Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia

 , and
, and
Abstract
1. Introduction
1.1. Description of Minimal Residual Disease
1.2. Genetic Descriptions of B-Cell Acute Lymphoblastic Leukemia and Minimal Residual Disease
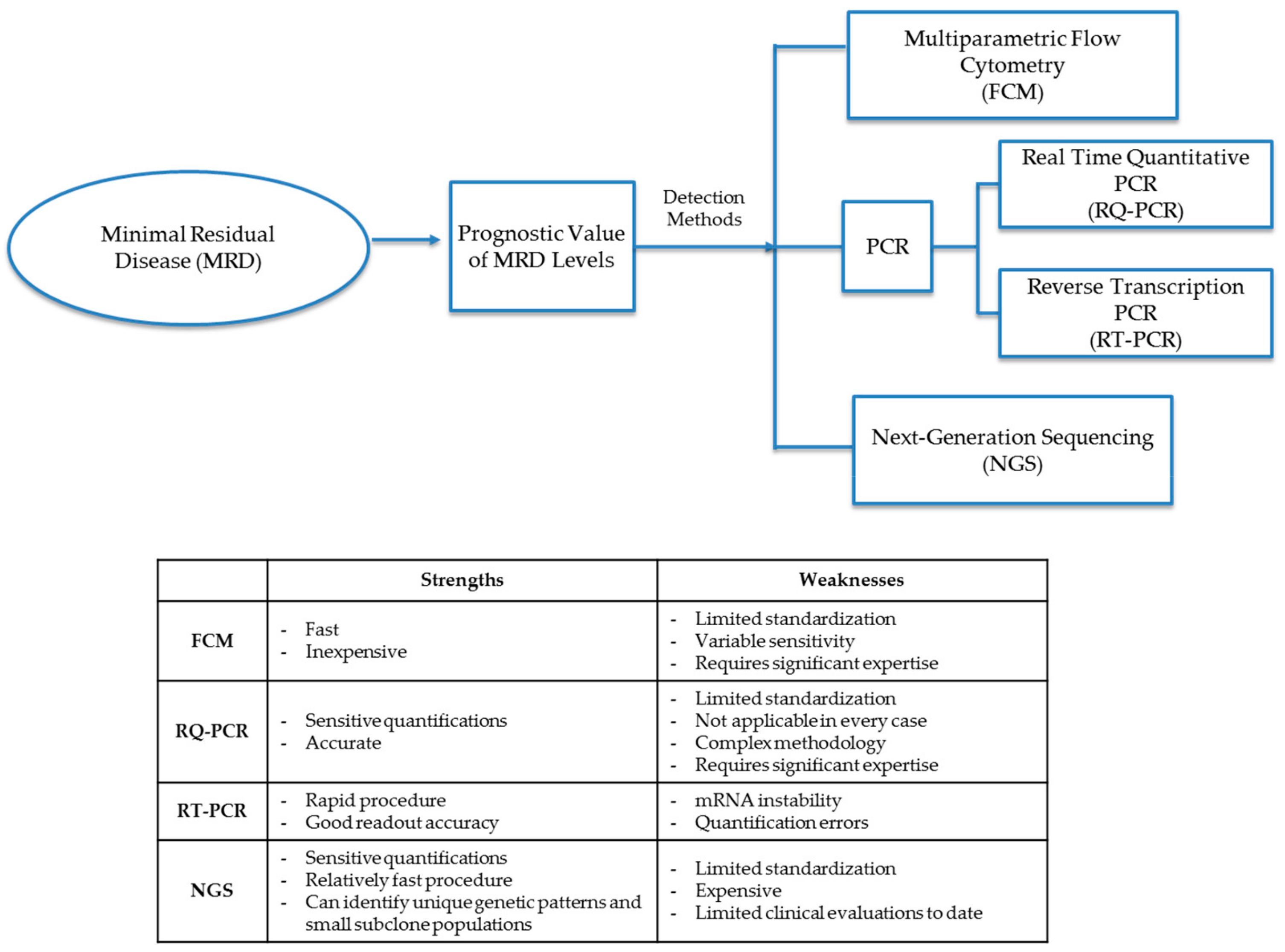
2. Prognostic Value of MRD
3. Phenotypic and Genetic Detection of MRD
3.1. Multiparametric Flow Cytometry
3.2. Polymerase Chain Reaction
3.3. Next-Generation Sequencing
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| B-ALL | B-cell acute lymphoblastic leukemia |
| dPCR | digital PCR |
| ddPCR | droplet digital PCR |
| FCM | flow cytometry |
| MRD | minimal residual disease |
| NGS | next-generation sequencing |
| PCR | polymerase chain reaction |
| RQ-PCR | real-time quantitative polymerase chain reaction |
| RT-PCR | reverse transcription polymerase chain reaction |
| T-ALL | T-cell acute lymphoblastic leukemia |
References
- Szczepanski, T.; Willemse, M.J.; Kamps, W.A.; van Wering, E.R.; Langerak, A.W.; van Dongen, J.J. Molecular discrimination between relapsed and secondary acute lymphoblastic leukemia: Proposal for an easy strategy. Med. Pediatr. Oncol. 2001, 36, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Brunson, A.; Paulus, J.K.; Tuscano, J.; Wun, T.; Keegan, T.H.M.; Jonas, B.A. Secondary acute lymphoblastic leukemia is a distinct clinical entity with prognostic significance. Blood Cancer J. 2017, 7, e605. [Google Scholar] [CrossRef] [PubMed]
- Campana, D. Minimal residual disease in acute lymphoblastic leukemia. Hematol. 2010 Am. Soc. Hematol. Educ. Program Book 2010, 2010, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, T. Why and how to quantify minimal residual disease in acute lymphoblastic leukemia? Leukemia 2007, 21, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Jabbour, E. Minimal Residual Disease in Acute Lymphoblastic Leukemia: How to Recognize and Treat It. Curr. Oncol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood J. Am. Soc. Hematol. 2015, 126, 964–971. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Wade, R.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Richards, S. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2013, 14, 199–209. [Google Scholar] [CrossRef]
- Yeoh, A.E.; Ariffin, H.; Chai, E.L.; Kwok, C.S.; Chan, Y.H.; Ponnudurai, K.; Campana, D.; Tan, P.L.; Chan, M.Y.; Kham, S.K.; et al. Minimal residual disease-guided treatment deintensification for children with acute lymphoblastic leukemia: Results from the Malaysia-Singapore acute lymphoblastic leukemia 2003 study. J. Clin. Oncol. 2012, 30, 2384–2392. [Google Scholar] [CrossRef]
- Pieters, R.; de Groot-Kruseman, H.; Van der Velden, V.; Fiocco, M.; van den Berg, H.; de Bont, E.; Egeler, R.M.; Hoogerbrugge, P.; Kaspers, G.; Van der Schoot, E.; et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J. Clin. Oncol. 2016, 34, 2591–2601. [Google Scholar] [CrossRef]
- Goulden, N.; Bader, P.; Van Der Velden, V.; Moppett, J.; Schilham, M.; Masden, H.O.; Krejci, O.; Kreyenberg, H.; Lankester, A.; Revesz, T.; et al. Minimal residual disease prior to stem cell transplant for childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2003, 122, 24–29. [Google Scholar] [CrossRef]
- Campana, D.; Leung, W. Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br. J. Haematol. 2013, 162, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.; Stella-Holowiecka, B.; Krawczyk-Kulis, M.; Gokbuget, N.; Hoelzer, D.; Doubek, M.; Mayer, J.; Piatkowska-Jakubas, B.; Skotnicki, A.B.; Dombret, H.; et al. Status of minimal residual disease determines outcome of autologous hematopoietic SCT in adult ALL. Bone Marrow Transplant. 2010, 45, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Kreyenberg, H.; Henze, G.H.; Eckert, C.; Reising, M.; Willasch, A.; Barth, A.; Borkhardt, A.; Peters, C.; Handgretinger, R.; et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: The ALL-REZ BFM Study Group. J. Clin. Oncol. 2009, 27, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Kreyenberg, H.; von Stackelberg, A.; Eckert, C.; Salzmann-Manrique, E.; Meisel, R.; Poetschger, U.; Stachel, D.; Schrappe, M.; Alten, J.; et al. Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: Results of the ALL-BFM-SCT 2003 trial. J. Clin. Oncol. 2015, 33, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, F.; Zecca, M.; Rossi, B.; Campeggio, M.; Magrin, E.; Giarin, E.; Buldini, B.; Songia, S.; Cazzaniga, G.; Mina, T.; et al. Pre- and post-transplant minimal residual disease predicts relapse occurrence in children with acute lymphoblastic leukaemia. Br. J. Haematol. 2018, 180, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 8th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Aster, J.C. Robbins and Cotran Pathologic Basis of Diseases, 8th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2010; pp. 602–603. [Google Scholar]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Cave, H.; van der Werff ten Bosch, J.; Suciu, S.; Guidal, C.; Waterkeyn, C.; Otten, J.; Bakkus, M.; Thielemans, K.; Grandchamp, B.; Vilmer, E. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer—Childhood Leukemia Cooperative Group. N. Engl. J. Med. 1998, 339, 591–598. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Behm, F.G.; Sanchez, J.; Boyett, J.M.; Hancock, M.L.; Raimondi, S.C.; Rubnitz, J.E.; Rivera, G.K.; Sandlund, J.T.; Pui, C.H.; et al. Immunological detection of minimal residual disease in children with acute lymphoblastic leukaemia. Lancet 1998, 351, 550–554. [Google Scholar] [CrossRef]
- van Dongen, J.J.; Seriu, T.; Panzer-Grumayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Bruggemann, M.; Kotrova, M. Minimal residual disease in adult ALL: Technical aspects and implications for correct clinical interpretation. Blood Adv. 2017, 1, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Stow, P.; Key, L.; Chen, X.; Pan, Q.; Neale, G.A.; Coustan-Smith, E.; Mullighan, C.G.; Zhou, Y.; Pui, C.H.; Campana, D. Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood 2010, 115, 4657–4663. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Sancho, J.; Hancock, M.L.; Boyett, J.M.; Behm, F.G.; Raimondi, S.C.; Sandlund, J.T.; Rivera, G.K.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 2000, 96, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Pei, D.; Coustan-Smith, E.; Jeha, S.; Cheng, C.; Bowman, W.P.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Inaba, H.; et al. Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: A prospective study. Lancet Oncol. 2015, 16, 465–474. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef]
- Ladetto, M.; Böttcher, S.; Kröger, N.; Pulsipher, M.A.; Bader, P. Methods and role of minimal residual disease after stem cell transplantation. Bone Marrow Transplant. 2019, 54, 681–690. [Google Scholar] [CrossRef]
- Kansagra, A.J.; Frey, N.V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Komanduri, K.V.; Gastineau, D.A.; et al. Clinical utilization of Chimeric Antigen Receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia (ALL)-an expert opinion from the European Society for Blood and Marrow Transplantation (EBMT) and the American Society for Blood and Marrow Transplantation (ASBMT). Bone Marrow Transplant. 2019, 25, e76–e85. [Google Scholar] [CrossRef]
- Takamatsu, H. Comparison of Minimal Residual Disease Detection by Multiparameter Flow Cytometry, ASO-qPCR, Droplet Digital PCR, and Deep Sequencing in Patients with Multiple Myeloma Who Underwent Autologous Stem Cell Transplantation. J. Clin. Med. 2017, 6, 91. [Google Scholar] [CrossRef]
- Salto-Tellez, M.; Shelat, S.G.; Benoit, B.; Rennert, H.; Carroll, M.; Leonard, D.G.; Nowell, P.; Bagg, A. Multiplex RT-PCR for the detection of leukemia-associated translocations: Validation and application to routine molecular diagnostic practice. J. Mol. Diagn. 2003, 5, 231–236. [Google Scholar] [CrossRef]
- Flohr, T.; Schrauder, A.; Cazzaniga, G.; Panzer-Grumayer, R.; van der Velden, V.; Fischer, S.; Stanulla, M.; Basso, G.; Niggli, F.K.; Schafer, B.W.; et al. Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia 2008, 22, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Demeke, T.; Dobnik, D. Critical assessment of digital PCR for the detection and quantification of genetically modified organisms. Anal. Bioanal. Chem. 2018, 410, 4039–4050. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Li, G.R.; Wang, R.J.; Yi, Y.T.; Yang, L.; Jiang, D.; Zhang, X.P.; Peng, Y. Application of next-generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin. J. Cancer 2012, 31, 463–470. [Google Scholar] [CrossRef]
- Kotrova, M.; Trka, J.; Kneba, M.; Bruggemann, M. Is Next-Generation Sequencing the way to go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia? Mol. Diagn. Ther. 2017, 21, 481–492. [Google Scholar] [CrossRef]
- Campana, D.; Coustan-Smith, E. Detection of minimal residual disease in acute leukemia by flow cytometry. Cytometry 1999, 38, 139–152. [Google Scholar] [CrossRef]
- Lucio, P.; Parreira, A.; van den Beemd, M.W.; van Lochem, E.G.; van Wering, E.R.; Baars, E.; Porwit-MacDonald, A.; Bjorklund, E.; Gaipa, G.; Biondi, A.; et al. Flow cytometric analysis of normal B cell differentiation: A frame of reference for the detection of minimal residual disease in precursor-B-ALL. Leukemia 1999, 13, 419–427. [Google Scholar] [CrossRef]
- van Dongen, J.J.; van der Velden, V.H.; Bruggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef]
- Chen, X.; Wood, B.L. Monitoring minimal residual disease in acute leukemia: Technical challenges and interpretive complexities. Blood Rev. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Ciudad, J.; San Miguel, J.F.; Lopez-Berges, M.C.; Garcia Marcos, M.A.; Gonzalez, M.; Vazquez, L.; del Canizo, M.C.; Lopez, A.; Van Dongen, J.J.; Orfao, A. Detection of abnormalities in B-cell differentiation pattern is a useful tool to predict relapse in precursor-B-ALL. Br. J. Haematol. 1999, 104, 695–705. [Google Scholar] [CrossRef]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrova, M.; Novakova, M.; Sonneveld, E.; et al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef]
- Denys, B.; van der Sluijs-Gelling, A.J.; Homburg, C.; van der Schoot, C.E.; de Haas, V.; Philippe, J.; Pieters, R.; van Dongen, J.J.; van der Velden, V.H. Improved flow cytometric detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia 2013, 27, 635–641. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Gaipa, G.; Ratei, R.; Veltroni, M.; Schumich, A.; Maglia, O.; Karawajew, L.; Benetello, A.; Potschger, U.; Husak, Z.; et al. Standardization of flow cytometric minimal residual disease evaluation in acute lymphoblastic leukemia: Multicentric assessment is feasible. Cytom. B Clin. Cytom. 2008, 74, 331–340. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.P.; Sartor, M.; Schumich, A.; et al. AIEOP-BFM consensus guidelines 2016 for flow cytometric immunophenotyping of Pediatric acute lymphoblastic leukemia. Cytom. B Clin. Cytom. 2018, 94, 82–93. [Google Scholar] [CrossRef]
- Bouriche, L.; Bernot, D.; Nivaggioni, V.; Arnoux, I.; Loosveld, M. Detection of Minimal Residual Disease in B Cell Acute Lymphoblastic Leukemia Using an Eight-Color Tube with Dried Antibody Reagents. Cytom. B Clin. Cytom. 2019, 96, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kalina, T.; Flores-Montero, J.; van der Velden, V.H.; Martin-Ayuso, M.; Bottcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonca, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- van Dongen, J.J.; Lhermitte, L.; Bottcher, S.; Almeida, J.; van der Velden, V.H.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lecrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Novakova, M.; Glier, H.; Brdickova, N.; Vlkova, M.; Santos, A.H.; Lima, M.; Roussel, M.; Flores-Montero, J.; Szczepanski, T.; Bottcher, S.; et al. How to make usage of the standardized EuroFlow 8-color protocols possible for instruments of different manufacturers. J. Immunol. Methods 2017, 475, 112388. [Google Scholar] [CrossRef]
- Grimwade, L.F.; Fuller, K.A.; Erber, W.N. Applications of imaging flow cytometry in the diagnostic assessment of acute leukaemia. Methods 2017, 112, 39–45. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Pullen, D.J.; Winick, N.; Martin, P.L.; Bowman, W.P.; Camitta, B. Comparison of diagnostic and relapse flow cytometry phenotypes in childhood acute lymphoblastic leukemia: Implications for residual disease detection: A report from the children’s oncology group. Cytom. B Clin. Cytom. 2005, 68, 18–24. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Subramanian Pg, P.G.; Ghogale, S.; Chatterjee, G.; Patkar, N.V.; Gupta, A.; Shukla, R.; Badrinath, Y.; Deshpande, N.; Narula, G.; et al. A High-Sensitivity 10-Color Flow Cytometric Minimal Residual Disease Assay in B-Lymphoblastic Leukemia/Lymphoma Can Easily Achieve the Sensitivity of 2-in-10(6) and Is Superior to Standard Minimal Residual Disease Assay: A Study of 622 Patients. Cytom. B Clin. Cytom. 2019. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, V.H.; Hochhaus, A.; Cazzaniga, G.; Szczepanski, T.; Gabert, J.; van Dongen, J.J. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: Principles, approaches, and laboratory aspects. Leukemia 2003, 17, 1013–1034. [Google Scholar] [CrossRef] [PubMed]
- Nunes, V.; Cazzaniga, G.; Biondi, A. An update on PCR use for minimal residual disease monitoring in acute lymphoblastic leukemia. Expert Rev. Mol. Diagn. 2017, 17, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, T.; Willemse, M.J.; Brinkhof, B.; van Wering, E.R.; van der Burg, M.; van Dongen, J.J. Comparative analysis of Ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B-ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood 2002, 99, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Reichel, M.; Gillert, E.; Breitenlohner, I.; Angermuller, S.; Fey, G.H.; Marschalek, R.; Repp, R.; Greil, J.; Beck, J.D. Rapid isolation of chromosomal breakpoints from patients with t(4;11) acute lymphoblastic leukemia: Implications for basic and clinical research. Leukemia 2001, 15, 286–288. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ladetto, M.; Bruggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef]
- Garand, R.; Beldjord, K.; Cave, H.; Fossat, C.; Arnoux, I.; Asnafi, V.; Bertrand, Y.; Boulland, M.L.; Brouzes, C.; Clappier, E.; et al. Flow cytometry and IG/TCR quantitative PCR for minimal residual disease quantitation in acute lymphoblastic leukemia: A French multicenter prospective study on behalf of the FRALLE, EORTC and GRAALL. Leukemia 2013, 27, 370–376. [Google Scholar] [CrossRef]
- Huang, Y.J.; Coustan-Smith, E.; Kao, H.W.; Liu, H.C.; Chen, S.H.; Hsiao, C.C.; Yang, C.P.; Jaing, T.H.; Yeh, T.C.; Kuo, M.C.; et al. Concordance of two approaches in monitoring of minimal residual disease in B-precursor acute lymphoblastic leukemia: Fusion transcripts and leukemia-associated immunophenotypes. J. Formos. Med. Assoc. 2017, 116, 774–781. [Google Scholar] [CrossRef]
- Pfeifer, H.; Cazzaniga, G.; van der Velden, V.H.J.; Cayuela, J.M.; Schafer, B.; Spinelli, O.; Akiki, S.; Avigad, S.; Bendit, I.; Borg, K.; et al. Standardisation and consensus guidelines for minimal residual disease assessment in Philadelphia-positive acute lymphoblastic leukemia (Ph + ALL) by real-time quantitative reverse transcriptase PCR of e1a2 BCR-ABL1. Leukemia 2019, 33, 1910–1922. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cave, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Gameiro, P.; Moreira, I.; Yetgin, S.; Papaioannou, M.; Potter, M.N.; Prentice, H.G.; Hoffbrand, A.V.; Foroni, L. Polymerase chain reaction (PCR)- and reverse transcription PCR-based minimal residual disease detection in long-term follow-up of childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2002, 119, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, J.; Manoj, M.G.; Kapoor, R. Detection of balanced translocations in acute lymphoblastic leukemia by a novel multiplex reverse transcriptase reverse transcription-polymerase chain reaction. J. Cancer Res. Ther. 2017, 13, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, V.H.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: Guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Schumich, A.; Maurer-Granofszky, M.; Attarbaschi, A.; Potschger, U.; Buldini, B.; Gaipa, G.; Karawajew, L.; Printz, D.; Ratei, R.; Conter, V.; et al. Flow-cytometric minimal residual disease monitoring in blood predicts relapse risk in pediatric B-cell precursor acute lymphoblastic leukemia in trial AIEOP-BFM-ALL 2000. Pediatr. Blood Cancer 2019, 66, e27590. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.; Owen, D.; Tsang, A.; Milner, R.; Vercauteren, S. The significance of peripheral blood minimal residual disease to predict early disease response in patients with B-cell acute lymphoblastic leukemia. Int. J. Lab Hematol. 2016, 38, 527–534. [Google Scholar] [CrossRef]
- Della Starza, I.; De Novi, L.A.; Santoro, A.; Salemi, D.; Tam, W.; Cavalli, M.; Menale, L.; Soscia, R.; Apicella, V.; Ilari, C.; et al. Digital droplet PCR and next-generation sequencing refine minimal residual disease monitoring in acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 2838–2840. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, X.; Dong, Z.; Lu, Y.C.; Sun, Y.; Liu, Y.; McCormack, R.; Gu, Y.; Liu, X. Highly Sensitive Droplet Digital PCR Method for Detection of EGFR-Activating Mutations in Plasma Cell-Free DNA from Patients with Advanced Non-Small Cell Lung Cancer. J. Mol. Diagn. 2015, 17, 265–272. [Google Scholar] [CrossRef]
- Villegas-Ruiz, V.; Olmos-Valdez, K.; Castro-Lopez, K.A.; Saucedo-Tepanecatl, V.E.; Ramirez-Chiquito, J.C.; Perez-Lopez, E.I.; Medina-Vera, I.; Juarez-Mendez, S. Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR. Genes 2019, 10, 376. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Minervini, A.; Francesco Minervini, C.; Anelli, L.; Zagaria, A.; Casieri, P.; Coccaro, N.; Cumbo, C.; Tota, G.; Impera, L.; Orsini, P.; et al. Droplet digital PCR analysis of NOTCH1 gene mutations in chronic lymphocytic leukemia. Oncotarget 2016, 7, 86469–86479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Casieri, P.; Tota, G.; Orsini, P.; Impera, L.; Minervini, A.; Minervini, C.F.; Cumbo, C.; et al. Droplet Digital PCR Is a Robust Tool for Monitoring Minimal Residual Disease in Adult Philadelphia-Positive Acute Lymphoblastic Leukemia. J. Mol. Diagn. 2018, 20, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Pulsipher, M.A.; Carlson, C.; Langholz, B.; Wall, D.A.; Schultz, K.R.; Bunin, N.; Kirsch, I.; Gastier-Foster, J.M.; Borowitz, M.; Desmarais, C.; et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 2015, 125, 3501–3508. [Google Scholar] [CrossRef]
- Bibault, J.E.; Figeac, M.; Helevaut, N.; Rodriguez, C.; Quief, S.; Sebda, S.; Renneville, A.; Nibourel, O.; Rousselot, P.; Gruson, B.; et al. Next-generation sequencing of FLT3 internal tandem duplications for minimal residual disease monitoring in acute myeloid leukemia. Oncotarget 2015, 6, 22812–22821. [Google Scholar] [CrossRef]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef]
- The EndRAD Trial: Eliminating Total Body Irradiation (TBI) for NGS-MRD Negative Children, Adolescents, and Young Adults with B-ALL. Available online: https://ClinicalTrials.gov/show/NCT03509961 (accessed on 27 April 2018).
- Cross, N.C.; White, H.E.; Ernst, T.; Welden, L.; Dietz, C.; Saglio, G.; Mahon, F.X.; Wong, C.C.; Zheng, D.; Wong, S.; et al. Development and evaluation of a secondary reference panel for BCR-ABL1 quantification on the International Scale. Leukemia 2016, 30, 1844–1852. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.; Domschke, K. Making sense of deep sequencing. Int. J. Neuropsychopharmacol. 2014, 17, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Watt, E.; Inglott, S.; Brooks, T.; Bartram, J.; Adams, S.P. Clinical benefit of a high-throughput sequencing approach for minimal residual disease in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2019, 66, e27787. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Sancho, J.; Hancock, M.L.; Razzouk, B.I.; Ribeiro, R.C.; Rivera, G.K.; Rubnitz, J.E.; Sandlund, J.T.; Pui, C.H.; Campana, D. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 2002, 100, 2399–2402. [Google Scholar] [CrossRef]
- van der Velden, V.H.; Jacobs, D.C.; Wijkhuijs, A.J.; Comans-Bitter, W.M.; Willemse, M.J.; Hahlen, K.; Kamps, W.A.; van Wering, E.R.; van Dongen, J.J. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia 2002, 16, 1432–1436. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Good Prognosis | Intermediate Prognosis | Poor Prognosis | Undetermined Prognosis |
|---|---|---|---|
| Hyperdiploid karyotypes | t(1;19); TCF3-PBX1 | Hypodiploid karyotypes | t(5;14); IL3-IGH* |
| t(12;21);ETV6-RUNX1 (TEL-AML1) | t(9;22); BCR-ABL | ||
| Philadelphia-like ALL | |||
| 11q23 MLL rearrangements |
| FCM * | Translocation PCR ** | Antigen Receptor PCR ** | Droplet Digital PCR ** | NGS *** | |
|---|---|---|---|---|---|
| Turnaround Time | 3–4 h [32] | 2–3 days [33] | Weeks [34] | 5–8 h [35] | ~1 week [36] |
| Cost Per Sample | ~$350 [32] | ~$500 [33] | ~$500 [32] | ~500 [32] | ~$1000 [32] |
| Standardization | Standardized in different consortia [37] | Limited standardization [37] | Limited standardization [37] | Limited Standardization [32] | Limited Standardization [37] |
| Use of Patient-Specific Reagent | No [37] | No [37] | Yes [37] | Yes [32] | No [37] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruse, A.; Abdel-Azim, N.; Kim, H.N.; Ruan, Y.; Phan, V.; Ogana, H.; Wang, W.; Lee, R.; Gang, E.J.; Khazal, S.; et al. Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1054. https://doi.org/10.3390/ijms21031054
Kruse A, Abdel-Azim N, Kim HN, Ruan Y, Phan V, Ogana H, Wang W, Lee R, Gang EJ, Khazal S, et al. Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2020; 21(3):1054. https://doi.org/10.3390/ijms21031054
Chicago/Turabian StyleKruse, Aaron, Nour Abdel-Azim, Hye Na Kim, Yongsheng Ruan, Valerie Phan, Heather Ogana, William Wang, Rachel Lee, Eun Ji Gang, Sajad Khazal, and et al. 2020. "Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 21, no. 3: 1054. https://doi.org/10.3390/ijms21031054
APA StyleKruse, A., Abdel-Azim, N., Kim, H. N., Ruan, Y., Phan, V., Ogana, H., Wang, W., Lee, R., Gang, E. J., Khazal, S., & Kim, Y.-M. (2020). Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 21(3), 1054. https://doi.org/10.3390/ijms21031054