissue functions via autocrine effects: ADIPOQ gene overexpression increases the number of adipocytes and adipose tissue mass [18].
WAT was once considered the main source of adiponectin; more recently it was reported that also BMAT secretes a large amount of adiponectin chiefly in the presence of cancer cells [30,31]. Though adiponectin is produced by adipocytes, circulating adiponectin levels in the obese are lower than in the non-obese (the so-called adiponectin paradox) [32] and the increase in serum adiponectin is associated with weight loss (i.e., in patients with anorexia nervosa or in animal models of caloric restriction) [33,34]. Adiponectin is considered a “good” adipokine because of its anti-inflammatory, anti-atherogenic, and insulin-sensitizing properties.
The relationship between adiponectin and bone cells is complex and not completely elucidated. Studies have reported an osteogenic property of adiponectin, as the adipokine is able to stimulate osteogenic differentiation of bone marrow mesenchymal stromal cells and adipose-derived stem cells vs. the osteogenic lineage [35,36].
Adiponectin and Breast Cancer/Bone Metastasis
Numerous studies have attempted to shed light on the role of adiponectin in carcinogenesis.
The in vitro exposure of cancer cell lines (e.g., breast, liver, colon, stomach, and endometrium) to adiponectin inhibits proliferation and induces apoptosis [37,38,39,40,41]. Though it has been reported that adiponectin negatively influences breast carcinogenesis [42], the question remains open and the answer probably depends on hormonal status (ER-PR expression in particular) [43]. Several in vivo and in vitro studies demonstrated that adiponectin in ER negative breast cancer suppresses cell proliferation, invasion, and migration and induces cell growth arrest and apoptosis [37,42,44,45,46], whereas in ER positive breast cancer it increases cell proliferation [47,48]. The review by Panno et al. explains the effect of adiponectin on ER positive and ER negative breast cancer cells [49].
It has also been reported that adiponectin inhibits the metastatic process via suppression of the adhesion, invasion, and migration of breast cancer cells through activation of the AMPK/S6K axis and upregulation of LKB1 [50]. Other studies reported that an increase in the level of globular adiponectin in tumour microenvironment autophagy supports the early stages of metastatic progression [51].
Adiponectin has been found in exosomes derived from adipocytes. Exosomes are extracellular vesicles that mediate cell-to-cell signalling in the tumour microenvironment. Exosomes from human adipose-derived mesenchymal stem cells induce proliferation and migration of breast cancer cells [52] and exosomes secreted by preadipocytes also regulate breast tumour stem cell formation and migration [53]. Further research is needed to discern the role of adiponectin in this context.
3.2. Leptin
Leptin, a hormone produced primarily by adipocytes, coordinates energy homeostasis by signalling from adipose tissue to the hypothalamus; its synthesis and plasma concentration increase proportionally to adipose tissue mass. Leptin acts by binding to leptin receptors (Ob-Rs) encoded by the LEPR gene and members of the family of class I cytokine receptors. Ob-R presents six isoforms (Ob-Ra, Ob-Rb, Ob-Rc, Ob-Rd, Ob-Re, and Ob-Rf) generated by alternative splicing of the gene. Leptin produces different effects in various organs by binding to central or peripheral receptors. As regards its effects on bone remodelling, central leptin receptors mediate bone loss, whereas the binding of leptin to peripheral receptors results in an increase in bone mass [54,55].
Leptin helps regulate bone health by modulating bone density and growth and adiposity. In the bone marrow, mature adipocytes release leptin, which in turn enhances the formation of BMAs by binding to Ob-R on bone marrow mesenchymal stem cells (BMSCs) to promote their adipogenesis [56]. More importantly, Ob-R (+) BMSCs are the main source of BMAs [57]. Bone cells, including osteoblasts, secrete low amounts of leptin [58,59]. It has been reported that, in the primary culture of human osteoblasts, leptin is expressed and secreted during the late maturation phase to promote bone mineralization [58]. Osteoblasts and adipocytes both originate from mesenchymal stem cells and share common markers.
3.2.1. Leptin and Breast Cancer
Studies using both in vivo and in vitro experimental models have extensively demonstrated the involvement of leptin in many aspects of breast cancer biology starting from the early stages of primary tumour to metastatic progression. In addition to adipose tissue, cancer cells can also secrete leptin and overexpress leptin receptors.
Studies on breast cancer patients have revealed an association between higher serum leptin levels and aggressive malignant tumour features. Leptin expression in normal breast tissue adjacent to ductal carcinoma and its absence in the breast tissue of healthy adults suggests that leptin is involved in the early stages of breast cancer tumorigenesis [60]. Nevertheless, while some studies found a negative correlation between serum leptin levels and breast cancer development in premenopausal women but a positive one in postmenopausal women [61], other studies reported no association between serum leptin and breast cancer development [62].
In their meta-analysis to evaluate the association between serum leptin levels and breast cancer risk [63], Gu et al. reported that the apparent discrepancies among the findings were related to the heterogeneity across the studies: different genetic backgrounds, different stages and types of breast cancer, different analytic methods to measure serum leptin levels, different menstrual and treatment status of breast cancer patients, different demographics and clinical characteristics.
Tumour tissue expression of leptin and/or of Ob-R has been found to correlate with aggressive tumour behaviour [64]. Adipocyte-secreted leptin and IL-6 have a paracrine effect on nearby breast cancer cells. One of the mechanisms through which leptin causes breast cancer progression is the regulation of breast cancer stem cell (CSC) activity by modulating the many signalling pathways (i.e., Notch, Wnt, mTOR, STAT3, HER2/Erb, and IGF pathways) and transcription factors (i.e., HIF-1 and NFκB) [65,66,67,68,69]. Leptin signalling in CSC is also involved in cancer recurrence and drug resistance.
Guo et al. reported a complex crosstalk between leptin, Notch, and Interleukin-1 (IL-1) (NILCO, Notch, IL-1, and leptin crosstalk outcome) that seems to be the integration of developmental, pro-inflammatory, and pro-angiogenic signals critical for leptin-induced cell proliferation/migration and regulation of VEGF/VEGFR-2 in breast cancer [65]. In obese breast cancer patients with often chronically elevated leptin levels, leptin NILCO signalling seems to provide a link between obesity and cancer [64]. The authors speculated that targeting NILCO may be a new pharmacological strategy to control breast cancer growth and angiogenesis [65,70].
Recently, Giordano C et al. found that leptin regulates exosome biogenesis and release in different models of breast cancer cells via the leptin receptor/Hsp90 axis. This additional mechanism in cell-to-cell communication is part of a biological process that is crucial for key events in breast cancer development and progression [71]. The authors suggested that impairing exosome secretion and interrupting the dangerous cell-to-cell crosstalk could counteract cancer progression. Furthermore, knowledge of the signals in the biogenesis of exosome in breast cancer (i.e., leptin signalling in obese patients) will be important for the design of new therapies [71].
As regards the tumour microenvironment within the mammary gland, leptin can modify the tumour to modulate migration of endothelial cells, angiogenesis, macrophage phenotypes and functions, and recruitment of monocytes and neutrophils. The interaction of leptin with cytokines can also influence breast cancer progression: leptin acts on both tumour cells and tumour stroma to secrete inflammatory cytokines such as IL-1, IL-6, TNF-α, and growth factors.
3.2.2. Leptin and Bone Metastasis
In addition to its effect on breast cancer growth, leptin may also play an important role in bone metastasis. A growing body of evidence shows that leptin may promote epithelial to mesenchymal transition (EMT) by up-regulating the expression of EMT-and metastasis-related genes (Twist2, Foxc2, Vim, Akt3, and Sox2) in the mouse mammary tumour virus (MMTV)-Wnt-1 transgenic mouse model of triple-negative breast cancer [72], highlighting the potential roles of leptin in metastasis development. Bowers and colleagues hypothesized that regulation of the EMT gene, via stimulation of the JAK2/STAT3 and/or PI3K/AKT pathways, may mediate the leptin-induced CSC/EMT-related phenotype.
Recently, Juárez-Cruz et al. hypothesized that leptin promotes FAK and Src activation in breast cancer cells and induces cell migration, metalloproteases secretion (MMP2 and MMP9), and invasion. They suggested that leptin is associated with the establishment of a more aggressive phenotype of cancer cells that leads to local invasion and to the formation of metastases [73].
Leptin is known to influence tumour-associated macrophages (TAMs), a critical component in the tumour microenvironment and a major source of inflammatory cytokines [74]. Stimulation of macrophage function by leptin leads to the release of pro-inflammatory cytokines, resulting in pro-inflammatory effects. Clinical evidence shows that the presence of macrophages within the tumour microenvironment correlates with poor prognosis in cancer patients. TAMs may play a major role in cancer progression, including EMT induction. Analysis of the transcriptome of TAMs in murine models of breast cancer indicated that an enrichment in macrophage transcripts is predictive of poor prognosis and reduced survival [75]. Cao and colleagues showed that leptin, by triggering M2 macrophage-related cytokine IL-18 production, contributes to tumour progression and the development of metastasis. These extremely important results add another link (leptin-Ob-R-IL-18) to the complex interaction between the tumour microenvironment and breast cancer cells that drives cancer progression [76].
The expression of leptin and/or Ob-R by tumour tissue indicates aggressive behaviour. Ishikawa et al. reported that distant metastasis was detected in patients with Ob-R-positive tumours overexpressing leptin, whereas it was absent in patients with tumours that lacked Ob-R expression or leptin overexpression [64].
He et al. found that adipocyte-derived IL-6 and leptin promote PLOD2 expression in breast cancer, an event that promotes breast cancer metastasis [77]. In addition, fat tissue accumulation may contribute to enhance the expression of PLOD2 [69]. The availability of PLOD2 is one way by which IL-6 and leptin may influence collagen re-organization to create a “highway” for cancer cell migration and invasion to promote metastasis [78].
Leptin can affect also the adhesion of cancer cells to the extracellular matrix (ECM) and the proteolysis of ECM components [79,80], essential steps in tumour metastasis. Tsai et al. identified a soluble intercellular adhesion molecule (sICAM)-1 as a downstream effector of leptin: the adhesion molecule is able to induce osteoclastogenesis. With their in vivo study, the authors showed that, in the bone microenvironment, leptin enhanced sICAM-1 production by tumour cells and induced bone erosion. Injection of ICAM-neutralizing antibody reduced the osteolytic process in tumour-bearing mice. These findings led the authors to speculate that sICAM could be a potential therapeutic target or diagnostic tool for bone metastases from lung and breast cancer [81].
4. The RNA-Binding Protein Src-Associated in Mitosis of 68 kDa (Sam68)
Src-associated in mitosis of 68 kDa (Sam68) was originally discovered as a protein that is physically associated with and phosphorylated by c-Src during mitosis [82]. Sam68, an RNA-binding protein (a STAR, signal transduction and activation of RNA family member) that links cellular signalling to RNA processing, is markedly expressed in breast cancer cell and tissues. Sam68 has been shown to exert a dual role as an RNA-binding protein and as a docking protein in different cellular contexts [83,84].
Sam68 presents protein–protein interaction motifs that enable it to function as a scaffold protein in multiple signal transduction pathways and in transcription complexes. Moreover, as an RNA-binding protein, Sam68 can modulate alternative splicing and translation of target mRNAs. Sam68 participates in the signal transduction of various receptors, including insulin, T-cell receptor, as well as EGF, HGF/Met, TNFR1, and leptin.
Studies have produced conflicting findings for Sam68 involvement in cancer; the discrepancies arise from the cellular context and the cell type considered. Sam68 was originally defined as a tumour suppressor protein [85,86], however, direct investigation of Sam68 expression and function in cancer cells suggest a pro-oncogenic role for the protein [87,88].
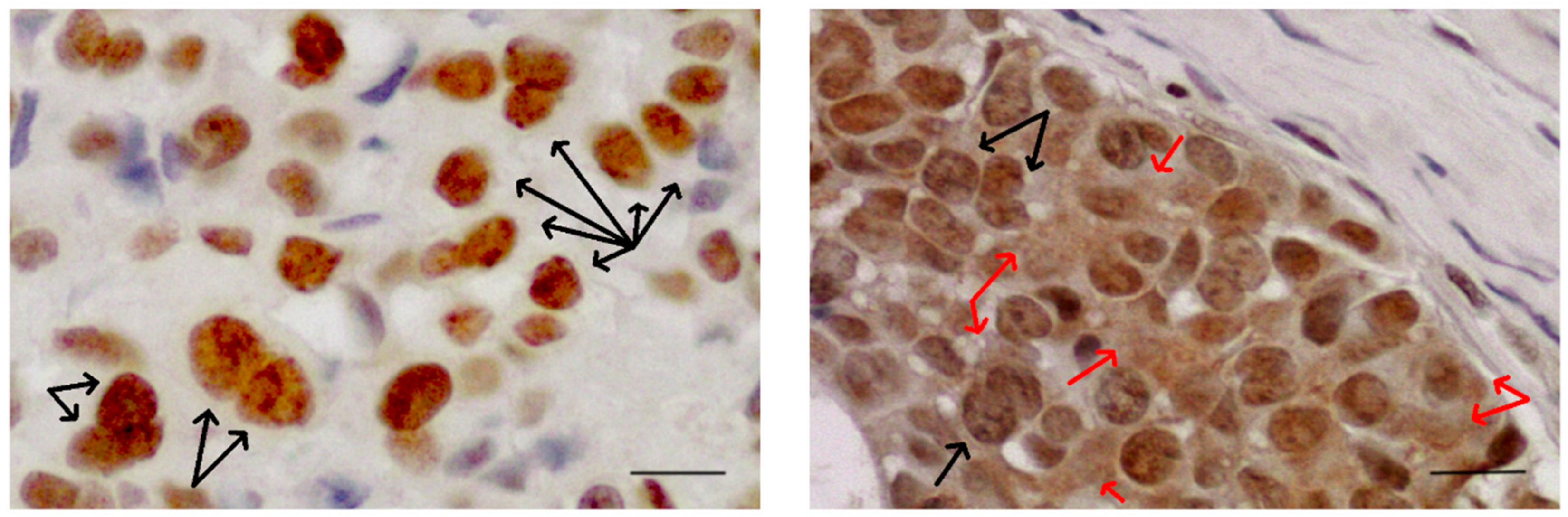
It is now known that Sam68 interacts with the SH3 domain of different members of the Src kinase family, like BRK [89], FYN [90], and Itk/Tec/BTK [91], all involved in different aspects of cell transformation. Sam68 is usually found in the nucleus where it is concentrated in distinct sub-nuclear structures (nuclear speckles) SLM/Sam68 nuclear bodies (SNBs, indicated by black arrows in Figure 3), structures that are enriched in cancer cells [92]. Sam68 in the cytoplasm is significantly associated with poor prognosis and progression in renal cell carcinoma [93], cervical cancer [94], and breast cancer [95].
Moreover, the deregulation of SRC and AKT pathways may be involved in the oncogenic function of Sam68 in the cytoplasm. The subcellular localization of Sam68 (cytoplasm or nucleus) reflects its different functions in various signalling pathways. It can contribute to neoplastic transformation or tumour progression through different molecular mechanisms in different cancer types or cellular contexts [96]. Sam68 in the cytoplasm in advanced tumour stages may contribute to neoplastic transformation through the translational enhancement of specific mRNAs [97].
The upregulation of Sam68 is reported in prostate cancer and in highly proliferative lesions of human breast cancer. Its downregulation seems to promote inhibition of cell proliferation and sensitization of cells to apoptosis induced by chemotherapeutic agents in prostate cancer [98], while silencing of Sam68 in breast cancer cells inhibits cancer proliferation. In their study on Sam68-null mice, Richard et al. found that the absence of Sam68 delays mammary tumorigenesis, metastasis, and cell migration, giving further evidence for the role of Sam68 in cell transformation [88]. Moreover, they demonstrated that activated Src and FAK kinases are present in Sam68-null mice but not wild type mice, suggesting that Sam68 may be a modulator of these pathways during cancer progression [88].
Sam68 is regulated at the post-translational level by phosphorylation [99], acetylation [100], methylation [101], and SUMO (small ubiquitin-like modifier) modification (SUMOylation, a post-translational modification analogous to ubiquitylation and involving the addition of SUMOs to proteins instead of ubiquitin) [102]. Such modifications can affect its subcellular localization, interaction with signalling proteins, as well as affinity for target RNAs [103,104]. Tyrosine phosphorylation regulates both the signal transduction and RNA-binding properties of Sam68 [89,105]. Tyrosine phosphorylation of Sam68 is elevated in human breast and prostate tumour tissues and cell lines [100,104,105,106]. Sam68 expression level and phosphorylation status play a role in tumorigenesis and indicate Sam68 as a proto-oncogene [107].
In human breast cancer cells, Sam68 participates in leptin receptor signalling by modulating the trophic effects of the hormone in cellular proliferation and growth. Leptin and insulin increase Sam68 expression in breast cancer cell lines, with an increase in its tyrosine phosphorylation [97]. Pérez-Pérez et al. demonstrated that Sam68 plays a role in leptin and insulin signal transduction pathways in three different breast carcinoma cell lines by activating MAPK and PI3K signalling pathways and partially modulating the expression of IRS1 [108]. Sam68 may be considered a point of convergence for various different signals involved in breast cancer growth and progression. Finally, Sam68 intervenes in EMT/Mesenchymal to epithelial transition (MET) programs. Because of the relevance of EMT/MET in the formation of metastases, Sam68 may play a role in cancer progression.
5. Alternative Splicing Regulates EMT: Role of Sam68
Alternative splicing of pre-mRNA is a major driver of protein diversity and affects more than 95% of multi-exonic coding human genes [109]. Upregulation of endogenous alternatively spliced tumorigenic variants is frequently observed in many cancers such as the pro-angiogenic splice variant of vascular endothelial growth factor (VEGF165), an EMT-inducing isoform of Rac1 (Rac1b), and the cancer-initiating form of protein kinase C (PKC) betaII [110,111,112,113].
Alternative splicing generates multiple mRNAs from a single transcript. It is fundamental for proteomic diversity and control of gene expression. In addition, it is involved in essential biological events such as development, cell differentiation, organogenesis, and response to environmental stimuli. The deregulation of alternative splicing leads to tumour biogenesis and progression [114,115].
Neoplastic cells positively exploit the biological flexibility that, via the production of splice variant proteins, promotes tumour growth and survival. Proteome remodelling by alternative splicing enables cancer cells to produce proteins that meet the needs of the tumour as it grows and spreads.
Many cancer-associated genes are regulated via alternative splicing, suggesting a significant role for this post-transcriptional regulatory mechanism in the production of oncogenes and tumour suppressors. Aberrant regulation of alternative splicing is emerging as a basic step in oncogenesis [116] and it is considered another hallmark feature of cancer among the growing list of tumour growth characteristics [117].
A critical step in cancer cell dissemination and progression is the transition towards a mesenchymal phenotype via the activation of EMT. EMT requires robust reprogramming of gene expression. Mounting evidence indicates an important contribution of alternative splicing regulation in EMT; the chief factors involved in EMT are regulated post-transcriptionally by alternative splicing. Tissue-specific alternative splicing regulators include the neuron specific RNA-binding proteins 1/2 (NOVA1/2), the polypyrimidine tract binding protein 2 (PTBP2), the serine/arginine repetitive matrix 4 protein (SRRM4), and members of the RBFOX, Muscle blind-like (MBNL) proteins, CUGBP Elav-like family (CELF), T-cell intracellular antigen (TIA), epithelial splicing regulatory protein (ESRP) and STAR families. Sam68 was the first “hub factor” to be identified that translates extracellular stimuli to pre-mRNA processing of specific target genes in the nucleus and causes specific RNA splicing decisions [118]. The CD44 gene is an example in which Sam68 exerts a link between signal transduction cascades and alternative splicing [119]. The alternative splicing generated CD44v, epithelial isoforms, or CD44s, mesenchymal isoform; the transition from CD44v to CD44s is thought to play a central role in EMT. Other Sam68/EMT/alternative splicing interactions were described by Valacca and colleagues, who reported that Sam68 might contribute to the malignant transformation of epithelial cancers by inducing the proto-oncogene SRSF1 (serine/arginine-rich splicing factor 1, historically known as SF2/ASF) and stimulating a post-transcriptionally regulated EMT [107]. Also, in mesenchymal cells Sam68 phosphorylation by the extracellular regulated kinase 1/2 (ERK1/2) increases the full-length transcript of SRSF1 (splicing factor), thus increasing SRSF1 protein levels that, in turn, drive a ΔRon (an oncogenic variant of constitutively active Ron) that triggers EMT [120]. Diffusible factors secreted by epithelial cells (human colon carcinoma-derived SW480 cells grown at high density) repress ERK1/2 activity (in SW480 cells grown at low density) and Sam68 phosphorylation, thus reducing SRSF1 protein levels and promoting the inclusion of Ron exon 11 and MET [107]. Anti-metastatic therapeutic strategies may be addressed to cancer-specific splicing variants involved in EMT [109].
Moreover, Sam68 regulates the apoptotic gene BCL-X via alternative splicing in cancer cells [105], leading to an alteration in the BCL-XS/BCL-XL ratio (pro-apoptotic and anti-apoptotic, respectively) [105]. Tyrosine phosphorylation of Sam68 may be one way that cancer cells protect against apoptosis.
6. Conclusions
BMAT appears to play a pivotal role in creating a microenvironment suitable for the colonization of bone by cancer cells via the release of numerous different factors. In addition, obesity mediates its effect on cancer progression via the dysregulation of adipokine release. Table 1 compares the role of the three molecules in the onset and progression of breast cancer. Leptin and adiponectin may exert antagonistic molecular effects on cancer progression. Further research into this aspect is particularly important because published data are sometimes conflicting. Leptin and adiponectin released into the tumor microenvironment at different times have opposite effects on tumor growth. Intracellular signals could favor the effect of one factor over the other. Indeed, leptin can intervene in different ways to sustain metastatic cells. The parallelism between leptin and Sam68 in their ability to promote cancer cells indicates that Sam68 is an intracellular mediator involved in leptin activity in cancer cells. The leptin-Sam68 axis appears to be essential for the growth and progression of breast cancer. Some observations may be relevant only for a particular cellular context. In vitro studies evaluating the effect of a microenvironment factor on tumoral cell lines do not take into account the simultaneous presence of other molecules, as occurs in vivo, that interact with each other. Advances in this field may open new insights into therapeutic opportunities for treating bone metastasis.
Funding
This work was supported by grants from the Ministero della Salute (Ricerca Corrente L4084, 2016; L4101, 2017).
Acknowledgments
I wish to thank Paola Bendinelli for her support and useful critique.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| MSC | Mesenchymal stem cell |
| STAR | Signal Transduction and Activation of RNA |
| WAT | White adipose tissue |
| BAT | Brown adipose tissue |
| BMAT | Bone marrow adipose tissue |
| cBMA | Constitutive bone marrow adipocytes |
| rBMA | Regulated bone marrow adipocytes |
| PLOD2 | 2-oxoglutarate 5-dioxygenase 2 |
| AMPK | Adenosine-monophosphate activated protein kinase |
| LKB1 | Liver kinase B1 |
| Ob-R | Leptin receptor |
| BMSC | Bone marrow mesenchymal stem cell |
| CSC | Cancer stem cell |
| NILCO | Notch, Interleukin-1, and leptin crosstalk outcome |
| EMT | Epithelial to mesenchymal transition |
| TAMs | Tumor-associate macrophages |
| ECM | Extracellular matrix |
| sICAM | Soluble intercellular adhesion molecules |
| Sam68 | Src-associated in mitosis 68 |
| MET | Mesenchymal to epithelial transition |
| SRSF1 | Serine/arginine-rich splicing factor 1 |
References
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; Defresne, M.P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighbouring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [Green Version]
- Brook, N.; Brook, E.; Dharmarajan, A.; Dass, C.R.; Chan, A. Breast cancer bone metastases: Pathogenesis and therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 96, 63–78. [Google Scholar] [CrossRef]
- Santini, D.; Tampellini, M.; Vincenzi, B.; Ibrahim, T.; Ortega, C.; Virzi, V.; Silvestris, N.; Berardi, R.; Masini, C.; Calipari, N.; et al. Natural history of bone metastasis in colorectal cancer: Final results of a large Italian bone metastases study. Ann. Oncol. 2012, 23, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Procopio, G.; Porta, C.; Ibrahim, T.; Barni, S.; Mazzara, C.; Fontana, A.; Berruti, A.; Berardi, R.; Vincenzi, B.; et al. Natural history of malignant bone disease in renal cancer: Final results of an Italian bone metastasis survey. PLoS ONE 2013, 8, e83026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulston, R.J.; Cawthorn, W.P. Bone marrow adipose tissue as an endocrine organ: Close to the bone? Horm. Mol. Biol. Clin. Investig. 2016, 28, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Cawthorn, W.P.; Scheller, E.L.; Parlee, S.D.; Pham, H.A.; Learman, B.S.; Redshaw, C.M.; Sulston, R.J.; Burr, A.A.; Das, A.K.; Simon, B.R.; et al. Expansion of Bone Marrow Adipose Tissue During Caloric Restriction Is Associated With Increased Circulating Glucocorticoids and Not With Hypoleptinemia. Endocrinology 2016, 157, 508–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blebea, J.S.; Houseni, M.; Torigian, D.A.; Fan, C.; Mavi, A.; Zhuge, Y.; Iwanaga, T.; Mishra, S.; Udupa, J.; Zhuang, J.; et al. Structural and functional imaging of normal bone marrow and evaluation of its age-related changes. Semin. Nucl. Med. 2007, 370, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.K.; Horowitz, M.C.; MacDougald, O.A.; Scheller, E.L.; Rodeheffer, M.S.; Rosen, C.J.; Klibanski, A. Marrow Fat and Bone—New Perspectives. J. Clin. Endocrinol. Metab. 2013, 98, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Scheller, E.L.; Doucette, C.R.; Learman, B.S.; Cawthorn, W.P.; Khandaker, S.; Schell, B.; Wu, B.; Ding, S.Y.; Bredella, M.A.; Fazeli, P.K.; et al. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun. 2015, 6, 7808. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.J.; Haren, N.; Ghali, O.; Clabaut, A.; Chauveau, C.; Hardouin, P.; Broux, O. Adipogenic RNAs are transferred in osteoblasts via bone marrow adipocytes-derived extracellular vesicles (EVs). BMC Cell Biol. 2015, 16, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; McDonald, L.T.; Russell, D.L.; Kelly, R.R.; Wilson, K.R.; Mehrotra, M.; Soloff, A.C.; LaRue, A.C. Hematopoietic stem cell-derived adipocytes and fibroblasts in the tumor microenvironment. World J. Stem Cells 2015, 7, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; He, Y.; Yu, X. Bone Marrow Adipocyte: An Intimate Partner with Tumor Cells in Bone Metastasis. Front. Endocrinol. 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Diedrich, J.D.; Heath, E.I.; Podgorski, I. Adipocyte-activated oxidative and ER stress pathways promote tumor survival in bone via upregulation of Heme Oxygenase 1 and Survivin. Sci. Rep. 2018, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.V.; Edwards, C.M. Adipokines, adiposity, and bone marrow adipocytes: Dangerous accomplices in multiple myeloma. J. Cell. Physiol. 2018, 233, 9159–9166. [Google Scholar] [CrossRef]
- Morris, E.V.; Edwards, C.M. Bone marrow adiposity and multiple myeloma. Bone 2019, 118, 42–46. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Irish, J.M.; Kotecha, N.; Nolan, G.P. Mapping normal and cancer cell signalling networks: Towards single-cell proteomics. Nat. Rev. Cancer 2006, 6, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodelling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zheng, S.; Wang, J.; Long, H.; Fang, L.; Wang, G.; Li, Z.; Que, T.; Liu, Y.; Li, Y.; et al. Hypoxia-induced PLOD2 promotes proliferation, migration and invasion via PI3K/Akt signaling in glioma. Oncotarget 2017, 8, 41947–41962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger-Mathason, T.S.; Zhang, M.; Qiu, Q.; Skuli, N.; Nakazawa, M.S.; Karakasheva, T.; Mucaj, V.; Shay, J.E.; Stangenberg, L.; Sadri, N.; et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013, 3, 1190–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Bajpai, S.; Wong, C.C.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol. Cancer Res. 2013, 11, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Terajima, M.; Yang, Y.; Sun, L.; Ahn, Y.H.; Pankova, D.; Puperi, D.S.; Watanabe, T.; Kim, M.P.; Blackmon, S.H.; et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Investig. 2015, 125, 1147–1162. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zhang, J.; Hu, G.; Liu, L.; Liang, W. Hypoxia and TGF-beta1 induced PLOD2 expression improve the migration and invasion of cervical cancer cells by promoting epithelial-to-mesenchymal transition (EMT) and focal adhesion formation. Cancer Cell Int. 2017, 17, 54. [Google Scholar] [CrossRef]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Beltowski, J. Adiponectin and resistin—New hormones of white adipose tissue. Med. Sci. Monit. 2003, 9, RA55–RA61. [Google Scholar]
- Cawthorn, W.P.; Scheller, E.L.; Learman, B.S.; Parlee, S.D.; Simon, B.R.; Mori, H.; Ning, X.; Bree, A.J.; Schell, B.; Broome, D.T.; et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014, 20, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Delporte, M.L.; Brichard, S.M.; Hermans, M.P.; Beguin, C.; Lambert, M. Hyperadiponectinaemia in anorexia nervosa. Clin. Endocrinol. 2003, 58, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Berg, A.H.; Rajala, M.W.; Klebanov, S.; Iyengar, P.; Jimenez-Chillaron, J.C.; Patti, M.E.; Klein, S.L.; Weinstein, R.S.; Scherer, P.E. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes 2003, 52, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, T.; Wu, Y.; Tang, Z. Role and mechanism of PTEN in adiponectin-induced osteogenesis in human bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2017, 483, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, Y.W.; Lu, H.; Guo, Y.; Tang, Z.H. Adiponectin enhances osteogenic differentiation in human adipose-derived stem cells by activating the APPL1-AMPK signalling pathway. Biochem. Biophys. Res. Commun. 2015, 461, 237–242. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, Y.Y.; Yu, B.Y.; Yang, B.S.; Cho, K.H.; Yoon, D.K.; Roh, Y.K. Adiponectin induces growth arrest and apoptosis of MDA-MB-231 breast cancer cell. Arch. Pharm. Res. 2005, 28, 1263–1269. [Google Scholar] [CrossRef]
- Dieudonne, M.N.; Bussiere, M.; Dos Santos, E.; Leneveu, M.C.; Giudicelli, Y.; Pecquery, R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem. Biophys. Res. Commun. 2006, 345, 271–279. [Google Scholar] [CrossRef]
- Saxena, N.K.; Fu, P.P.; Nagalingam, A.; Wang, J.; Handy, J.; Cohen, C.; Tighiouart, M.; Sharma, D.; Anania, F.A. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology 2010, 139, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kitayama, J.; Yamauchi, T.; Kadowaki, T.; Maki, T.; Miyato, H.; Yamashita, H.; Nagawa, H. Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci. 2007, 98, 1120–1127. [Google Scholar] [CrossRef]
- Cong, L.; Gasser, J.; Zhao, J.; Yang, B.; Li, F.; Zhao, A.Z. Human adiponectin inhibits cell growth and induces apoptosis in human endometrial carcinoma cells, HEC-1-A and RL95 2. Endocr. Relat. Cancer 2007, 14, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.E.; Nkhata, K.J.; Mizuno, N.K.; Ray, A.; Cleary, M.P. Effects of adiponectin on breast cancer cell growth and signaling. Br. J. Cancer 2008, 98, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zazzo, E.; Polito, R.; Bartollino, S.; Nigro, E.; Porcile, C.; Bianco, A.; Daniele, A.; Moncharmont, B. Adiponectin as Link Factor between Adipose Tissue and Cancer. Int. J. Mol. Sci. 2019, 20, E839. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lam, J.B.; Lam, K.S.; Liu, J.; Lam, M.C.; Hoo, R.L.; Wu, D.; Cooper, G.J.; Xu, A. Adiponectin modulates the glycogen synthase kinase-3β/β-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006, 66, 11462–11470. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, E.; Benaitreau, D.; Dieudonne, M.N.; Leneveu, M.C.; Serazin, V.; Giudicelli, Y.; Pecquery, R. Adiponectin mediates an antiproliferative response in human MDA-MB 231 breast cancer cells. Oncol. Rep. 2008, 20, 971–977. [Google Scholar]
- Nakayama, S.; Miyoshi, Y.; Ishihara, H.; Noguchi, S. Growth-inhibitory effect of adiponectin via adiponectin receptor 1 on human breast cancer cells through inhibition of s-phase entry without inducing apoptosis. Breast Cancer Res. Treat. 2008, 112, 405–410. [Google Scholar] [CrossRef]
- Mauro, L.; Pellegrino, M.; De Amicis, F.; Ricchio, E.; Giordano, F.; Rizza, P.; Catalano, S.; Bonofiglio, D.; Sisci, D.; Panno, M.L.; et al. Evidences that estrogen receptor α interferes with adiponectin effects on breast cancer cell growth. Cell Cycle 2014, 13, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, L.; Pellegrino, M.; Giordano, F.; Ricchio, E.; Rizza, P.; De Amicis, F.; Catalano, S.; Bonofiglio, D.; Panno, M.L.; Andò, S. Estrogen receptor-α drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J. 2015, 29, 2150–2160. [Google Scholar] [CrossRef] [Green Version]
- Panno, M.L.; Naimo, G.D.; Spina, E.; Andò, S.; Mauro, L. Different molecular signaling sustaining adiponectin action in breast cancer. Curr. Opin. Pharmacol. 2016, 31, 1–7. [Google Scholar] [CrossRef]
- Saxena, N.K.; Sharma, D. Metastasis suppression by adiponectin: LKB1 rises up to the challenge. Cell Adhes. Migr. 2010, 4, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Libby, E.F.; Frost, A.R.; Demark-Wahnefried, W.; Hurst, D.R. Linking adiponectin and autophagy in the regulation of breast cancer metastasis. J. Mol. Med. 2014, 92, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef]
- Gernapudi, R.; Yao, Y.; Zhang, Y.; Wolfson, B.; Roy, S.; Duru, N.; Eades, G.; Yang, P.; Zhou, Q. Targeting exosomes from preadipocytes inhibits preadipocyte to cancer stem cell signaling in early-stage breast cancer. Breast Cancer Res. Treat. 2015, 150, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.T.; Kalra, S.P.; Wong, C.P.; Philbrick, K.A.; Lindenmaier, L.B.; Boghossian, S.; Iwaniec, U.T. Peripheral leptin regulates bone formation. J. Bone Miner. Res. 2013, 28, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.O.; Yue, R.; Murphy, M.M.; Peyer, J.G.; Morrison, S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014, 15, 154–168. [Google Scholar] [CrossRef] [Green Version]
- Reseland, J.E.; Syversen, U.; Bakke, I.; Qvigstad, G.; Eide, L.G.; Hjertner, O.; Gordeladze, J.O.; Drevon, C.A. Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J. Bone Miner. Res. 2001, 16, 1426–1433. [Google Scholar] [CrossRef]
- Köllmer, M.; Buhrman, J.S.; Zhang, Y.; Gemeinhart, R.A. Markers Are Shared Between Adipogenic and Osteogenic Differentiated Mesenchymal Stem Cells. J. Dev. Biol. Tissue Eng. 2013, 5, 18–25. [Google Scholar] [CrossRef]
- Caldefie-Chézet, F.; Damez, M.; de Latour, M.; Konska, G.; Mishellani, F.; Fusillier, C.; Guerry, M.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin: A proliferative factor for breast cancer? Study on human ductal carcinoma. Biochem. Biophys. Res. Commun. 2005, 334, 737–741. [Google Scholar] [CrossRef]
- Assiri, A.M.; Kamel, H.F. Evaluation of diagnostic and predictive value of serum adipokines: Leptin, resistin and visfatin in postmenopausal breast cancer. Obes Res. Clin. Pract. 2016, 10, 442–453. [Google Scholar] [CrossRef]
- Aliustaoglu, M.; Bilici, A.; Gumus, M.; Colak, A.T.; Baloglu, G.; Irmak, R.; Seker, M.; Ustaalioglu, B.B.; Salman, T.; Sonmez, B.; et al. Preoperative serum leptin levels in patients with breast cancer. Med. Oncol. 2010, 27, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, C.D.; Cao, C.; Cai, L.R.; Li, D.H.; Zheng, Y.Z. Association of serum leptin with breast cancer: A meta-analysis. Medicine 2019, 98, e14094. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS ONE 2011, 6, e21467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Pratt, M.A.; Tibbo, E.; Robertson, S.J.; Jansson, D.; Hurst, K.; Perez-Iratxeta, C.; Lau, R.; Niu, M.Y. The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene 2009, 28, 2710–2722. [Google Scholar] [CrossRef] [Green Version]
- Knight, B.B.; Oprea-Ilies, G.M.; Nagalingam, A.; Yang, L.; Cohen, C.; Saxena, N.K.; Sharma, D. Survivin upregulation, dependent on leptin-EGFR-Notch1 axis, is essential for leptin-induced migration of breast carcinoma cells. Endocr. Relat. Cancer 2011, 18, 413–428. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liu, M.; Wang, G.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim. Biophys. Acta 2012, 1825, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. 2016, 6, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Gelsomino, L.; Barone, I.; Panza, S.; Augimeri, G.; Bonofiglio, D.; Rovito, D.; Naimo, G.D.; Leggio, A.; Catalano, S.; et al. Leptin Modulates Exosome Biogenesis in Breast Cancer Cells: An Additional Mechanism in Cell-to-Cell Communication. J. Clin. Med. 2019, 8, E1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; deGraffenried, L.A.; Hursting, S.D. Leptin Signaling Mediates Obesity-Associated CSC Enrichment and EMT in Preclinical TNBC Models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.A.; Ortuño-Pineda, C.; Moreno-Godinez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src- dependent manner in breast cancer cells. Endocr. Connect. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojalvo, L.S.; King, W.; Cox, D.; Pollard, J.W. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am. J. Pathol. 2009, 174, 1048–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar] [CrossRef] [Green Version]
- He, J.Y.; Wei, X.H.; Li, S.J.; Liu, Y.; Hu, H.L.; Li, Z.Z.; Kuang, X.H.; Wang, L.; Shi, X.; Yuan, S.T.; et al. Adipocyte-derived IL-6 and leptin promote breast cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal. 2018, 16, 100. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Pang, M.; Hou, X.; Yuan, S.; Sun, L. PLOD2 in cancer research. Biomed. Pharmacother. 2017, 90, 670–676. [Google Scholar] [CrossRef]
- Ghasemi, A.; Saeidi, J.; Azimi-Nejad, M.; Hashemy, S.I. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell Oncol. 2019, 42, 243–260. [Google Scholar] [CrossRef]
- Joseph-Silverstein, J.; Silverstein, R.L. Cell adhesion molecules: An overview. Cancer Investig. 1998, 16, 176–182. [Google Scholar] [CrossRef]
- Tsai, C.F.; Chen, J.H.; Wu, C.T.; Chang, P.C.; Wang, S.L.; Yeh, W.L. Induction of osteoclast-like cell formation by leptin-induced soluble intercellular adhesion molecule secreted from cancer cells. Ther. Adv. Med. Oncol. 2019, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Shalloway, D. An RNA-binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature 1994, 368, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Frisone, P.; Pradella, D.; Di Matteo, A.; Belloni, E.; Ghigna, C.; Paronetto, M.P. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. Biomed. Res. Int. 2015, 2015, 528954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroni, P.; Citterio, L.; Piccoletti, R.; Bendinelli, P. Sam68 and ERKs regulate leptin-induced expression of OB-Rb mRNA in C2C12 myotubes. Mol. Cell. Endocrinol. 2009, 309, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Li, L.; Nisson, P.E.; Gruber, C.; Jessee, J.; Cohen, S.N. Neoplastic transformation and tumorigenesis associated with sam68 protein deficiency in cultured murine fibroblasts. J. Biol. Chem. 2000, 275, 40195–40201. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.J.; Resnick, R.J.; Shalloway, D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biol. 2004, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Richard, S.; Torabi, N.; Franco, G.V.; Tremblay, G.A.; Chen, T.; Vogel, G.; Morel, M.; Cléroux, P.; Forget-Richard, A.; Komarova, S.; et al. Ablation of the Sam68 RNA binding protein protects mice from age-related bone loss. PLoS Genet 2005, 1, e74. [Google Scholar] [CrossRef]
- Richard, S.; Vogel, G.; Huot, M.E.; Guo, T.; Muller, W.J.; Lukong, K.E. Sam68 haploinsufficiency delays onset of mammary tumorigenesis and metastasis. Oncogene 2008, 27, 548–556. [Google Scholar] [CrossRef]
- Derry, J.J.; Richard, S.; Valderrama Carvajal, H.; Ye, X.; Vasioukhin, V.; Cochrane, A.W.; Chen, T.; Tyner, A.L. Sik (BRK) phosphorylates Sam68 in the nucleus and negatively regulates its RNA binding ability. Mol. Cell Biol. 2000, 20, 6114–6126. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Venables, J.P.; Elliott, D.J.; Geremia, R.; Rossi, P.; Sette, C. Tr-kit promotes the formation of a multimolecular complex composed by Fyn, PLCgamma1 and Sam68. Oncogene 2003, 22, 8707–8715. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, A.H.; Bunnell, S.C.; Feng, S.; Berg, L.J.; Schreiber, S.L. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature 1997, 385, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Boisvert, F.M.; Bazett-Jones, D.P.; Richard, S. A role for the GSG domain in localizing Sam68 to novel nuclear structures in cancer cell lines. Mol. Biol. Cell 1999, 10, 3015–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, J.; Zheng, H.; Yu, C.; Chen, J.; Liu, Z.; Li, M.; Zeng, M.; Zhou, F.; Song, L. Expression and cytoplasmic localization of SAM68 is a significant and independent prognostic marker for renal cell carcinoma. Cancer Epidemiol. Prev. Biomark. 2009, 18, 2685–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yu, C.P.; Zhong, Y.; Liu, T.J.; Huang, Q.D.; Zhao, X.H.; Huang, H.; Tu, H.; Jiang, S.; Zhang, Y.; et al. Sam68 expression and cytoplasmic localization is correlated with lymph node metastasis as well as prognosis in patients with early-stage cervical cancer. Ann. Oncol. 2012, 23, 638–646. [Google Scholar] [CrossRef]
- Song, L.; Wang, L.; Li, Y.; Xiong, H.; Wu, J.; Li, J.; Li, M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. J. Pathol. 2010, 222, 227–237. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Z.; He, B.; Liu, J.; Li, S.; Zhou, L.; Pan, C.; Yu, Z.; Xu, Z. Sam68 is a novel marker for aggressive neuroblastoma. Onco Targets Ther. 2013, 6, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Bielli, P.; Busà, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [Green Version]
- Busà, R.; Paronetto, M.P.; Farini, D.; Pierantozzi, E.; Botti, F.; Angelini, D.F.; Attisani, F.; Vespasiani, G.; Sette, C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 2007, 26, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Richard, S. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 2003, 1653, 73–86. [Google Scholar] [CrossRef]
- Babic, I.; Jakymiw, A.; Fujita, D.J. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene 2004, 23, 3781–3789. [Google Scholar] [CrossRef] [Green Version]
- Côté, J.; Boisvert, F.M.; Boulanger, M.C.; Bedford, M.T.; Richard, S. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell 2003, 14, 274–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babic, I.; Cherry, E.; Fujita, D.J. SUMO modification of Sam68 enhances its ability to repress cyclin D1 expression and inhibits its ability to induce apoptosis. Oncogene 2006, 25, 4955–4964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, C. Post-translational regulation of star proteins and effects on their biological functions. Adv. Exp. Med. Biol. 2010, 693, 54–66. [Google Scholar]
- Lukong, K.E.; Larocque, D.; Tyner, A.L.; Richard, S. Tyrosine phosphorylation of sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. J. Biol. Chem. 2005, 280, 38639–38647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paronetto, M.P.; Farini, D.; Sammarco, I.; Maturo, G.; Vespasiani, G.; Geremia, R.; Rossi, P.; Sette, C. Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am. J. Pathol. 2004, 164, 1243–1251. [Google Scholar] [CrossRef]
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; de la Cruz, L.; Virizuela, J.A.; Sánchez-Margalet, V. Sam68 Mediates the Activation of Insulin and Leptin Signalling in Breast Cancer Cells. PLoS ONE 2016, 11, e0158218. [Google Scholar] [CrossRef]
- Biamonti, G.; Bonomi, S.; Gallo, S.; Ghigna, C. Making alternative splicing decisions during epithelial-to-mesenchymal transition (EMT). Cell. Mol. Life Sci. 2012, 69, 2515–2526. [Google Scholar] [CrossRef]
- Da Costa, P.J.; Menezes, J.; Romao, L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int. J. Biochem. Cell Biol. 2017, 91, 168–175. [Google Scholar] [CrossRef]
- Goncalves, V.; Pereira, J.F.S.; Jordan, P. Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelisch, F.; Khauv, D.; Risso, G.; Stallings-Mann, M.; Blaustein, M.; Quadrana, L.; Radisky, D.C.; Srebrow, A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J. Cell Biochem. 2012, 113, 2319–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokmen-Polar, Y.; Murray, N.R.; Velasco, M.A.; Gatalica, Z.; Fields, A.P. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 2001, 61, 1375–1381. [Google Scholar]
- Martínez-Montiel, N.; Rosas-Murrieta, N.; Martínez-Contreras, R. Alternative splicing regulation: Implications in cancer diagnosis and treatment. Med. Clin. 2015, 144, 317–323. [Google Scholar] [CrossRef]
- Sebestyén, E.; Zawisza, M.; Eyras, E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015, 43, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Matter, N.; Herrlich, P.; König, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef]
- Cheng, C.; Sharp, P.A. Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol. Cell Biol. 2006, 26, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Dogan, S.; Hu, X.; Zhang, Y.; Maihle, N.J.; Grande, J.P.; Cleary, M.P. Effects of high-fat diet and/or body weight on mammary tumor leptin and apoptosis signaling pathways in MMTV-TGF-alpha mice. Breast Cancer Res. 2007, 9, R91. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.D.H.; Idrees, F.; Ahsan, M.; Khanam, A.; Sultan, N.; Akhter, N. Association of serum leptin with serum estradiol in relation to breast carcinogenesis: A comparative case-control study between pre- and postmenopausal women. Turk. J. Med. Sci. 2018, 48, 305–310. [Google Scholar] [CrossRef]
- Liu, J.; Lam, J.B.; Chow, K.H.; Xu, A.; Lam, K.S.; Moon, R.T.; Wang, Y. Adiponectin stimulates Wnt inhibitory factor-1 expression through epigenetic regulations involving the transcription factor specificity protein 1. Carcinogenesis 2008, 29, 2195–2202. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.B.; Chow, K.H.; Xu, A.; Lam, K.S.; Liu, J.; Wong, N.S.; Moon, R.T.; Shepherd, P.R.; Cooper, G.J.; Wang, Y. Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyVT mice by modulation of phosphatase and tensin homolog activities. PLoS ONE 2009, 4, e4968. [Google Scholar] [CrossRef]
- Lukong, K.E.; Richard, S. Targeting the RNA-binding protein Sam68 as a treatment for cancer? Future Oncol. 2007, 3, 539–544. [Google Scholar] [CrossRef]
- Chen, C.; Chang, Y.C.; Liu, C.L.; Chang, K.J.; Guo, I.C. Leptin-induced growth of human ZR-75-1 breast cancer cells is associated with up-regulation of cyclin D1 and c-Myc and down-regulation of tumor suppressor p53 and p21WAF1/CIP1. Breast Cancer Res. Treat. 2006, 98, 121–132. [Google Scholar] [CrossRef]
- Perera, C.N.; Spalding, H.S.; Mohammed, S.I.; Camarillo, I.G. Identification of proteins secreted from leptin stimulated MCF-7 breast cancer cells: A dual proteomic approach. Exp. Biol. Med. 2008, 233, 708–720. [Google Scholar] [CrossRef]
- Okumura, M.; Yamamoto, M.; Sakuma, H.; Kojima, T.; Maruyama, T.; Jamali, M.; Cooper, D.R.; Yasuda, K. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: Reciprocal involvement of PKC-alpha and PPAR expression. Biochim. Biophys. Acta 2002, 1592, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Liu, Y.; Cui, S. Adiponectin induces breast cancer cell migration and growth factor expression. Cell Biochem. Biophys. 2014, 70, 1239–1245. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, Q.; Su, M.; Ji, F.; Wang, N.; Zhong, C.; Jiang, Y.; Liu, Y.; Zhang, Z.; Yang, J.; et al. Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell. Oncol. 2017, 40, 537–547. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Libby, F.E.; Liu, J.; Li, Y.I.; Lewis, M.J.; Demark-Wahnefried, W.; Hurst, D.R. Globular adiponectin enhances invasion in human breast cancer cells. Oncol. Lett. 2016, 11, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, A.; Lange, C.A. Met receptors induce Sam68-dependent cell migration by activation of alternate extracellular signal-regulated kinase family members. J. Biol. Chem. 2011, 286, 21062–21072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.K.; Parish, C.R.; Wong, M.L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017, 12, e0178454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta 2011, 1815, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Delort, L.; Jardé, T.; Dubois, V.; Vasson, M.P.; Caldefie-Chézet, F. New insights into anticarcinogenic properties of adiponectin: A potential therapeutic approach in breast cancer? Vitam. Horm. 2012, 90, 397–417. [Google Scholar] [CrossRef]
- Denzel, M.S.; Hebbard, L.W.; Shostak, G.; Shapiro, L.; Cardiff, R.D.; Ranscht, B. Adiponectin deficiency limits tumor vascularization in the MMTV-PyV-mT mouse model of mammary cancer. Clin. Cancer Res. 2009, 15, 3256–3264. [Google Scholar] [CrossRef] [Green Version]
- Landskroner-Eiger, S.; Qian, B.; Muise, E.S.; Nawrocki, A.R.; Berger, J.P.; Fine, E.J.; Koba, W.; Deng, Y.; Pollard, J.W.; Scherer, P.E. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin. Cancer Res. 2009, 15, 3265–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Yu, J.; Guo, H.; Song, H.; Chen, S. Upregulation of survivin by leptin/STAT3 signaling in MCF-7 cells. Biochem. Biophys. Res. Commun. 2008, 368, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Cheng, J.; Chen, L.; Xu, H.; Li, Q.; Pang, T. Sam68 affects cell proliferation and apoptosis of human adult T-acute lymphoblastic leukemia cells via AKT/mTOR signal pathway. Leuk. Res. 2016, 46, 1–9. [Google Scholar] [CrossRef]
- Esper, R.M.; Dame, M.; McClintock, S.; Holt, P.R.; Dannenberg, A.J.; Wicha, M.S.; Brenner, D.E. Leptin and Adiponectin Modulate the Self-renewal of Normal Human Breast Epithelial Stem Cells. Cancer Prev. Res. 2015, 8, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Benoit, Y.D.; Mitchell, R.R.; Risueño, R.M.; Orlando, L.; Tanasijevic, B.; Boyd, A.L.; Aslostovar, L.; Salci, K.R.; Shapovalova, Z.; Russell, J.; et al. Sam68 Allows Selective Targeting of Human Cancer Stem Cells. Cell Chem. Biol. 2017, 24, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Song, Z.; Liao, Z.; Liu, Z.; Sun, H.; Lei, B.; Chen, W.; Dang, C. PKM2 promotes stemness of breast cancer cell by through Wnt/β-catenin pathway. Tumor Biol. 2016, 37, 4223–4234. [Google Scholar] [CrossRef] [PubMed]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Mendoza-Catalán, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling Pathways Induced by Leptin during Epithelial-Mesenchymal Transition in Breast Cancer. Int. J. Mol. Sci. 2018, 19, E3493. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 166. [Google Scholar] [CrossRef] [Green Version]
- Cui, E.; Guo, H.; Shen, M.; Yu, H.; Gu, D.; Mao, W.; Wang, X. Adiponectin inhibits migration and invasion by reversing epithelial-mesenchymal transition in non-small cell lung carcinoma. Oncol. Rep. 2018, 40, 1330–1338. [Google Scholar] [CrossRef]
- Tan, W.; Wang, L.; Ma, Q.; Qi, M.; Lu, N.; Zhang, L.; Han, B. Adiponectin as a potential tumor suppressor inhibiting epithelial-to-mesenchymal transition but frequently silenced in prostate cancer by promoter methylation. Prostate 2015, 75, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of regulated (rBMA) and constitutive (cBMA) bone marrow adipocytes in the mouse tibia. Original figure courtesy of the author.
Figure 1.
Distribution of regulated (rBMA) and constitutive (cBMA) bone marrow adipocytes in the mouse tibia. Original figure courtesy of the author.
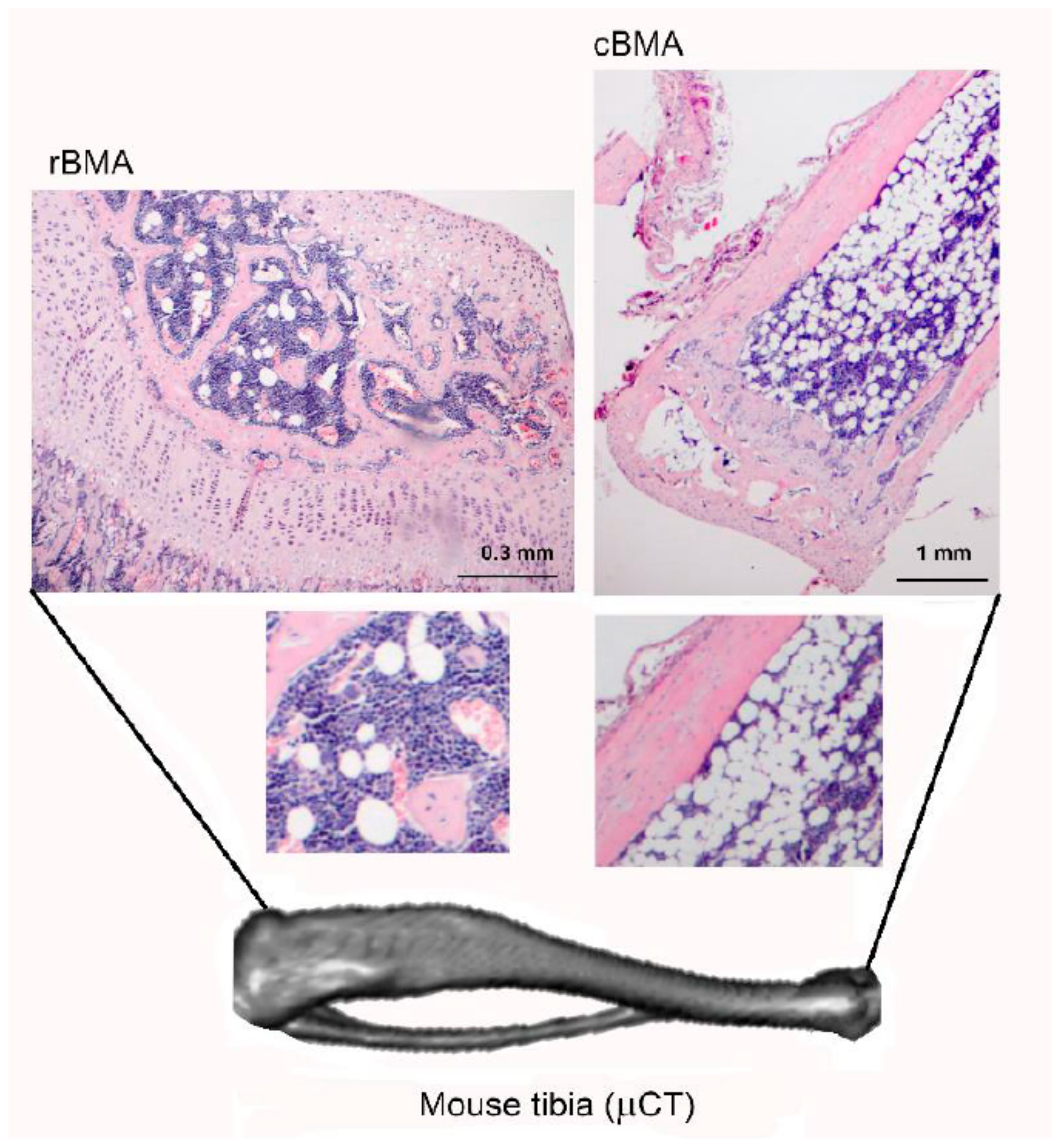
Figure 2.
Potential contribution of BMAT to bone metastatic growth.

Figure 3.
Sam68 is found in distinct sub-nuclear structures SLM/Sam68 nuclear bodies (SNBs, black arrows) enriched in cancer cells. Sam68 in cytoplasm (red arrows) is significantly associated with poor prognosis and progression in different types of tumours. Images of bone metastases from breast cancer. Scale bar= 10μm. Original figure courtesy of the author.
Figure 3.
Sam68 is found in distinct sub-nuclear structures SLM/Sam68 nuclear bodies (SNBs, black arrows) enriched in cancer cells. Sam68 in cytoplasm (red arrows) is significantly associated with poor prognosis and progression in different types of tumours. Images of bone metastases from breast cancer. Scale bar= 10μm. Original figure courtesy of the author.

{kind=link}
{kind=link}
{kind=link}
Table 1.
The role of leptin, adiponectin, and Sam68 in breast cancer: (+) positive regulation (activation), (-) negative regulation (inhibition), (+/-) positive or negative regulation depending on phosphorylation status.
Table 1.
The role of leptin, adiponectin, and Sam68 in breast cancer: (+) positive regulation (activation), (-) negative regulation (inhibition), (+/-) positive or negative regulation depending on phosphorylation status.
| Breast Cancer Progress | Leptin | Adiponectin | Sam68 |
|---|---|---|---|
| Tumourigenesis | + [60,121,122] | − [123,124,125] | + [94,125] |
| Tumour progression | + [126,127,128] | − [42,50,129] or + [51,130] | + [96,125] |
| Cancer cell invasion | + [131,132] | − [50,74] or + [130,133] | + [134] |
| Metastasis | + [72,135] | − [50] or + [51] | + [134] |
| Angiogenesis | + [65,136,137] | − [138] or + [139,140] | ? |
| Tumour cell apoptosis | − [127,141] | − [38] or + [138] | + [112,142] |
| Cancer stem cells (CSC) | + [69,72,135] | − [143] | + [144] |
| EMT | + [145,146,147,148] | ? (in breast cancer) − (in lung and prostate carcinoma [149,150]) | +/− [107,151] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maroni, P. Leptin, Adiponectin, and Sam68 in Bone Metastasis from Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1051. https://doi.org/10.3390/ijms21031051
AMA Style
Maroni P. Leptin, Adiponectin, and Sam68 in Bone Metastasis from Breast Cancer. International Journal of Molecular Sciences. 2020; 21(3):1051. https://doi.org/10.3390/ijms21031051
Chicago/Turabian StyleMaroni, Paola. 2020. "Leptin, Adiponectin, and Sam68 in Bone Metastasis from Breast Cancer" International Journal of Molecular Sciences 21, no. 3: 1051. https://doi.org/10.3390/ijms21031051
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.