Abstract
Cutaneous fibrosis results from suboptimal wound healing following significant tissue injury such as severe burns, trauma, and major surgeries. Pathologic skin fibrosis results in scars that are disfiguring, limit normal movement, and prevent patient recovery and reintegration into society. While various therapeutic strategies have been used to accelerate wound healing and decrease the incidence of scarring, recent studies have targeted the molecular regulators of each phase of wound healing, including the inflammatory, proliferative, and remodeling phases. Here, we reviewed the most recent literature elucidating molecular pathways that can be targeted to reduce fibrosis with a particular focus on post-burn scarring. Current research targeting inflammatory mediators, the epithelial to mesenchymal transition, and regulators of myofibroblast differentiation shows promising results. However, a multimodal approach addressing all three phases of wound healing may provide the best therapeutic outcome.
Keywords:
fibrosis; wound healing; burn; hypertrophic scarring; myofibroblasts; macrophages; inflammation; EMT 1. Introduction
Fibrosis is a natural process to restore tissue function during healthy wound healing. When pathological, fibrosis can result in detrimental scarring and dysfunctional tissue. These scars result from increased and prolonged inflammation, leading to suboptimal wound healing. In burns, the hypermetabolic stress response manifests as a sustained surge of inflammatory cytokines [1] that affect the migration of immune cells, including neutrophils and macrophages, to the wound area. Tissue damage following burn induces the activation of various damage-associated molecules (DAMPs) that signal immune cells to migrate to the wound. Initial inflammation activates multiple cell populations in the dermis and epidermis to aid in wound re-epithelization and closure [2]. Among the inflammatory cytokines, transforming growth factor-beta (TGF-β) enables differentiation of fibroblasts to myofibroblasts to assist with wound coverage. However, the persistent activation of TGF-β signaling provides an erroneous signal to myofibroblasts for continuous extracellular matrix (ECM) production leading to pathological scarring [3]. Importantly, almost all fibrotic diseases exhibit one underlying phenomenon—disarray of three distinct but overlapping phases of wound healing. The molecular mediators regulating the wound healing phases are summarized in Figure 1. While various therapeutic approaches are used to target different stages of wound healing to prevent scar development, none of these therapies have been entirely successful. This review recapitulates the most recent advancements to study the underlying mechanism of fibrosis. We analyzed the current literature on the role of various inflammatory markers in triggering the immune response following injury to protect against infections and begin the healing process. We dissected the role of epigenetics and transcriptional regulation in the expression or repression of fibrosis genes. We also analyzed the current knowledge of the mechanisms underlying myofibroblasts differentiation. Finally, we report ongoing clinical trials aiming to treat pathologic fibrosis utilizing biological and pharmacological tools.
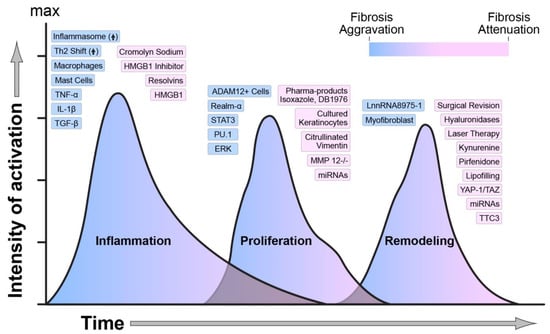
Figure 1.
Regulators of wound healing and scarring. The temporal activation, overlap, and intensity of activation of each phase of wound healing are regulated by numerous molecular, biological, and mechanical factors. The figure indicates how each one of these factors is modulating wound healing towards an aggravation or an attenuation of fibrosis. Blue indicates activation, and pink indicates attenuation of fibrosis.
2. Interacting Wound Healing Phases
It is recognized that increased and sustained inflammation leads to suboptimal wound healing and subsequent fibrotic development. Independent of the cause of injury, wound healing is characterized by three established phases that are synchronized in time and activity until complete wound closure [4]. The three stages are namely the inflammatory, proliferation, and remodeling phase [5]. Current studies have updated the roles of numerous molecular pathways in regulating each phase of wound healing, as discussed in this review.
3. Inflammation in the Healing Wound
In adults, tissue damage prompts the inflammatory phase of wound healing and is classically defined with the temporal activation and recruitment of various cells of innate and adaptive immunity [4]. Inflammation involves robust mechanisms allowing tissues to repair the damage, regenerate, and ultimately heal to restore function. However, not all inflammatory processes inevitably result in fibrotic pathophysiology. It is well documented that most fetal tissues heal without fibrosis, and various investigations have sought to elucidate this scarless fetal phenotype [6,7,8].
It was previously thought that the amniotic environment within the mammalian placenta was beneficial for scarless healing. The intrauterine setting is a naturally sterile compartment bathed in abundant growth factors, is free of pathogens, and lacks stimuli to initiate inflammation; thus, tissues were hypothesized to heal without fibrosis. Indeed, initial investigations demonstrated scarless fetal wound healing in several amniote species, including rats, mice, pigs, sheep, and humans [6,7,9]. These studies showed inherent tissue structure differences and environmental molecular variances between fetal and adult wounds that ultimately determined scarless healing capacity [10,11,12,13]. Significantly, experiments demonstrated that adult skin, when placed in a fetal environment and then subsequently wounded, healed with scar formation [14], underscoring the intrinsic differences between adult and fetal wound healing. As investigations into scarless pathophysiology evolved, data demonstrated that fetal wound healing is also a function of wound defect size and gestational age; larger defect size or more advanced gestation age resulted in a higher probability of scar formation [8,15]. Gestational age was significant because it demonstrated that immune system maturation allows for a more robust response and eventually results in some level of fibrosis, further supporting the necessity of the inflammatory response in fibrotic pathogenesis. However, to initiate inflammatory reactions, factors from tissue damage must first be present.
3.1. Inflammation Arises Due to Tissue Damage
Tissue damage results both in necrotic and apoptotic cell death, in turn releasing direct factors and chemoattractant molecules that initiate the recruitment of immune cells. Traumatic injuries, such as deep thermal burns, along with other ischemic, toxic, or allergic insults, result in cell death culminating in the release of inflammatory stimuli derived from cytosolic proteins and various organelles including nuclear and mitochondrial nucleic acids [1,16]. Collectively, these are known as danger-associated molecular patterns (DAMPs), which in turn bind to their associated pattern-recognition receptors (PRRs), recruiting immune cells and driving initial inflammation [17]. DAMPs are one of the primary signaling molecules that activate the immune system to prevent infection of injured tissue [17].
The high-mobility group box-1 protein (HMGB1) is one of the first DAMP proteins described and as such, one of the best-characterized. Interestingly, HMGB1 has been shown to follow secretory pathways by actively stressed cells as well [2,18]. By adjusting oxidation states, HMGB1 binds to specific PRRs in adjacent cells, further directing exclusive signaling, immune cell recruitment, and may have direct impacts on fibrogenesis. Studies have demonstrated that HMGB1 acts as a chemoattractant for monocytes and that HMGB1 signaling can reprogram tissue macrophages to a more profibrotic M1 phenotype, which is further discussed in Section 4.2 [19,20]. Other investigations show that HMGB1 can bind to the receptor for advanced glycation end products (RAGE), activating a fibrotic signaling cascade and directly upregulating collagen production [21]. Through exclusionary signaling and inflammatory polarization, HMGB1 has been suggested as a biomarker in active fibrotic diseases, including hepatic and cutaneous fibrosis [22,23,24]. As such an essential inflammatory factor linked to fibrogenesis, several studies have shown pharmacological HMGB1 inhibition to limit fibrosis in a rat model of non-alcoholic steatohepatitis and cardiac fibrosis [25,26]. This evidence further suggests that HMGB1 may become a critical target to prevent pathological scarring in the future.
Another interesting phenomenon involving the neutrophil cell population can occur during the initial inflammatory phase in many pathologies. Neutrophils are one of the first cell populations found in damaged tissue. Classically, neutrophils function to engulf potential pathogens or secrete anti-microbial proteins to prevent wound infection. However, a third neutrophil function has been elucidated over the past decade describing a unique cellular process that actively emits nuclear material to signal other inflammatory cells and sequester pathogens. The extruded nuclear material contains higher-order chromatin that clumps together forming fibrous strand meshes resembling a net and thus came to be known as neutrophil extracellular traps (NETs). NET formation is exclusive to neutrophils and separate from necrotic or apoptotic chromatin release; therefore, the extraordinary process was termed NETosis. Uncondensed nucleosome complexes released during NETosis include histone cores with associated dsDNA. NETs formed during this inflammatory process have also been linked to fibrotic pathologies. Increased NET formation has been observed in acute kidney injury (AKI) in humans; investigators showed that by enhancing (endothelial cell) tubular necrosis, NETs further exacerbated AKI fibrotic progression [27]. Another recent investigation has shown enhanced NET formation in the blood of cystic fibrosis patients that was precipitated by aberrant and delayed neutrophil apoptosis [28], further linking this unique process to fibrotic pathogenesis. Additionally, individual histones may release from NETs and themselves serve as robust DAMPs that vigorously protract excessive inflammation. Several studies have shown histones to interact with toll-like receptors that activate pro-inflammatory signaling and have proved cytotoxic to endothelial cells and myocytes [29,30]. Furthermore, it has been demonstrated that the acute inflammatory phase C-reactive protein can limit the cytotoxicity of circulating free histones through competitive inhibition of lysosome binding [31], lending further insight into histone DAMP signaling. These studies all suggest that limiting NETosis during inflammatory processes could have the potential to abrogate subsequent fibrosis as well.
Damaged tissues also release nuclear and mitochondrial nucleic acids through necrosis and serve as potent DAMPs that illicit the inflammatory response, activating immune cells and leading to the production of pro-inflammatory cytokines including potent fibrogenic initiators interleukin-1beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and transforming growth factor-beta (TGF-β), which further direct the adaptive immune response [32,33,34,35]. In recent years, studies have demonstrated that extracellular DNA fragments, either free or contained in extracellular vesicles, are phagocytized by tissue macrophages. Once phagocytized, DNA can stimulate the intracellular multiprotein complex receptor of innate immunity known as the inflammasome through interaction with AIM2 (absent in melanoma 2), an accessory protein required for inflammasome function [36,37]. Inflammasome activation is known to upregulate the production of pro-inflammatory IL-1β, IL-18, and TGF-β cytokines and induces fibrotic signaling [38,39,40]. This link to fibrosis further suggests that targeting inflammasome activation during the inflammatory phase could be a potential therapy to reduce fibrogenesis and warrants further investigation.
Additionally, damaged endothelium releases complement, which is a potent chemoattractant for neutrophils. Infiltrating neutrophils substantially increase the necrotic load and further contribute to a pro-inflammatory cycle through TNF-α and IL-1α/β production. Other factors known as alarmins are released from stressed and irreparably damaged cells following tissue insult and contribute directly to immune cell migration to the wound [34,41]. By DAMP production and through alarmin release, tissue damage ultimately results in the recruitment and activation of inflammatory cells tasked with the primary goal to moderate damage and begin the repair process.
3.2. Inflammatory Progression
IL-25 and IL-33 are inflammatory alarmins released by damaged epithelial cells and have been shown to induce a Th2 shift in CD4+ populations, which in turn is thought to promote fibrogenesis [41,42,43]. Representative Th2 cytokines include IL-4, IL-5, IL-10, and IL-13. IL-4 and IL-13 have overlapping signaling and are known to promote collagen production via STAT pathways [44,45] and produce significantly higher expression of extracellular matrix proteins in several fibrotic pathologies [46,47,48,49]. Burns resulting in severe fibrosis have also been shown to promote a Th2 shift and concomitantly increased Th2 cytokines [50,51]. Conversely, these same profibrotic Th2 cytokines inhibit the expression of many pro-inflammatory mediators, including TNF-α, IL-1, and IL-6, all known fibrogenic initiators, meaning Th2 pathway inhibition necessarily upregulates pro-inflammatory Th1 profiles. Ideally, CD4+ subsets must be balanced for an adequate inflammatory response and recruitment of other immune cell populations.
Mast cells may be another vital, yet overlooked, immune cell population needed as critical effectors in wound healing processes. Apart from their known role in allergy and anaphylaxis, several investigations have now shown mast cells’ growing importance in fibrogenesis and progression. Mast cells exert their immune influence through a process of regulated exocytosis known as degranulation. Following degranulation, mast cells release preformed mediators such as histamine that contribute directly to vasodilation and vascular permeability during inflammation. Histamine metabolites have been found significantly elevated in adult burn patients [52,53], and the multifaceted vasoactive amine has been shown to contribute to myofibroblast conversion with subsequent upregulation of collagen and other ECM components [54,55]. Histamine inhibition was also shown to mitigate fibrotic progression in mouse models of fibrotic cholangitis, underscoring its fibrogenic capacity [56,57]. Secreted mast cell serine proteases such as tryptase and chymase as well are known to upregulate collagen production and stimulate fibroblast proliferation and myofibroblast differentiation [58,59,60]. Our research group has also shown significantly increased mast cell densities in hypertrophic scars (HS) of severely burned pediatric survivors as well as burn-injured red Duroc pigs [61,62]. Furthermore, pharmacologic mast cell stabilization through topical application of cromolyn sodium or systemic administration of ketotifen can mitigate fibrotic progression in vivo [63,64,65]. The current data demonstrate the importance of mast cells during inflammation and highlight this versatile cell’s fibrotic capabilities. Despite the plethora of cellular and molecular participants described, resolving inflammation is critical to continuing the wound healing cascade.
Natural killer (NK) cells act as a liaison between the innate and adaptive immune systems. These cells sense stressed cells via their CD944/NKG2A receptors in humans and play an active role in tissue homeostasis following injury. The role of natural killer T (NKT) cells as negative regulators of wound healing has been extensively reviewed [4,66]. The absence of NKT cells in a mouse model of excisional wound injury resulted in accelerated wound healing and decreased risk of infection [67], while in wild type animals, NKT cells were expressed in the early phase of wound healing and regulated other immune cells including neutrophils and macrophages in addition to TGF-b secretion and collagen deposition [67]. Tanno et al. also reported that Jα18KO mice deficient in invariant NKT (iNKT) displayed a delay in wound closure by decreasing collagen deposition, myofibroblasts differentiation, and angiogenesis [68]. The same group has shown that iNKT cells promote skin wound healing by reducing the prolonged inflammatory response after injury [69]. Besides wound healing, these cells play an active role in other skin diseases like atopic dermatitis [70]. Taken together, these studies demonstrate that natural killer cells play an active role in all phases of wound healing including inflammation, proliferation, and tissue remodeling.
3.3. Resolving Inflammation to Initiate the Proliferation Phase of Wound Healing
Resolution is the final and essential stage of the wound healing inflammatory cascade. It is an active process directed by several factors designed to provide apoptotic or chemotactic stimuli to inflammatory cells that ultimately shifts progress toward the proliferation phase. Numerous mediators have been identified to control resolution; among the classes of factors best described are the resolvins (Rv). Rvs are bioactive lipid compounds derived from the arachidonic acid metabolic pathway and, as their name implies, are rapidly generated during inflammation resolution [71]. Phenotypic anti-inflammatory macrophages show upregulation of multiple RVs that have been shown to inhibit neutrophil chemotaxis and prevent entry into wound sites via cytoskeletal perturbation, and importantly, dysregulation of their resolving function protracts the inflammatory phase, further promoting fibrotic progression [72,73]. Thus, recent studies have found that exogenous application of Rvs, specifically the RvD1 lipid mediator, can significantly limit inflammation and prevent a fibrotic phenotype in both in vivo mouse models and in vitro human models of inflammatory disease [74,75,76]. Clinically, significantly higher Rv gene expression is seen after 28 days in trauma patients with uncomplicated recoveries; conversely, lower Rv gene expression was measured in complicated traumatic recoveries [77]. Similar results have also been found following severe burns. Recently, Inoue and colleagues showed postburn hepatic and renal damage was significantly reduced following intravenous administration of RvD2 [78]. Correspondingly, Bohr and colleagues [79] demonstrated RvD2 administration could prevent thrombosis following a severe burn in rats. As deep thermal injuries typically result in painful fibrosis and hypertrophic scarring, further investigations into Rv therapies are warranted.
Regulating pro- and anti-inflammatory conditions are vitally important in the healing wound, and failure of mechanisms to equilibrate this delicate balance may result in a prolonged and chronic inflammatory state, ultimately leading to fibrotic pathology. However, inflammation is a necessary process to heal damaged tissue. Limiting its detrimental impacts and resolving inflammation to initiate the proliferation phase of wound healing should be a significant research focus moving forward.
4. Proliferation in the Healing Wound
The dynamic progression from the inflammatory phase to the proliferative phase is one of the vital steps of wound healing. Accumulating evidence shows that prolongation or delay in the inflammatory phase has adverse effects on the remaining stages of wound healing, resulting in fibrosis [5,80]. The increase in specific cytokines and growth factors during initial inflammation drives the proliferation phase. However, prolongation in the inflammatory phase gives rise to fibrotic tissues that can manifest as cancers in some organs [81] and hypertrophic scars in the skin [82,83]. Thus, the inflammatory phase is tightly controlled and connected at cellular and molecular levels with the proliferative phase of wound healing.
The following sections discuss current understanding of the role of vital cells involved in the proliferation phase and highlight recent advancements made at pharmacological and biological levels as well as interventions that modulate these cells for a favorable wound healing scenario. Following skin injury, the platelet-rich plasma (PRP)-derived growth factor matrix provides an ideal platform for vital cells like keratinocytes, endothelial cells, fibroblasts, epidermal stem cells, and macrophages to begin proliferating, giving rise to immature granulation tissue [84,85]. The granulation tissue in the wound bed is vital to wound closure and is filled with new capillaries and connective tissue. The new capillaries and endothelial cells help with the formation of new blood vessels also referred to as angiogenesis. This process ensures that nutrients are supplied to the new granulation tissue.
4.1. Keratinocytes
Keratinocytes are the major cells of the epidermis. These cells produce structural and biologically active proteins and peptides that protect against various pathogens [86]. As the most peripheral cells of the skin, keratinocytes are one of the first cell populations to respond to a skin injury [87]. Over the last two decades, many therapeutic interventions have been investigated to modulate the proliferation of keratinocytes and expedite wound healing after burn. Cultured keratinocytes have evolved over the previous three decades from sheets [88], to inlay within different tissue-engineered products [89], to spray systems [90]. These methods showed promising results to accelerate wound healing and decrease fibrosis but have inherent drawbacks, with graft rejection being the most prominent [91]. Pharmacological stimulators like glycitin and phytochemical, 4′,6,7-trimethoxy-isoflavone (TMF) [92], icariin [93], and epigallocatechin-3-gallate [94] improved wound healing in different disease models by activating keratinocytes and fibroblasts in vitro and in vivo. Similarly, pharmacological activation of the Nrf2–IL-36γ pathway accelerated keratinocyte proliferation and expression of keratinocyte mitogens in fibroblasts, paving a new mechanistic path for keratinocyte modulation [95].
MicroRNA (miRNA) modulation was shown to regulate keratinocytes in various pathological scenarios. Overexpression of novel miRNA (seq-915_x4024) in keratinocytes improved skin regeneration and reduced scar formation by targeting TGF-β1/Smad signaling in a full-thickness excision mouse model [96]. Similarly, miR-132 is shown to regulate the transition from the inflammatory to the proliferative phase by dampening inflammation via STAT3 and ERK signaling as well as by activating the proliferation of keratinocytes [97]. Silencing miR-132 exacerbated inflammation, prolonged wound healing, and decreased keratinocyte proliferation in mouse and human ex vivo wound models [97]. However, miR-181b inhibited the proliferation of keratinocytes in psoriasis model via the TLR4 pathway [98].
4.2. Macrophages
As myeloid progenitors, macrophages are expanded in the liver and then distributed almost throughout the entire body as mature tissue-resident macrophages expressing the surface proteins F4/80 [99,100,101]. Macrophages, located at the basal and supra-basal layer of skin where they maintain tissue homeostasis, are commonly known as the Langerhans cells (LC) [102]. Following an acute cutaneous injury, neutrophils, tissue-resident macrophages, and hematopoietic monocytes arrive at the wound site by following the chemotactic gradient where they are predominantly activated via endotoxins secreted by bacteria, growth factors, or by interferon-gamma secreted by fibroblasts and natural killer cells [103]. PAMPs and DAMPs generated by damaged tissues also attract immune cells to the wound site. Macrophages are among the few active immune cells that are found from the beginning to the end of wound healing; thus, their intensity of activation, differentiation, and clearance defines the outcome of the wound [104]. As one of the most plastic cells of hematopoietic origin, macrophages constitute more than half of the cell population in granulation tissue [105]. Macrophages have significant roles in the development of fibrosis in many disease pathologies [106]. While macrophage presence and activity in the wound is vital, over-activation is pathologic as they prolong the proliferation and remodeling phases of wound healing, resulting in fibrotic tissue formation.
An increasing number of pharmacological and biological therapeutic agents have been developed to modulate macrophage polarization for improved wound healing outcomes. Macrophages are the primary producers of matrix metalloproteinases (MMPs), which have a significant role in creating a platform for new cells to grow and expand, while also transforming excess ECM during the remodeling phase [107]. MMP-12, a major macrophage-secreted elastase, has been linked to a fibrotic phenotype during the proliferation phase. Interestingly, MMP-12-/- mice show decreased expression of profibrotic markers like pSMAD2, TGF-β1, PDGF-BB in the skin and liver [108]. Contrarily, MMP-10-/- mice demonstrated that MMP-10 directs collagen production via macrophage modulation of MMP-13, further highlighting the pleiotropic effects of MMPs in wound healing [109].
Knipper and colleagues reported that interleukin-4 receptor α (IL-4Rα) is vital for macrophage activation and maturation, and plays an active role in tissue repair and the formation of granulation tissue [110]. IL-4Rα also promotes a pro-fibrotic phenotype by modulating collagen fibril compilation [110]. Relm-α was identified as an important mediator of IL-4Rα in macrophages as well. Macrophage activation resulted in a persistent production of lysyl hydroxylase 2 (LH2) in fibroblasts, a condition observed in aberrant human skin fibrosis [110].
Various modulators of macrophage behavior have shown promising results in improving systemic skin sclerosis (SSc), which is defined by aberrant fibrosis and vascular abnormalities. Among these modulators is the adipokine visfatin [111], the Friend leukemia virus integration 1 (Fli1) [112], and citrullinated vimentin [113]. Uderhardt and colleagues described a novel macrophage mechanism termed “cloaking” [114]. Cloaking is a process where tissue-resident macrophages respond to injury by extending their membranes to surround the wound, thus preventing tissue neutrophils from infiltrating the wound. Cloaking was not observed in macrophage-depleted animals resulting in compromised homeostasis and extensive tissue damage [114].
In our studies, we observed a significant increase in M2 phenotype macrophages in skin and scar biopsies obtained from burn survivors compared to controls [115]. Based on their secretory profile, surface antigen presentation, and differential temporal presence in the wound, macrophages are categorized into two phenotypes: M1 and M2 (Table 1).

Table 1.
Difference between M1 and M2 macrophages.
Rather than a research focus to diminish macrophage presence in a healing wound, molecular targeting, cellular reprogramming, or modulating macrophages via pharmacological or biological mediators hold a better promise to prevent fibrosis following tissue injury.
4.3. Endothelial Cells
Aberrant and excessive microvessel formation is one of the salient features of fibrosis [116]. Tissue injury is generally followed by the transient loss of vascular structures resulting in a hypoxic state and activation of hypoxia-inducible factor-1 (HIF-1) signaling. The hypoxic environment further intensifies angiogenesis to satisfy the increased oxygen demand, thus promoting endothelial cell proliferation [117]. The newly formed vessels are irregular and dysfunctional, which further exacerbates inflammation. Several studies have targeted the endothelial cell population for better therapeutic outcomes in fibrotic disease conditions. Gao et al. [118] reported that leucine-rich-alpha-2-glycoprotein 1 (LRG-1) mediated biomechanical forces to improve angiogenesis. Overexpression of LRG-1 was reported in hypertrophic scars, and depletion of LRG-1 in a mice model inhibited active angiogenesis and lessened skin fibrosis [118]. Using Cdh5-creERt2/ROSA-YFP mice for vascular-specific tracking, Patel and colleagues [119] concluded that the loss of notch signaling in the transgenic mice resulted in the rapid transition of endothelial cells to mesenchymal cells (EndMT), which in turn increased fibrosis, stimulated scar tissue formation, and delayed wound healing. Xu et al. [120] used Cdc42 deficient mice to determine the role of macrophages in wound healing. Cdc42 is a Rho family protein that plays a role in microvasculature permeability. They showed that knockdown of Cdc42 significantly increases the macrophage population, induces fibrosis, and substantially delays wound healing [120]. Dulauroy et al. [121] reported that the anchored membrane metalloprotease ADAM12 expression is upregulated during skin and muscle injury. Expression of ADAM12 during injury resulted in a profibrotic state with increased interstitial collagen deposition, while genetic ablation of ADAM12 expressing cells resulted in significantly less fibrosis and better wound healing. Aberrant endothelial cell proliferation and distribution is one of the hallmarks of fibrosis, and effectively targeting molecular pathways to reduce or inhibit the expression of endothelial cells following injury holds a promising future in attenuating fibrosis.
4.4. Mechanosensors in Fibrosis
Dynamic and positive mechanosensory feedback mechanisms drive cells towards a profibrotic phenotype by regulating the cell’s behavior and transforming the mechanical forces into biochemical signals, with the outcome being excess ECM accumulation [122,123,124]. The harmonic combination of three main physical forces, including pressure [125], stretch/strain [125], and shear [126] holds fibroblasts together in a three-dimensional structure composed of specific ratios of various ECM components. Injury disrupts the composition of the ECM, and the ratio of type III/type I collagen increases during scar development [127]. In controlled preclinical models of hypertrophic scarring, the application of pressure garment therapy improves physical appearance and reduces the total collagen content in scars by more than 50%, while the transcript of both type I and III collagen decreased by more than 40-fold [128]. Velasquez and colleagues [129] reported that the small molecule isoxazole modulates myocardin-related transcription factors (MRTFs) to regulate cellular cytoskeleton dynamics. Isoxazole accelerated wound closure and attenuated inflammation via MRTF pathway activation. These studies suggest that interventions to modulate mechanosensory mechanisms can potentially decrease fibrosis and scarring.
4.5. Fibroblast Role in Fibrosis
Regardless of the type of tissue injury, fibroblasts are of mesenchymal origin and play a significant role in maintaining tissue integrity by proliferating [130], differentiating [131], and collaborating with other cells [132] both in homeostasis and disease states. They modulate fibrosis by producing the components of the ECM. In the skin, fibroblasts primarily respond to injury by covering the insult with a granulation tissue during the proliferation phase and by differentiating into a myofibroblast phenotype during the remodeling phase, driven primarily by TGF-β/Smad signaling, which is further discussed in Section 5.2 of this review.
To establish a consensus regarding the origin of myofibroblasts, Driskell and colleagues used lineage tracing and reported two distinct fibroblast populations in the upper and lower dermis [133]. Papillary dermal fibroblasts are responsible for hair growth and arrector pili muscle formation, while the reticular dermal fibroblasts play a significant role in ECM production [133]. Similarly, using the Engrailed-1 (En1) gene for embryonic lineage mapping, Jiang et al. [134] followed the fate of two subsequent populations of fibroblasts in a mice model of skin wound healing; En-1 lineage-past fibroblast (EPFs) and En1-lineage-naive fibroblasts (ENFs). The authors reported a dynamic replacement of ENF progenitors by EPFs as the wound progressed and pointed to an increase in the expression of EPFs as a significant source of scarring. Besides Engrailed-1, the transcription factor PU.1 was reported to control tissue fibrosis [134]. Genetic ablation of PU.1 expressing gene SPI1, using CRISPR–Cas9 system, resulted in lower collagen production, but the expression of α-smooth muscle actin (α-SMA) and F-actin were not changed. However, overexpression of PU.1 in fibroblasts induced a profibrotic phenotype characterized by increased collagen, α-SMA, and F-actin production. Inhibition of PU.1 by an anti-fibrotic pharmacological mediator, DB1976, prevented skin fibrosis [135]. Although the role of fibroblasts in tissue repair is widely accepted, debate continues about their specific characterization. Robust characterization of fibroblasts based on surface protein expression, functional roles, and tissue niche will aid in developing novel treatments for fibrotic disorders.
4.6. Role of the Fascia in Wound Closure and Fibrosis
Current investigations are revealing a unique role for the subcutaneous fascia in wound closure and scarring. Utilizing fate mapping and live imaging, researchers traced the rise of embryonic Engrailed-1 positive fibroblasts from the fascia to the wound bed and subsequently to the skin surface [136]. Blood vessels, macrophages, and peripheral nerves are embedded in this raised jelly-like matrix, which may explain the morbidities of itch and pain emanating from some scars. This is a new line of investigation that may be worth pursuing to gain better insights into the pathophysiology of fibrosis.
5. Remodeling the Wound
The remodeling phase of wound healing starts by the end of the proliferation phase where wound reepithelization through keratinocytes and ECM deposition by the fibroblasts and endothelial cells occurs. In normal wounds, this phase lasts weeks to months and is characterized by wound contraction and scar maturation. In burns, the remodeling phase is protracted due to prolonged inflammation as detailed above.
During the remodeling phase of wound healing, the skin/scar acquires an ultimate morphology that mostly depends on the final organization of collagen fibers. In normal scars, the collagen fibers are small in parallel bundles. In hypertrophic scars, the collagen fibers are thin, more abundant and cross-linked [137]. During the remodeling phase, myofibroblasts also secrete Decorin; a protein that regulates collagen fibrogenesis by presenting as a “C” shape localized between the collagen fibrils to assure a uniform spatial fibril arrangement [138]. The fate of myofibroblasts during remodeling determines whether the wound closes and continues to develop a hypertrophic scar. In non-hypertrophic scars, myofibroblasts surrounded by fibrillar collagen may cause adverse effects leading to cell cycle arrest [139] or loss in the ability to adhere and thus undergo apoptosis [3].
As mentioned above, mechanical tension and increased inflammatory cytokines concentration, like TGF-β, drive fibroblast differentiation into myofibroblasts by the end of the granulation phase [137]. Myofibroblasts express high levels of α-smooth muscle actin (SMA), stress fibers, and contribute significantly to wound contraction [140]. Myofibroblasts also produce substantially more collagen than their regular counterparts. Collagen III in the ECM is replaced by collagen I, which has higher tensile strength but takes longer to deposit. Collagen organization is also altered in hypertrophic scars, and the healed skin can only achieve ~80% of the original tensile strength [141]. In burns, excessive and prolonged inflammation leads to excessive pathologic collagen deposition and fibrosis. Therefore, the attenuation of the inflammatory response can minimize aberrant collagen production.
5.1. Myofibroblasts and Apoptosis
During the wound healing process, skin fibroblasts that expanded in the proliferation phase now acquire a contractile phenotype by expressing high levels of the motile α-SMA protein, which aids fibroblast migration and ultimate wound closure. These fibroblasts, now termed myofibroblasts, were first described by Gabbiani et al. [131] and participate in the wound healing process by depositing substantial amounts of ECM proteins including collagen, elastin, and hyaluronic acid. Myofibroblasts are activated by inflammatory cytokines. TGF-β acts as the critical cytokine in epithelial−mesenchymal transition (EMT) and myofibroblast differentiation. In healthy wound healing, myofibroblast populations should dissipate when the wound is closed, mainly through apoptosis, or revert to quiescent fibroblasts [137,142]. In fibrotic conditions, myofibroblast apoptosis is delayed and the cells continue to express collagen and other ECM components. In burns, the prolonged inflammatory response over-activates myofibroblasts leading to overexpression of various components of the ECM and, therefore, the development of hypertrophic scars. Recent studies have targeted fibrosis by inducing myofibroblast apoptosis [143,144] including the use of gene therapy [144] and cuprous oxide nanoparticles [145].
New research has focused on ways to accelerate myofibroblast apoptosis or inhibit the transformation of fibroblasts into myofibroblasts [146]. The anti-fibrotic drug pirfenidone reduces the profibrotic and contractile phenotype of differentiated human dermal myofibroblasts [146]. Pirfenidone is a pyridine (5-methyl-1-phenyl-2-(1H)-pyridone) that is FDA-approved for the treatment of idiopathic pulmonary fibrosis [147,148]. Pirfenidone reduces hypertrophic scarring following a burn, animal bites, and in diabetic ulcer wounds [149,150,151,152] and a study by Wells and Leung [146] have shown that pirfenidone may reduce fibrosis through inhibition of fibroblast differentiation into myofibroblasts. Additional evidence has demonstrated that Pirfenidone can reduce the expression of αSMA and the contractile activity of fibroblasts while concurrently increasing the mRNA expression of MMP-1 [146].
New approaches are using regenerative therapies to reprogram myofibroblast differentiation in hypertrophic scarring [153,154]. Adipocytes have been shown to modulate myofibroblast differentiation through the activation of the bone morphogenetic protein -2 (BMP-2) and BMP-4 [154]. In fact, adipocytes initiate tissue remodeling by secreting BMP-4 to activate the signaling of nuclear receptor PPAR-y [153]. The role of PPAR in fibrosis and scarring is detailed further in Section 5.6 of this review.
5.2. Collagen Degradation and the Role of MMPs
Matrix metalloproteases (MMPs) are endopeptidases using calcium or zinc in their active sites. They are involved in the degradation of collagen and other ECM components. MMPs are grouped into four categories: collagenases, gelatinases, stromelysins, and membrane-type MMPs. The collagenases include MMP-1, MMP-2, MMP-8, and MMP-13. MMPs are essential regulators of collagen degradation that have been shown to cleave collagen I and III in scar tissue. MMP activation may reduce excessive collagen deposition and subsequent scarring. However, over-activation of MMPs may impede wound closure, increase the risk of infection, and further prolong the inflammatory phase.
Various studies have shown that MMPs modulate skin regeneration and hypertrophic scarring post-burn. Clinically, plasma levels of both MMP-2 and MMP-9 were significantly increased in patients that received a dermal regeneration Integra graft [155]. Using the Red Duroc pig model of hypertrophic scarring, DeBruler and colleagues [156] have shown that MMP-2 protein expression peaked four weeks after burn and subsequent split-thickness autographs. Using the same model, others have shown that MMPs are differentially regulated in response to compression therapy, with MMP-7 being the most downregulated [157]. Pharmacological approaches have been used to suppress collagen production by increasing MMPs as well [158]. The anti-inflammatory drug methotrexate reduces MMP-1 through the MAPK pathway [158], and kynurenine was also shown to increase MMP-1 and -3 expressions to improve wound healing in vivo through the same pathway [159]. MMP activity is also regulated by the tissue inhibitors of metalloproteinase (TIMPs). TIMP-1 and MMP-1 proteins are highly expressed in patients with extensive post-burn hypertrophic scars [160].
5.3. Role of the Ubiquitin-Proteasome System in Fibrosis
Various studies have addressed the role of ubiquitination and deubiquitination in idiopathic pulmonary fibrosis [161] as reviewed by Li et al. [162]. However, the role of this pathway in myofibroblast differentiation during scar formation is not well understood. We have reported that the basal levels of β2-AR ubiquitination were higher in hypertrophic scar fibroblasts from burn patients compared to non-burn skin fibroblasts, suggesting accelerated receptor degradation [163]. Recent studies have elucidated the role of the ubiquitination in both normal tissue repair and fibrosis and identified specific ubiquitin ligases in this process. Tetratricopeptide repeat domain 3 (TTC3), a new ubiquitin E3 ligase, positively regulates TGF-β1-induced epithelial to mesenchymal transition and myofibroblast differentiation [164]. TTC3 induces the ubiquitination and proteasome-dependent degradation of the Smad ubiquitination regulatory factor 2 (SMURF2) [164]. This leads to a suppression of SMAD2/3, which in turn activates TTC3 [164]. SMURF2, mainly localized in the nucleus, binds to SMAD7, translocates out of the nucleus, and induces the turnover of TGF-β receptors [165]. Our group has previously shown that the increased expression of SMURF2 is involved in the progression of hypertrophic scarring after burn [166]. Modulation of another ubiquitin 3 ligase, Mitsugumin 53 or MG53, was shown to be essential to the regulation of wound healing and scarring [167]. MG53 is a member of the tripartite-motif (TRIM) family of proteins that plays an essential role in the cell membrane repair machinery. MG53 deficient mice with excisional wounds display delayed wound healing and abnormal scarring and significant defects in skin architecture and collagen overproduction [167].
5.4. The Role of Plasminogen Activation Inhibitors and MicroRNAs in Wound Healing and Scarring
Recent studies have pointed to new players that can reduce HS if targeted at the right time. Among those is plasminogen activator inhibitor-1 (PAI-1). PAI-1 is a serine protease inhibitor that functions as the principal inhibitor of the tissue plasminogen activator (tPA) and urokinase (uPA) [168], the activators of plasminogen, and hence controls fibrinolysis. PAI-1 protein expression is increased in cancer [169], obesity [170], and metabolic syndrome [171,172]. Increased levels of PAI-1 can lead to an increased risk of thrombosis and atherosclerosis [172]. PAI-1 controls tissue homeostasis by regulating the proteolytic activities of uPA/tPA/plasmin/MMP [173], therefore regulating the degradation of collagen and other ECM components [173]. A very recent report showed that PAI-1 regulates fibrosis by mediating the effects of mast cells on fibroblasts, therefore regulating collagen secretion by these cells [174]. These data also connect with and underline the important role of the immune response in the regulation of fibrosis.
The role of non-coding RNAs, including miRNA and long non-coding RNAs (lncRNAs) in the regulation of wound healing and scarring, is well-established and was extensively reviewed by Herter and Landén [175]. PAI-1 also seems to drive collagen degradation through microRNAs. In fact, Collagen I generation in hypertrophic scars is regulated by MiR-10 and MiR-181C targeting of PAI-1 and uPA, respectively [176]. Pang et al. [177] have shown that miR-152-p reduces keloid scar fibroblast proliferation and migration and promotes apoptosis through the Smad2/3 pathway. Similarly, MiR-23b inhibits the proliferation and migration of heat-denatured fibroblasts by directly targeting Smad3 and Notch1 signaling pathways [178]. MiR-181b reduces the expression of Decorin by dermal fibroblasts, and its inhibition increases Decorin production, destabilizes the formation of collagen bundles, and prevents scarring [179]. The same study identified MiR-181b as a potential therapeutic target to reduce fibrosis [179]. MiR-148b also targets TGF-β signaling to regulate EMT and angiogenesis during wound healing [180]. Together, these studies open new avenues for the therapeutic application of miRNAs in tissue repair, acceleration of wound healing, and reduction of scarring.
Besides miRNAs, long non-coding RNAs (lncRNAs) are also involved in wound healing and scarring by modulating TGF-β-induced fibrosis [181,182]. LncRNAs are a class of RNAs longer than 200 nucleotides without protein-coding capacity. LncRNAs are involved in the pathophysiology and development of human hypertrophic scars, and high throughput screening has shown that lncRNAs are differentially expressed in human hypertrophic scars [183]. A microarray study showed that 6104 lncRNAs and 2952 mRNAs were differentially expressed in hypertrophic scars compared to normal fibroblasts [182]. Among the lncRNA, expression of AC067945.2 was shown to decrease in hypertrophic scar tissues [184]. Overexpression of AC067945.2 slightly stimulated early apoptosis in normal skin fibroblasts without affecting cell proliferation [184]. Co-expression analysis has shown that the expression of lncRNAs NR_125715 and NR_046402 in hypertrophic scar fibroblasts correlated with TGF‑β2 and POLD1 mRNA expression [182]. LncRNA8975-1 is increased in HS fibroblasts and regulates collagen production [185]. The same group identified 1871 lncRNAs that are differentially expressed between regressive and mature scars including lncRNA8975-1, AC097662.2, and RP11-586K2.1 [185].
5.5. Epigenetic Regulation of Wound Healing
Persistent expression of pro-fibrotic genes after wound resolution results in fibrosis [186], and accumulating evidence suggests a role for epigenetics in this process [187]. Various studies have demonstrated that pathologically profibrotic fibroblasts are epigenetically altered [188,189,190]. Epigenetics is one of the most reliable mechanisms to transfer the gene function from mother to daughter cells without any changes in the DNA sequences. This process is carried out in two different ways via DNA methylation or histone modifications. Aberrant hypermethylation of certain genes facilitates profibrotic gene activation and drives fibrogenesis. Fibroblasts from scars and keloids express high levels of DNA methyltransferase 1 (DNMT1) compared to normal skin fibroblasts [76]. Following treatment with a methylase inhibitor, 5-aza-2-deoxycytidine, the expression of DNMT1, and TGF-β were decreased while Smad7 mRNA transcript levels and apoptosis markers were increased in the hypertrophic scar fibroblasts [76].
Histone methylation and acetylation are both implicated in profibrotic diseases. Histone deacetylases are upregulated in normal and keloid scars [190]. Inhibition of histone deacetylation, using trichostatin A (TSA), blocks TGF-b mediated myofibroblast differentiation [191] and collagen expression in scar fibroblasts [189]. TSA was also shown to reduce hypertrophic scarring in the rabbit ear excision model [192]. Together, these data suggest a potential therapeutic application of histone deacetylase inhibitors in the reduction of fibrosis and scarring.
Histone methylation is mediated by methyltransferases and demethylases, and its effects on gene activation or repression depend on the extent of methylation and position of this modification in the histone proteins [193]. Historically, activation of gene expression is linked to trimethylation of histone 3 at the lysine 4 (H3K4me3) and dimethylation or tri-methylation at lysine 79 of histone 3 (H3K79me2/3) [194]. Gene repression, however, is linked to trimethylation at lysine 9 (H3K9me3), lysine 27 (H3K27me3), and lysine 20 (H3K20me3) [194]. The role of histone methylation in fibrotic diseases is well-established [195]. In the skin, expression of H3K27 histone demethylases is the most upregulated during wound repair [196]. TSA and 5-aza-deoxycytidine were shown to reverse epigenetic repression of the Fli1 gene [197], a gene involved in the regulation of collagen expression [198], and to reduce collagen expression in scleroderma fibroblasts [197]. A time-course scarring study of engrailed positive fibroblasts (EPFs), a precursor of activated fibroblasts, demonstrated that transposase-accessible chromatin sequencing (ATAC-seq) is functionally distinct in these fibroblasts and expresses major differences in genes associated with fibrosis [199]. These findings strongly support a role of epigenetic regulation of gene expression in the fibroblast phenotype and open the doors for novel potential therapeutic targets.
5.6. Transcriptional Regulation of Myofibroblasts Differentiation, Contraction, and Wound Healing
Bellavia et al. [200] extensively reviewed the role of transcription factors in wound healing and re-epithelization. Many transcription factors were shown to play a role in wound healing and scarring. That includes the activator protein-1 (AP-1) transcription factor family members including Jun, Fos, and ATF dimers and Calveolins [201]. The myocardin-related transcription factors (MRTFs) regulate myofibroblasts’ contractility by associating with serum response factor (SRF) and activating genes implicated in cytoskeletal dynamics [129]. The Hippo members Yes-associated protein 1 (YAP1) and transcriptional coactivator with PDZ-binding motif (TAZ) are activated by tissue stiffness and TGF-β1 [202]. Knockdown of YAP-1 abrogates TGF-β-induced expression of SMA stress fibers and collagen type I deposition [202]. In a full-thickness, skin wound model, knockdown of YAP/TAZ delays wound closure and modulates the expression of various components of TGF-β signaling including Smad-2, p21, and Smad-7 [164]. While some data suggest that Hippo member transcription factors YAP-1/TAZ contribute to the contractile phenotype of myofibroblasts in the full-thickness burn model, their role in scar development after burn is less understood. The transcriptional modulation of metabolic reprogramming also regulates the contractile phenotype of myofibroblasts as shown by genetic and pharmacologic approaches that aimed to impair mitochondrial biogenesis or glycolysis [203].
Nuclear receptors also play a role in wound healing. Among those, the estrogen receptors (ERs) contribute to cutaneous aging, delayed or impaired wound healing, and increased risk of infection [204,205]. Decreased estrogen levels with age reduce both types I and type III collagen and shift the collagen III/I ratio in the skin [206]. Estrogen receptors are involved in all three phases of wound healing; they regulate the inflammation phase by modulating the infiltration of immune cells to the wound bed [207]. Most immune cells including macrophages, monocytes, neutrophils, dendritic cells, and mast cells express ERs, further implicating these nuclear receptors in regulating the inflammatory phase of wound healing [207,208,209,210]. More importantly, estrogens were shown to alter macrophage polarization between a pro-inflammatory M1 to an anti-inflammatory M2 phenotype [211,212]. ERs are also expressed in fibroblasts, endothelial, epithelial cells, and keratinocytes, therefore modulating the proliferative phase of wound healing [207]. By reducing inflammation and modulating the proliferation phase of wound healing, ERs contribute to accelerated wound closure. Simultaneously, ERs reduce aberrant collagen deposition and subsequent scaring by inhibiting TGF-beta signaling by accelerating the degradation of Smad 2/3 [213].
Another group of nuclear receptors that regulate wound healing is the peroxisome proliferator-activated receptors (PPARs). The role of PPARs in skin wound healing was extensively reviewed by Yin and Smith [214]. Skin injury stimulates the expression of PPAR-α and PPAR-β the site of the wound [215,216] and regulates wound healing by antagonizing the effects of TGF-β and TNF-α [215]. Expression of PPAR-γ is also induced by skin injury [216] and is known to suppress the profibrogenic response to injury by antagonizing TGF-beta signaling [217]. PPAR-γ is usually bound to the retinoid X receptor (RXR) and its corepressors. After receptor activation, the PPAR-γ RXR complex detaches from the repressors and binds the PPAR-γ response elements to activate gene expression [218]. PPAR-γ reduces fibroproliferative responses by competing with Smad 2/3 for its cofactor p300 [219]. These effects are not observed in the PPAR-γ knockout mice, which exhibit a profibrotic skin phenotype [219].
6. Clinical Trials to Reduce Fibrosis
With the skyrocketing increase in the economic burden of fibrosis and skin-related diseases, there has been a significant increase in the number of clinical trials aiming to treat fibrotic-related morbidities, specifically the ones related to lung and liver fibrosis [220,221]. Currently, there are no FDA-approved treatments for skin-related fibrosis. According to clinicaltrials.gov, 43 registered interventional clinical trials were primarily funded by industries in the last 10 years. These interventional studies include biological mediators like EXC 001 [222], an antisense oligonucleotide that targets connective tissue growth factor, tilapia skin for surgical wound dressing [222], fat lipofilling [223], and stem cell injection [224]. Other trials have used pharmacological mediators such as PF-06473871 [225] and RXI-109 [226] to ameliorate hypertrophic scars and P144 to treat skin fibrosis by blocking the interaction between TGF-β1 type III and TGF-β1 receptors (Acces on26th Nov, 2019, https://clinicaltrials.gov/ct2/show/NCT00574613).
Although TGF-β is regarded as the master regulator of fibrosis [227], clinical trials targeting the TGF-β signaling pathway have yet to produce a therapeutically beneficial outcome in treating fibrosis-related morbidities. Using anti-TGF-β antibodies, two trials, CAT-152 using a dose of 100 micrograms in 100 microliters (TGF-β2 > TGF-β3, 2002) [228] and CAT-192 using a dose of 5 mg/kg (TGF-β1, 2007) [229], failed to show promising results in treating patients with conjunctival fibrosis undergoing trabeculectomy and cutaneous SSc, respectively. Unfortunately, four deaths and significant adverse side effects were reported in the study using CAT-192, compared to the placebo [229]. Besides TGF-β signaling, physical interventions like non-ablative fractional laser therapy [223], gel-creams, and silicone garment therapy [230] for preventing hypertrophic scarring have also been reported.
7. Molecular Modulation of Hypertrophic Scarring Following Severe Burns
Post-burn hypertrophic scars are the epitome of wound healing phases in disarray. These scars are serious complications of deep thermal burns and remain the biggest challenge to patient recovery [231]. Burn survivors’ scars are raised, pruritic, prevent normal function, and severely reduce the quality of life. Traditionally, hypertrophic scarring is assessed using various scales including the Vancouver Scar Scale (VSS) or the modified patient and observer scar assessment scale (POSAS) [232]. Recent methods for scar assessment include the use of a three-dimensional camera [233]. Three-dimensional stereo-photogrammetry was recently introduced as a reliable method for assessing scar volume in clinics [234]. The location of scars on the face and other visible areas affects the patient self-esteem and prevents total reintegration into society [235,236]. While the incidence of hypertrophic scars is high following deep thermal injury, it is difficult to predict which patients will scar and those that will not. Equivalently, patients respond differently to similar therapeutic approaches, warranting the need to understand mechanisms underlying hypertrophic scarring after burn and the differential response to treatments.
Various types of surgical and non-surgical strategies are used for the treatment of hypertrophic scars, including the application of dynamic and static mechanical forces, pressure garments [237], and light-based therapies including the CO2 ablative fractional laser [238,239]. Administration of various injectable and topical treatments including corticosteroids, immune modulators, hyaluronidases, and cell-based therapies [240,241,242,243] have demonstrated limited efficacy. Injection of uncultured centrifuged adipose tissue in burn survivors has resolved pain, restored movement, and significantly reduced scar hypertrophy [244]. The fat tissue in the above study was harvested by liposuction using the Coleman technique to minimize tissue processing and contains adipocytes, SVF, and blood cells. However, none of these treatments are entirely effective in fully attenuating hypertrophic scars and associated morbidities, making surgical revisions a last resort for these patients. All evidence further supports the introduction of novel research initiatives to prevent scar development and progression.
Conclusions
Skin fibrosis and scarring involve an imbalance of various mediators throughout wound healing. Efficacious therapies should include a multidimensional approach targeting all three phases of wound healing, starting by moderating the inflammatory response, limiting the excessive proliferation of myeloid cells and myofibroblasts, and applying physical and biological interventions during wound remodeling.
Author Contributions
Conceptualization—A.E.A., J.W.J., A.P.; methodology—A.E.A., J.W.J., A.P., literature search and investigation—A.E.A., J.W.J., A.P.; writing—original draft preparation—A.E.A., J.W.J., A.P.; writing—review and editing—A.E.A., J.W.J., A.P.; project administration—A.E.A. All authors have read and agreed to the published version of the manuscript.
Funding
We apologize to all colleagues whose important findings could not be cited owing to space limitations. JWJ is funded by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM008256. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
The authors would like to thank Eileen Figueroa and Steve Schuenke from the Department of Surgery at the University of Texas Medical Branch, for their assistance in the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AIM2 | absent in melanoma 2 |
| AP-1 | activator protein-1 |
| BMP | bone morphogenetic protein |
| DAMPs | danger-associated molecular patterns |
| DNMT1 | DNA methyltransferase 1 |
| ECM | extracellular matrix |
| EMT | epithelial−mesenchymal transition |
| En1 | Engrailed-1 |
| EndMT | endothelial cells to mesenchymal cells |
| ERs | estrogen receptor |
| Fli1 | Friend leukemia virus integration 1 |
| HIF-1 | hypoxia-inducible factor-1 |
| HMGB1 | high-mobility group box-1 |
| HS | hypertrophic scars |
| IL-1β | initiators interleukin-1beta |
| IL-4Rα | interleukin-4 receptor α |
| LC | Langerhans cells |
| LH2 | lysyl hydroxylase 2 |
| lncRNAs | long non-coding RNAs |
| LRG-1 | leucine-rich-alpha-2-glycoprotein 1 |
| miRNA | microRNA |
| MMP | matrix metalloprotease |
| MRTFs | myocardin-related transcription factors |
| NETs | neutrophil extracellular traps |
| NK | natural killer |
| PAI-1 | plasminogen activator inhibitor-1 |
| POSAS | patient and observer scar assessment scale |
| PPAR | peroxisome proliferator-activated receptor |
| PRP | platelet-rich plasma |
| PRRs | pattern-recognition receptors |
| RAGE | receptor for advanced glycation end |
| Rv | resolvins |
| RXR | retinoid X receptor |
| SMURF | Smad ubiquitination regulatory factor 2 |
| SSc | systemic skin sclerosis |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TGF-β | transforming growth factor-beta |
| TIMP | tissue inhibitors of metalloproteinase |
| TMF | trimethoxy-isoflavone |
| TNF-α | tumor necrosis factor-alpha |
| TRIM | tripartite-motif |
| TSA | trichostatin A |
| TTC3 | tetratricopeptide repeat domain 3 |
| VSS | Vancouver Scar Scale |
| YAP1 | Yes-associated protein 1 |
| α-SMA | α-smooth muscle actin |
References
- Jeschke, M.G.; Gauglitz, G.G.; Kulp, G.A.; Finnerty, C.C.; Williams, F.N.; Kraft, R.; Suman, O.E.; Mlcak, R.P.; Herndon, D.N. Long-Term persistance of the pathophysiologic response to severe burn injury. PLoS ONE 2011, 6, e21245. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-Mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef]
- Sarrazy, V.; Billet, F.; Micallef, L.; Coulomb, B.; Desmouliere, A. Mechanisms of pathological scarring: Role of myofibroblasts and current developments. Wound Repair Regen. 2011, 19 (Suppl. 1), s10–s15. [Google Scholar] [CrossRef]
- Canedo-Dorantes, L.; Canedo-Ayala, M. Skin acute wound healing: A comprehensive review. Int. J. Inflam. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.L.; Marshall, C.D.; Barnes, L.A.; Murphy, M.P.; Ransom, R.C.; Longaker, M.T. Scarless wound healing: Transitioning from fetal research to regenerative healing. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e309. [Google Scholar] [CrossRef]
- Walmsley, G.G.; Hu, M.S.; Hong, W.X.; Maan, Z.N.; Lorenz, H.P.; Longaker, M.T. A mouse fetal skin model of scarless wound repair. J. Vis. Exp. 2015, 95, 52297. [Google Scholar] [CrossRef] [PubMed]
- Larson, B.J.; Longaker, M.T.; Lorenz, H.P. Scarless fetal wound healing: A basic science review. Plast. Reconstr. Surg. 2010, 126, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Colwell, A.S.; Longaker, M.T.; Lorenz, H.P. Mammalian fetal organ regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 93, 83–100. [Google Scholar] [PubMed]
- Longaker, M.T.; Chiu, E.S.; Adzick, N.S.; Stern, M.; Harrison, M.R.; Stern, R. Studies in fetal wound healing. V. A prolonged presence of hyaluronic acid characterizes fetal wound fluid. Ann. Surg. 1991, 213, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Longaker, M.T.; Chiu, E.S.; Harrison, M.R.; Crombleholme, T.M.; Langer, J.C.; Duncan, B.W.; Adzick, N.S.; Verrier, E.D.; Stern, R. Studies in fetal wound healing. IV. Hyaluronic acid-stimulating activity distinguishes fetal wound fluid from adult wound fluid. Ann. Surg. 1989, 210, 667–672. [Google Scholar] [CrossRef]
- Longaker, M.T.; Whitby, D.J.; Adzick, N.S.; Crombleholme, T.M.; Langer, J.C.; Duncan, B.W.; Bradley, S.M.; Stern, R.; Ferguson, M.W.; Harrison, M.R. Studies in fetal wound healing, VI. Second and early third trimester fetal wounds demonstrate rapid collagen deposition without scar formation. J. Pediatr. Surg. 1990, 25, 63–68. [Google Scholar] [CrossRef]
- Longaker, M.T.; Whitby, D.J.; Ferguson, M.W.; Harrison, M.R.; Crombleholme, T.M.; Langer, J.C.; Cochrum, K.C.; Verrier, E.D.; Stern, R. Studies in fetal wound healing: III. Early deposition of fibronectin distinguishes fetal from adult wound healing. J. Pediatr. Surg. 1989, 24, 799–805. [Google Scholar] [CrossRef]
- Longaker, M.T.; Whitby, D.J.; Ferguson, M.W.; Lorenz, H.P.; Harrison, M.R.; Adzick, N.S. Adult skin wounds in the fetal environment heal with scar formation. Ann. Surg. 1994, 219, 65–72. [Google Scholar] [CrossRef]
- Cass, D.L.; Bullard, K.M.; Sylvester, K.G.; Yang, E.Y.; Longaker, M.T.; Adzick, N.S. Wound size and gestational age modulate scar formation in fetal wound repair. J. Pediatr. Surg. 1997, 32, 411–415. [Google Scholar] [CrossRef]
- Szczesny, B.; Brunyanszki, A.; Ahmad, A.; Olah, G.; Porter, C.; Toliver-Kinsky, T.; Sidossis, L.; Herndon, D.N.; Szabo, C. Time-Dependent and organ-specific changes in mitochondrial function, mitochondrial DNA integrity, oxidative stress and mononuclear cell infiltration in a mouse model of burn injury. PLoS ONE 2015, 10, e0143730. [Google Scholar] [CrossRef]
- D’Arpa, P.; Leung, K.P. Toll-Like receptor signaling in burn wound healing and scarring. Adv. Wound Care (New Rochelle) 2017, 6, 330–343. [Google Scholar] [CrossRef]
- Li, L.C.; Gao, J.; Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef]
- El Gazzar, M. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm. Res. 2007, 56, 162–167. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, P.; Yu, Y.; Lu, H.; Liu, Y.; Ni, P.; Su, X.; Wang, D.; Liu, Y.; Wang, J.; et al. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Sci. Rep. 2016, 6, 21884. [Google Scholar] [CrossRef]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Dela Cruz, R.; Theise, N.; Nieto, N. High mobility group Box-1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, A.; Uyanik, M.H.; Cerrah, S.; Altas, S.; Dursun, H.; Demir, M.; Uslu, H. Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol. 2010, 23, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Bucova, M.; Majernikova, B.; Durmanova, V.; Cudrakova, D.; Gmitterova, K.; Lisa, I.; Klimova, E.; Kluckova, K.; Buc, M. HMGB1 as a potential new marker of disease activity in patients with multiple sclerosis. Neurol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074. [Google Scholar] [CrossRef]
- Wu, R.N.; Yu, T.Y.; Zhou, J.C.; Li, M.; Gao, H.K.; Zhao, C.; Dong, R.Q.; Peng, D.; Hu, Z.W.; Zhang, X.W.; et al. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2-mediated autophagy suppression in myocardial fibroblasts. Int. J. Cardiol. 2018, 267, 156–162. [Google Scholar] [CrossRef]
- Zeng, W.; Shan, W.; Gao, L.; Gao, D.; Hu, Y.; Wang, G.; Zhang, N.; Li, Z.; Tian, X.; Xu, W.; et al. Inhibition of HMGB1 release via salvianolic acid B-mediated SIRT1 up-regulation protects rats against non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 16013. [Google Scholar] [CrossRef]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef]
- Vogel, B.; Shinagawa, H.; Hofmann, U.; Ertl, G.; Frantz, S. Acute DNase1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin. Basic Res. Cardiol. 2015, 110, 15. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Dart, C.; Wang, S.S.; Thachil, J.; Guan, Y.; Wang, G.; Toh, C.H. Human CRP defends against the toxicity of circulating histones. J. Immunol. 2013, 191, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Houston, S.A.; Sherwood, E.; Casulli, J.; Travis, M.A. Regulation of innate and adaptive immunity by TGFbeta. Adv. Immunol. 2017, 134, 137–233. [Google Scholar] [PubMed]
- Magna, M.; Pisetsky, D.S. The alarmin properties of DNA and DNA-associated nuclear proteins. Clin. Ther. 2016, 38, 1029–1041. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of endogenous nucleic acids by the innate immune system. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Artlett, C.M. The role of the NLRP3 inflammasome in fibrosis. Open Rheumatol. J. 2012, 6, 80–86. [Google Scholar] [CrossRef]
- Erlich, Z.; Shlomovitz, I.; Edry-Botzer, L.; Cohen, H.; Frank, D.; Wang, H.; Lew, A.M.; Lawlor, K.E.; Zhan, Y.; Vince, J.E.; et al. Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat. Immunol. 2019, 20, 397–406. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Imitazione, P.; Somma, P.; Rega, R.; Saccomanno, A.; Aquino, R.P.; Pinto, A.; Sorrentino, R. AIM2 inflammasome activation leads to IL-1alpha and TGF-beta release from exacerbated chronic obstructive pulmonary disease-derived peripheral blood mononuclear cells. Front. Pharmacol. 2019, 10, 257. [Google Scholar] [CrossRef]
- Scott, I.C.; Majithiya, J.B.; Sanden, C.; Thornton, P.; Sanders, P.N.; Moore, T.; Guscott, M.; Corkill, D.J.; Erjefalt, J.S.; Cohen, E.S. Interleukin-33 is activated by allergen-and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci. Rep. 2018, 8, 3363. [Google Scholar] [CrossRef] [PubMed]
- Oshio, T.; Komine, M.; Tsuda, H.; Tominaga, S.I.; Saito, H.; Nakae, S.; Ohtsuki, M. Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice. J. Dermatol. Sci. 2017, 85, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gieseck, R.L., III; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Aoudjehane, L.; Pissaia, A., Jr.; Scatton, O.; Podevin, P.; Massault, P.P.; Chouzenoux, S.; Soubrane, O.; Calmus, Y.; Conti, F. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab. Invest. 2008, 88, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, Z.; Yan, J.; Wang, Y.; Hu, Z.; Mitch, W.E.; Wang, Y. The IL-4 receptor alpha has a critical role in bone marrow-derived fibroblast activation and renal fibrosis. Kidney Int. 2017, 92, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Fujitsu, Y.; Fukuda, K.; Kumagai, N.; Nishida, T. IL-4-induced cell proliferation and production of extracellular matrix proteins in human conjunctival fibroblasts. Exp. Eye Res. 2003, 76, 107–114. [Google Scholar] [CrossRef]
- Postlethwaite, A.E.; Holness, M.A.; Katai, H.; Raghow, R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J. Clin. Invest. 1992, 90, 1479–1485. [Google Scholar] [CrossRef]
- Sugimoto, R.; Enjoji, M.; Nakamuta, M.; Ohta, S.; Kohjima, M.; Fukushima, M.; Kuniyoshi, M.; Arimura, E.; Morizono, S.; Kotoh, K.; et al. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int. 2005, 25, 420–428. [Google Scholar] [CrossRef]
- Peng, H.; Sarwar, Z.; Yang, X.P.; Peterson, E.L.; Xu, J.; Janic, B.; Rhaleb, N.; Carretero, O.A.; Rhaleb, N.E. Profibrotic role for interleukin-4 in cardiac remodeling and dysfunction. Hypertension 2015, 66, 582–589. [Google Scholar] [CrossRef]
- Rani, M.; Schwacha, M.G. The composition of T-cell subsets are altered in the burn wound early after injury. PLoS ONE 2017, 12, e0179015. [Google Scholar] [CrossRef]
- Rani, M.; Zhang, Q.; Schwacha, M.G. Burn wound gammadelta T-cells support a Th2 and Th17 immune response. J. Burn Care Res. 2014, 35, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tredget, E.E.; Iwashina, T.; Scott, P.G.; Ghahary, A. Determination of plasma Ntau-methylhistamine In Vivo by isotope dilution using benchtop gas chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1997, 694, 1–9. [Google Scholar] [CrossRef]
- Tredget, E.E.; Levi, B.; Donelan, M.B. Biology and principles of scar management and burn reconstruction. Surg. Clin. N. Am. 2014, 94, 793–815. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Yamauchi, Y.; Mikami, Y.; Sakamoto, M.; Jo, T.; Nakajima, J.; Takizawa, H.; Nagase, T.; Kohyama, T. Histamine induces human lung fibroblast-mediated collagen gel contraction via histamine H1 receptor. Exp. Lung Res. 2014, 40, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Jordana, M.; Befus, A.D.; Newhouse, M.T.; Bienenstock, J.; Gauldie, J. Effect of histamine on proliferation of normal human adult lung fibroblasts. Thorax 1988, 43, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(-/-) mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Karstens, W.; Jones, H.; DeMorrow, S.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Alpini, G.; et al. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2(-/-) mice and human cholangiocarcinoma tumorigenesis. Hepatology 2018, 68, 1042–1056. [Google Scholar] [CrossRef]
- Bagher, M.; Larsson-Callerfelt, A.K.; Rosmark, O.; Hallgren, O.; Bjermer, L.; Westergren-Thorsson, G. Mast cells and mast cell tryptase enhance migration of human lung fibroblasts through protease-activated receptor 2. Cell Commun. Signal. 2018, 16, 59. [Google Scholar] [CrossRef]
- Chen, H.; Xu, Y.; Yang, G.; Zhang, Q.; Huang, X.; Yu, L.; Dong, X. Mast cell chymase promotes hypertrophic scar fibroblast proliferation and collagen synthesis by activating TGF-beta1/Smads signaling pathway. Exp. Ther. Med. 2017, 14, 4438–4442. [Google Scholar]
- White, M.J.; Galvis-Carvajal, E.; Gomer, R.H. A brief exposure to tryptase or thrombin potentiates fibrocyte differentiation in the presence of serum or serum amyloid p. J. Immunol. 2015, 194, 142–150. [Google Scholar]
- Jay, J.; Prasai, A.; El Ayadi, A.; Herndon, D.; Finnerty, C.C. Protease-Activated Receptor-2 knockdown attenuates the fibrotic phenotype in postburn hypertrophic scar fibroblasts. Wound Repair Regen. 2018, 26, A15–A16. [Google Scholar]
- Jay, J.; Prasai, A.; El Ayadi, A.; Wetzel, M.; Herndon, D.; Finnerty, C.C. Mast cell tryptase induction of postburn fibrosis via protease-activated receptor-2. Wound Repair Regen. 2017, 25, A7–A8. [Google Scholar]
- Gallant-Behm, C.L.; Hildebrand, K.A.; Hart, D.A. The mast cell stabilizer ketotifen prevents development of excessive skin wound contraction and fibrosis in red Duroc pigs. Wound Repair Regen. 2008, 16, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.W.; Prasai, A.; El Ayadi, A.; Sommerhalder, C.; Popp, D.; Ross, E.; Blears, E.; Foncerrada-Ortega, G.; Tapking, C.; Herndon, D.N.; et al. Topical cromolyn sodium reduces post-burn hypertrophic scars in female red Duroc pigs. Wound Repair Regen. 2019, 27, A1–A40. [Google Scholar]
- Monument, M.J.; Hart, D.A.; Befus, A.D.; Salo, P.T.; Zhang, M.; Hildebrand, K.A. The mast cell stabilizer ketotifen fumarate lessens contracture severity and myofibroblast hyperplasia: A study of a rabbit model of posttraumatic joint contractures. J. Bone Jt. Surg. Am. 2010, 92, 1468–1477. [Google Scholar] [CrossRef]
- Brubaker, A.L.; Schneider, D.F.; Kovacs, E.J. Neutrophils and natural killer T cells as negative regulators of wound healing. Expert Rev. Dermatol. 2011, 6, 5–8. [Google Scholar] [CrossRef]
- Schneider, D.F.; Palmer, J.L.; Tulley, J.M.; Speicher, J.T.; Kovacs, E.J.; Gamelli, R.L.; Faunce, D.E. A novel role for NKT cells in cutaneous wound repair. J. Surg. Res. 2011, 168, 325–333. [Google Scholar] [CrossRef]
- Tanno, H.; Kawakami, K.; Ritsu, M.; Kanno, E.; Suzuki, A.; Kamimatsuno, R.; Takagi, N.; Miyasaka, T.; Ishii, K.; Imai, Y.; et al. Contribution of invariant natural killer T cells to skin wound healing. Am. J. Pathol. 2015, 185, 3248–3257. [Google Scholar] [CrossRef]
- Tanno, H.; Kawakami, K.; Kanno, E.; Suzuki, A.; Takagi, N.; Yamamoto, H.; Ishii, K.; Imai, Y.; Maruyama, R.; Tachi, M. Invariant NKT cells promote skin wound healing by preventing a prolonged neutrophilic inflammatory response. Wound Repair Regen. 2017, 25, 805–815. [Google Scholar] [CrossRef]
- Katsuta, M.; Takigawa, Y.; Kimishima, M.; Inaoka, M.; Takahashi, R.; Shiohara, T. NK cells and gamma delta+ T cells are phenotypically and functionally defective due to preferential apoptosis in patients with atopic dermatitis. J. Immunol. 2006, 176, 7736–7744. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.M.; Thatcher, T.H.; Colas, R.A.; Serhan, C.N.; Phipps, R.P.; Sime, P.J. Resolvin D1 reduces emphysema and chronic inflammation. Am. J. Pathol. 2015, 185, 3189–3201. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, L.; Xia, H.; Chen, L.; Cui, S.; Wang, Y.; Zhou, T.; Xiong, W.; Song, L.; Li, S.; et al. Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1013–L1024. [Google Scholar] [CrossRef]
- Orr, S.K.; Butler, K.L.; Hayden, D.; Tompkins, R.G.; Serhan, C.N.; Irimia, D. Gene expression of proresolving lipid mediator pathways is associated with clinical outcomes in trauma patients. Crit. Care Med. 2015, 43, 2642–2650. [Google Scholar] [CrossRef]
- Inoue, Y.; Yu, Y.M.; Kurihara, T.; Vasilyev, A.; Ibrahim, A.; Oklu, R.; Zhao, G.; Nair, A.V.; Brown, D.; Fischman, A.J.; et al. Kidney and liver injuries after major burns in rats are prevented by Resolvin D2. Crit. Care Med. 2016, 44, e241–e252. [Google Scholar] [CrossRef]
- Bohr, S.; Patel, S.J.; Sarin, D.; Irimia, D.; Yarmush, M.L.; Berthiaume, F. Resolvin D2 prevents secondary thrombosis and necrosis in a mouse burn wound model. Wound Repair Regen. 2013, 21, 35–43. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in chronic wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Rybinski, B.; Franco-Barraza, J.; Cukierman, E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol. Genom. 2014, 46, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef]
- Grey, J.E.; Enoch, S.; Harding, K.G. Wound assessment. BMJ 2006, 332, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Bragulla, H.H.; Homberger, D.G. Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J. Anat. 2009, 214, 516–559. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in wound healing: A aomprehensive review. Adv. Wound Care (New Rochelle) 2014, 3, 445–464. [Google Scholar] [CrossRef]
- Gallico, G.G., III; O’Connor, N.E.; Compton, C.C.; Kehinde, O.; Green, H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N. Engl. J. Med. 1984, 311, 448–451. [Google Scholar] [CrossRef]
- Scuderi, N.; Anniboletti, T.; Carlesimo, B.; Onesti, M.G. Clinical application of autologous three-cellular cultured skin substitutes based on esterified hyaluronic acid scaffold: Our experience. In Vivo 2009, 23, 991–1003. [Google Scholar]
- Sood, R.; Roggy, D.E.; Zieger, M.J.; Nazim, M.; Hartman, B.C.; Gibbs, J.T. A comparative study of spray keratinocytes and autologous meshed split-thickness skin graft in the treatment of acute burn injuries. Wounds 2015, 27, 31–40. [Google Scholar]
- Dixit, S.; Baganizi, D.R.; Sahu, R.; Dosunmu, E.; Chaudhari, A.; Vig, K.; Pillai, S.R.; Singh, S.R.; Dennis, V.A. Immunological challenges associated with artificial skin grafts: Available solutions and stem cells in future design of synthetic skin. J. Biol. Eng. 2017, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.Y.; Lim, Y.; Koh, D.; Huh, J.S.; Hyun, C.; Kim, Y.M.; Cho, M. TMF and glycitin act synergistically on keratinocytes and fibroblasts to promote wound healing and anti-scarring activity. Exp. Mol. Med. 2017, 49, e302. [Google Scholar] [CrossRef] [PubMed]
- Mi, B.; Liu, J.; Liu, G.; Zhou, W.; Liu, Y.; Hu, L.; Xiong, L.; Ye, S.; Wu, Y. Icariin promotes wound healing by enhancing the migration and proliferation of keratinocytes via the AKT and ERK signaling pathway. Int. J. Mol. Med. 2018, 42, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Siddiqui, I.A.; Adhami, V.M.; Esnault, S.; Bharali, D.J.; Babatunde, A.S.; Adame, S.; Massey, R.J.; Wood, G.S.; Longley, B.J.; et al. Chitosan-Based nanoformulated (-)-epigallocatechin-3-gallate (EGCG) modulates human keratinocyte-induced responses and alleviates imiquimod-induced murine psoriasiform dermatitis. Int. J. Nanomed. 2018, 13, 4189–4206. [Google Scholar] [CrossRef]
- Kurinna, S.; Muzumdar, S.; Kohler, U.A.; Kockmann, T.; Auf dem Keller, U.; Schafer, M.; Werner, S. Autocrine and paracrine regulation of keratinocyte proliferation through a novel Nrf2-IL-36gamma pathway. J. Immunol. 2016, 196, 4663–4670. [Google Scholar] [CrossRef]
- Zhao, F.; Lang, H.; Wang, Z.; Zhang, T.; Zhang, D.; Wang, R.; Lin, X.; Liu, X.; Shi, P.; Pang, X. Human novel MicroRNA Seq-915_x4024 in Keratinocytes contributes to skin regeneration by suppressing scar formation. Mol. Ther. Nucleic Acids 2019, 14, 410–423. [Google Scholar] [CrossRef]
- Li, D.; Wang, A.; Liu, X.; Meisgen, F.; Grunler, J.; Botusan, I.R.; Narayanan, S.; Erikci, E.; Li, X.; Blomqvist, L.; et al. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J. Clin. Invest. 2015, 125, 3008–3026. [Google Scholar] [CrossRef]
- Feng, C.; Bai, M.; Yu, N.Z.; Wang, X.J.; Liu, Z. MicroRNA-181b negatively regulates the proliferation of human epidermal keratinocytes in psoriasis through targeting TLR4. J. Cell. Mol. Med. 2017, 21, 278–285. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Austyn, J.M.; Gordon, S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 1981, 11, 805–815. [Google Scholar] [CrossRef]
- Merad, M.; Manz, M.G.; Karsunky, H.; Wagers, A.; Peters, W.; Charo, I.; Weissman, I.L.; Cyster, J.G.; Engleman, E.G. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 2002, 3, 1135–1141. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Sen, C.K.; Singh, K.; Das, A.; Ghatak, S.; Rhea, B.; Blackstone, B.; Powell, H.M.; Khanna, S.; Roy, S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018, 9, 936. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef]
- Stawski, L.; Haines, P.; Fine, A.; Rudnicka, L.; Trojanowska, M. MMP-12 deficiency attenuates angiotensin II-induced vascular injury, M2 macrophage accumulation, and skin and heart fibrosis. PLoS ONE 2014, 9, e109763. [Google Scholar] [CrossRef]
- Rohani, M.G.; McMahan, R.S.; Razumova, M.V.; Hertz, A.L.; Cieslewicz, M.; Pun, S.H.; Regnier, M.; Wang, Y.; Birkland, T.P.; Parks, W.C. MMP-10 regulates collagenolytic activity of alternatively activated resident macrophages. J. Invest. Dermatol. 2015, 135, 2377–2384. [Google Scholar] [CrossRef]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maass, T.; Wagener, R.; Krieg, T.; Sutherland, T.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 receptor alpha signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity 2015, 43, 803–816. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Shibata, S.; Noda, S.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; et al. A possible contribution of visfatin to the resolution of skin sclerosis in patients with diffuse cutaneous systemic sclerosis via a direct anti-fibrotic effect on dermal fibroblasts and Th1 polarization of the immune response. Rheumatology 2013, 52, 1239–1244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taniguchi, T.; Miyagawa, T.; Toyama, S.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; et al. CXCL13 produced by macrophages due to Fli1 deficiency may contribute to the development of tissue fibrosis, vasculopathy and immune activation in systemic sclerosis. Exp. Dermatol. 2018, 27, 1030–1037. [Google Scholar] [CrossRef]
- Siebuhr, A.S.; Juhl, P.; Bay-Jensen, A.C.; Karsdal, M.A.; Franchimont, N.; Chavez, J.C. Citrullinated vimentin and biglycan protein fingerprints as candidate serological biomarkers for disease activity in systemic sclerosis: A pilot study. Biomarkers 2019, 24, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lammermann, T.; Germain, R.N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 2019, 177, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Prasai, A.; El Ayadi, A.; Jay, J.; Herndon, D.N.; Finnerty, C.C. Changes in Macrophage Phenotype in Hypertrophic Scarring Population. Wound Healing Society. Available online: https://woundheal.org/Meeting/Program/2018/P.LB11.cgi (accessed on 6 November 2019).
- van der Veer, W.M.; Niessen, F.B.; Ferreira, J.A.; Zwiers, P.J.; de Jong, E.H.; Middelkoop, E.; Molema, G. Time course of the angiogenic response during normotrophic and hypertrophic scar formation in humans. Wound Repair Regen. 2011, 19, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-Inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, J.; Xie, Z.; Wang, J.; Ho, C.K.; Zhang, Y.; Li, Q. Mechanical strain promotes skin fibrosis through LRG-1 induction mediated by ELK1 and ERK signalling. Commun. Biol. 2019, 2, 359. [Google Scholar] [CrossRef]
- Patel, J.; Baz, B.; Wong, H.Y.; Lee, J.S.; Khosrotehrani, K. Accelerated endothelial to mesenchymal transition increased fibrosis via deleting notch signaling in wound vasculature. J. Invest. Dermatol. 2018, 138, 1166–1175. [Google Scholar] [CrossRef]
- Xu, M.; Lv, J.; Wang, P.; Liao, Y.; Li, Y.; Zhao, W.; Zen, J.; Dong, Z.; Guo, Z.; Bo, X.; et al. Vascular endothelial Cdc42 deficiency delays skin wound-healing processes by increasing IL-1beta and TNF-alpha expression. Am. J. Transl. Res. 2019, 11, 257–268. [Google Scholar]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular mechanotransduction: From tension to function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Invest. 2018, 128, 74–84. [Google Scholar] [CrossRef]
- Kessler, D.; Dethlefsen, S.; Haase, I.; Plomann, M.; Hirche, F.; Krieg, T.; Eckes, B. Fibroblasts in mechanically stressed collagen lattices assume a “synthetic” phenotype. J. Biol. Chem. 2001, 276, 36575–36585. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Invest. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Oliveira, G.V.; Hawkins, H.K.; Chinkes, D.; Burke, A.; Tavares, A.L.; Ramos-e-Silva, M.; Albrecht, T.B.; Kitten, G.T.; Herndon, D.N. Hypertrophic versus non hypertrophic scars compared by immunohistochemistry and laser confocal microscopy: Type I and III collagens. Int. Wound J. 2009, 6, 445–452. [Google Scholar] [CrossRef]
- Tejiram, S.; Zhang, J.; Travis, T.E.; Carney, B.C.; Alkhalil, A.; Moffatt, L.T.; Johnson, L.S.; Shupp, J.W. Compression therapy affects collagen type balance in hypertrophic scar. J. Surg. Res. 2016, 201, 299–305. [Google Scholar] [CrossRef]
- Velasquez, L.S.; Sutherland, L.B.; Liu, Z.; Grinnell, F.; Kamm, K.E.; Schneider, J.W.; Olson, E.N.; Small, E.M. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl. Acad. Sci. USA 2013, 110, 16850–16855. [Google Scholar] [CrossRef]
- Atamas, S.P.; Luzina, I.G.; Dai, H.; Wilt, S.G.; White, B. Synergy between CD40 ligation and IL-4 on fibroblast proliferation involves IL-4 receptor signaling. J. Immunol. 2002, 168, 1139–1145. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-Fibroblast interactions in wound healing. J. Invest. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef]
- Jiang, D.; Correa-Gallegos, D.; Christ, S.; Stefanska, A.; Liu, J.; Ramesh, P.; Rajendran, V.; De Santis, M.M.; Wagner, D.E.; Rinkevich, Y. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat. Cell Biol. 2018, 20, 422–431. [Google Scholar] [CrossRef]
- Wohlfahrt, T.; Rauber, S.; Uebe, S.; Luber, M.; Soare, A.; Ekici, A.; Weber, S.; Matei, A.E.; Chen, C.W.; Maier, C.; et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 2019, 566, 344–349. [Google Scholar] [CrossRef]
- Correa-Gallegos, D.; Jiang, D.; Christ, S.; Ramesh, P.; Ye, H.; Wannemacher, J.; Kalgudde Gopal, S.; Yu, Q.; Aichler, M.; Walch, A.; et al. Patch repair of deep wounds by mobilized fascia. Nature 2019. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.J.; Liu, Y.; Zhang, X.; Li, Y.Y.; Xu, W.S. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns 2007, 33, 634–641. [Google Scholar] [CrossRef]
- Koyama, H.; Raines, E.W.; Bornfeldt, K.E.; Roberts, J.M.; Ross, R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell 1996, 87, 1069–1078. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Schilling, J.A. Wound healing. Surg. Clin. N. Am. 1976, 56, 859–874. [Google Scholar] [CrossRef]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar]
- Larson, S.A.; Dolivo, D.M.; Dominko, T. Artesunate inhibits myofibroblast formation via induction of apoptosis and antagonism of pro-fibrotic gene expression in human dermal fibroblasts. Cell Biol. Int. 2019, 43, 1317–1322. [Google Scholar] [CrossRef]
- Gupta, S.; Fink, M.K.; Ghosh, A.; Tripathi, R.; Sinha, P.R.; Sharma, A.; Hesemann, N.P.; Chaurasia, S.S.; Giuliano, E.A.; Mohan, R.R. Novel combination BMP7 and HGF gene therapy instigates selective myofibroblast apoptosis and reduces corneal haze In Vivo. Invest. Ophthalmol. Vis. Sci. 2018, 59, 1045–1057. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, D.; Song, H.; Shu, F.; Wei, P.; Yang, X.; Zhong, C.; Wang, X.; Muller, W.E.; Zheng, Y.; et al. Cuprous oxide nanoparticles reduces hypertrophic scarring by inducing fibroblast apoptosis. Int. J. Nanomed. 2019, 14, 5989–6000. [Google Scholar] [CrossRef]
- Wells, A.R.; Leung, K.P. Pirfenidone attenuates the profibrotic contractile phenotype of differentiated human dermal myofibroblasts. Biochem. Biophys. Res. Commun. 2019, in press. [Google Scholar] [CrossRef]
- Adams, T.N.; Eiswirth, C.; Newton, C.A.; Battaile, J.T. Pirfenidone for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 374–376. [Google Scholar] [CrossRef]
- Droitcourt, C.; Adamski, H.; Polat, A.; Polard, E.; Kerjouan, M.; Arnouat, B.; Le Garrec, M.; Oger, E.; Dupuy, A.; Jouneau, S. Pirfenidone photosensitization in patients with idiopathic pulmonary fibrosis: A case series. Br. J. Dermatol. 2018, 178, e222–e223. [Google Scholar] [CrossRef]
- Armendariz-Borunda, J.; Lyra-Gonzalez, I.; Medina-Preciado, D.; Gonzalez-Garcia, I.; Martinez-Fong, D.; Miranda, R.A.; Magana-Castro, R.; Pena-Santoyo, P.; Garcia-Rocha, S.; Bautista, C.A.; et al. A controlled clinical trial with pirfenidone in the treatment of pathological skin scarring caused by burns in pediatric patients. Ann. Plast. Surg. 2012, 68, 22–28. [Google Scholar] [CrossRef]
- Freshwater, M.F. Re: A controlled clinical trial with pirfenidone in the treatment of pathological skin scarring caused by burns in pediatric patients. Ann. Plast. Surg. 2012, 69, 111. [Google Scholar] [CrossRef]
- Cantu-Cantu, M.Z.; Lyra-Gonzalez, I.; Armendariz-Borunda, J. Coadjuvant treatment with surgery and pirfenidone in severe facial trauma due to dog bite. J. Craniofac. Surg. 2013, 24, 675–678. [Google Scholar] [CrossRef]
- Janka-Zires, M.; Almeda-Valdes, P.; Uribe-Wiechers, A.C.; Juarez-Comboni, S.C.; Lopez-Gutierrez, J.; Escobar-Jimenez, J.J.; Gomez-Perez, F.J. Topical administration of pirfenidone increases healing of chronic diabetic foot ulcers: A randomized crossover study. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef]
- Hoerst, K.; van den Broek, L.; Sachse, C.; Klein, O.; von Fritschen, U.; Gibbs, S.; Hedtrich, S. Regenerative potential of adipocytes in hypertrophic scars is mediated by myofibroblast reprogramming. J. Mol. Med. 2019, 97, 761–775. [Google Scholar] [CrossRef]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef]
- Nessler, M.B.; Puchala, J.; Chrapusta, A.; Nessler, K.; Drukala, J. Levels of plasma matrix metalloproteinases (MMP-2 and MMP-9) in response to INTEGRA(R) dermal regeneration template implantation. Med. Sci. Monit. 2014, 20, 91–96. [Google Scholar]
- DeBruler, D.M.; Blackstone, B.N.; McFarland, K.L.; Baumann, M.E.; Supp, D.M.; Bailey, J.K.; Powell, H.M. Effect of skin graft thickness on scar development in a porcine burn model. Burns 2018, 44, 917–930. [Google Scholar] [CrossRef]
- Travis, T.E.; Ghassemi, P.; Prindeze, N.J.; Moffatt, L.T.; Carney, B.C.; Alkhalil, A.; Ramella-Roman, J.C.; Shupp, J.W. Matrix metalloproteinases are differentially regulated and responsive to compression therapy in a red Duroc model of hypertrophic scar. Eplasty 2018, 18, e1. [Google Scholar]
- Nabai, L.; Kilani, R.T.; Aminuddin, F.; Li, Y.; Ghahary, A. Methotrexate modulates the expression of MMP-1 and type 1 collagen in dermal fibroblast. Mol. Cell. Biochem. 2015, 409, 213–224. [Google Scholar] [CrossRef]
- Li, Y.; Kilani, R.T.; Rahmani-Neishaboor, E.; Jalili, R.B.; Ghahary, A. Kynurenine increases matrix metalloproteinase-1 and-3 expression in cultured dermal fibroblasts and improves scarring In Vivo. J. Invest. Dermatol. 2014, 134, 643–650. [Google Scholar] [CrossRef]
- Ulrich, D.; Ulrich, F.; Unglaub, F.; Piatkowski, A.; Pallua, N. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with different types of scars and keloids. J. Plast. Reconstr. Aesthet. Surg. 2010, 63, 1015–1021. [Google Scholar] [CrossRef]
- Liu, S.S.; Lv, X.X.; Liu, C.; Qi, J.; Li, Y.X.; Wei, X.P.; Li, K.; Hua, F.; Cui, B.; Zhang, X.W.; et al. Targeting degradation of the transcription factor C/EBPbeta reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme A20 in macrophages. Immunity 2019, 51, 522–534. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.; Shang, D.; Kass, D.J.; Zhao, Y. Ubiquitination and deubiquitination emerge as players in idiopathic pulmonary fibrosis pathogenesis and treatment. JCI Insight 2018, 3, e120362. [Google Scholar] [CrossRef] [PubMed]
- El Ayadi, A.; Prasai, A.; Wang, Y.; Herndon, D.N.; Finnerty, C.C. Beta-Adrenergic receptor trafficking, degradation, and cell surface expression are altered in dermal fibroblasts from hypertrophic scars. J. Invest. Dermatol. 2018, 138, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ham, S.; Lee, Y.; Suh, G.Y.; Lee, Y.S. TTC3 contributes to TGF-beta1-induced epithelial-mesenchymal transition and myofibroblast differentiation, potentially through SMURF2 ubiquitylation and degradation. Cell Death Dis. 2019, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Finnerty, C.C.; He, J.; Herndon, D.N. Smad ubiquitination regulatory factor 2 expression is enhanced in hypertrophic scar fibroblasts from burned children. Burns 2012, 38, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Duann, P.; Lin, P.H.; Zhao, L.; Fan, Z.; Tan, T.; Zhou, X.; Sun, M.; Fu, M.; Orange, M.; et al. Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J. Biol. Chem. 2015, 290, 24592–24603. [Google Scholar] [CrossRef] [PubMed]
- Leake, D.; Doerr, T.D.; Scott, G. Expression of urokinase-type plasminogen activator and its receptor in keloids. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 1334–1338. [Google Scholar] [CrossRef]
- Harbeck, N.; Kates, R.E.; Gauger, K.; Willems, A.; Kiechle, M.; Magdolen, V.; Schmitt, M. Urokinase-Type plasminogen activator (uPA) and its inhibitor PAI-I: Novel tumor-derived factors with a high prognostic and predictive impact in breast cancer. Thromb. Haemost. 2004, 91, 450–456. [Google Scholar]
- Alessi, M.C.; Poggi, M.; Juhan-Vague, I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr. Opin. Lipidol. 2007, 18, 240–245. [Google Scholar] [CrossRef]
- Zahran, A.M.; Sayed, S.K.; Abd El Hafeez, H.A.; Khalifa, W.A.; Mohamed, N.A.; Hetta, H.F. Circulating microparticle subpopulation in metabolic syndrome: Relation to oxidative stress and coagulation markers. Diabetes Metab. Syndr. Obes. 2019, 12, 485–493. [Google Scholar] [CrossRef]
- Basurto, L.; Diaz, A.; Rodriguez, A.; Robledo, A.; Vega, S.; Garcia-Vega, J.; Martinez-Murillo, C. Circulating levels of plasminogen activator inhibitor-1 are associated with metabolic syndrome rather than with menopause. Gynecol. Endocrinol. 2019, 26, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Pincha, N.; Hajam, E.Y.; Badarinath, K.; Batta, S.P.R.; Masudi, T.; Dey, R.; Andreasen, P.; Kawakami, T.; Samuel, R.; George, R.; et al. PAI1 mediates fibroblast-mast cell interactions in skin fibrosis. J. Clin. Invest. 2018, 128, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Herter, E.K.; Xu Landen, N. Non-Coding RNAs: New players in skin wound healing. Adv. Wound Care 2017, 6, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, H.Y.; Bai, W.D.; Su, L.L.; Liu, J.Q.; Cai, W.X.; Zhao, B.; Gao, J.X.; Han, S.C.; Li, J.; et al. MiR-10a and miR-181c regulate collagen type I generation in hypertrophic scars by targeting PAI-1 and uPA. FEBS Lett. 2015, 589, 380–389. [Google Scholar] [CrossRef]
- Pang, Q.; Wang, Y.; Xu, M.; Xu, J.; Xu, S.; Shen, Y.; Lei, R. MicroRNA-152-5p inhibits proliferation and migration and promotes apoptosis by regulating expression of Smad3 in human keloid fibroblasts. BMB Rep. 2019, 52, 202–207. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, J.; Zhao, J.; Zhang, P.; Huang, X. MicroRNA-23b inhibits the proliferation and migration of heat-denatured fibroblasts by targeting Smad3. PLoS ONE 2015, 10, e0131867. [Google Scholar] [CrossRef]
- Kwan, P.; Ding, J.; Tredget, E.E. MicroRNA 181b regulates decorin production by dermal fibroblasts and may be a potential therapy for hypertrophic scar. PLoS ONE 2015, 10, e0123054. [Google Scholar] [CrossRef]
- Miscianinov, V.; Martello, A.; Rose, L.; Parish, E.; Cathcart, B.; Mitic, T.; Gray, G.A.; Meloni, M.; Al Haj Zen, A.; Caporali, A. MicroRNA-148b targets the TGF-beta pathway to regulate angiogenesis and endothelial-to-mesenchymal transition during skin wound healing. Mol. Ther. 2018, 26, 1996–2007. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Li, Q.; Yan, H.; Zhou, B.; Gao, Y. Non-Coding RNAs: The new insight on hypertrophic scar. J. Cell. Biochem. 2017, 118, 1965–1968. [Google Scholar] [CrossRef]
- Tu, L.; Huang, Q.; Fu, S.; Liu, D. Aberrantly expressed long noncoding RNAs in hypertrophic scar fibroblasts In Vitro: A microarray study. Int. J. Mol. Med. 2018, 41, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Liu, D.; Huang, H. Highthroughput sequencing reveals differentially expressed lncRNAs and circRNAs, and their associated functional network, in human hypertrophic scars. Mol. Med. Rep. 2018, 18, 5669–5682. [Google Scholar] [PubMed]
- Chen, L.; Li, J.; Li, Q.; Li, X.; Gao, Y.; Hua, X.; Zhou, B. Overexpression of LncRNA AC067945.2 down-regulates collagen expression in skin fibroblasts and possibly correlates with the VEGF and Wnt signalling pathways. Cell. Physiol. Biochem. 2018, 45, 761–771. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Cao, C.; Yan, H.; Zhou, B.; Gao, Y.; Li, Q. The long non-coding RNA LncRNA8975-1 is upregulated in hypertrophic scar fibroblasts and controls collagen expression. Cell. Physiol. Biochem. 2016, 40, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, M.; Bachem, M.; Anders, D.; Trischler, G.; Faehling, M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 2005, 183, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Zeisberg, M. The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J. Pathol. 2013, 229, 264–273. [Google Scholar] [CrossRef]
- Russell, S.B.; Russell, J.D.; Trupin, K.M.; Gayden, A.E.; Opalenik, S.R.; Nanney, L.B.; Broquist, A.H.; Raju, L.; Williams, S.M. Epigenetically altered wound healing in keloid fibroblasts. J. Invest. Dermatol. 2010, 130, 2489–2496. [Google Scholar] [CrossRef]
- Rombouts, K.; Niki, T.; Greenwel, P.; Vandermonde, A.; Wielant, A.; Hellemans, K.; De Bleser, P.; Yoshida, M.; Schuppan, D.; Rojkind, M.; et al. A histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp. Cell Res. 2002, 278, 184–197. [Google Scholar] [CrossRef]
- Fitzgerald O’Connor, E.J.; Badshah, I., I; Addae, L.Y.; Kundasamy, P.; Thanabalasingam, S.; Abioye, D.; Soldin, M.; Shaw, T.J. Histone deacetylase 2 is upregulated in normal and keloid scars. J. Invest. Dermatol. 2012, 132, 1293–1296. [Google Scholar] [CrossRef]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef]
- Diao, J.S.; Xia, W.S.; Yi, C.G.; Yang, Y.; Zhang, X.; Xia, W.; Shu, M.G.; Wang, Y.M.; Gui, L.; Guo, S.Z. Histone deacetylase inhibitor reduces hypertrophic scarring in a rabbit ear model. Plast. Reconstr. Surg. 2013, 132, 61e–69e. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Reddy, M.A.; Yuan, H.; Lanting, L.; Kato, M.; Natarajan, R. Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 2010, 21, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Martin, P. Epigenetic reprogramming during wound healing: Loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 2009, 10, 881–886. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, P.S.; Kahaleh, B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279. [Google Scholar] [CrossRef]
- Asano, Y.; Bujor, A.M.; Trojanowska, M. The impact of Fli1 deficiency on the pathogenesis of systemic sclerosis. J. Dermatol. Sci. 2010, 59, 153–162. [Google Scholar] [CrossRef]
- Moore, A.L.; Marshall, C.D.; Litzenburger, U.; Barnes, L.; Ransom, R.C.; Hu, M.; Leavitt, T.; Chang, H.Y.; Longaker, M.T. Epigenetic analysis of scar forming fibroblasts reveals key differences in genes associated with fibrosis. J. Am. Coll. Surg. 2017, 225, S200–S201. [Google Scholar] [CrossRef][Green Version]
- Bellavia, G.; Fasanaro, P.; Melchionna, R.; Capogrossi, M.C.; Napolitano, M. Transcriptional control of skin reepithelialization. J. Dermatol. Sci. 2014, 73, 3–9. [Google Scholar] [CrossRef]
- Tourkina, E.; Richard, M.; Gooz, P.; Bonner, M.; Pannu, J.; Harley, R.; Bernatchez, P.N.; Sessa, W.C.; Silver, R.M.; Hoffman, S. Antifibrotic properties of caveolin-1 scaffolding domain In Vitro and In Vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L843–L861. [Google Scholar] [CrossRef]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 Is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Ravi, S.; Xie, N.; Persons, B.P.; Rangarajan, S.; Zmijewski, J.W.; Mitra, K.; Liu, G.; Darley-Usmar, V.M.; et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem. 2015, 290, 25427–25438. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. The role of estrogen in cutaneous ageing and repair. Maturitas 2017, 103, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Urai, T.; Asano, K.; Nakajima, Y.; Nakatani, T. Evaluation of effects of topical estradiol benzoate application on cutaneous wound healing in ovariectomized female mice. PLoS ONE 2016, 11, e0163560. [Google Scholar] [CrossRef] [PubMed]
- Affinito, P.; Palomba, S.; Sorrentino, C.; Di Carlo, C.; Bifulco, G.; Arienzo, M.P.; Nappi, C. Effects of postmenopausal hypoestrogenism on skin collagen. Maturitas 1999, 33, 239–247. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Gubbels Bupp, M.R. Sex, the aging immune system, and chronic disease. Cell. Immunol. 2015, 294, 102–110. [Google Scholar] [CrossRef]
- Murphy, A.J.; Guyre, P.M.; Wira, C.R.; Pioli, P.A. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS ONE 2009, 4, e5539. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Greenwell-Wild, T.; Horan, M.A.; Wahl, S.M.; Ferguson, M.W. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am. J. Pathol. 1999, 155, 1137–1146. [Google Scholar] [CrossRef]
- Horng, H.C.; Chang, W.H.; Yeh, C.C.; Huang, B.S.; Chang, C.P.; Chen, Y.J.; Tsui, K.H.; Wang, P.H. Estrogen effects on wound healing. Int. J. Mol. Sci. 2017, 18, 2325. [Google Scholar] [CrossRef]
- Dou, C.; Ding, N.; Zhao, C.; Hou, T.; Kang, F.; Cao, Z.; Liu, C.; Bai, Y.; Dai, Q.; Ma, Q.; et al. Estrogen deficiency-mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. J. Bone Miner. Res. 2018, 33, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A.; et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J. Biol. Chem. 2010, 285, 14747–14755. [Google Scholar] [CrossRef]
- Yin, K.; Smith, A.G. Nuclear receptor function in skin health and disease: Therapeutic opportunities in the orphan and adopted receptor classes. Cell. Mol. Life Sci. 2016, 73, 3789–3800. [Google Scholar] [CrossRef] [PubMed]
- Icre, G.; Wahli, W.; Michalik, L. Functions of the peroxisome proliferator-activated receptor (PPAR) alpha and beta in skin homeostasis, epithelial repair, and morphogenesis. J. Investig. Dermatol. Symp. Proc. 2006, 11, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Wahli, W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Invest. 2006, 116, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Bhattacharyya, S.; Varga, J. Peroxisome proliferator-activated receptor gamma: Innate protection from excessive fibrogenesis and potential therapeutic target in systemic sclerosis. Curr. Opin. Rheumatol. 2010, 22, 671–676. [Google Scholar] [CrossRef]
- Collino, M.; Patel, N.S.; Thiemermann, C. PPARs as new therapeutic targets for the treatment of cerebral ischemia/reperfusion injury. Ther. Adv. Cardiovasc. Dis. 2008, 2, 179–197. [Google Scholar] [CrossRef]
- Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Turner, N.A.; Porter, K.E. Peroxisome proliferator-activated receptor gamma-independent effects of thiazolidinediones on human cardiac myofibroblast function. Clin. Exp. Pharmacol. Physiol. 2009, 36, 478–486. [Google Scholar] [CrossRef]
- Lim, H.W.; Collins, S.A.B.; Resneck, J.S., Jr.; Bolognia, J.L.; Hodge, J.A.; Rohrer, T.A.; Van Beek, M.J.; Margolis, D.J.; Sober, A.J.; Weinstock, M.A.; et al. The burden of skin disease in the United States. J. Am. Acad. Dermatol. 2017, 76, 958–972. [Google Scholar] [CrossRef]
- Tsou, P.S.; Haak, A.J.; Khanna, D.; Neubig, R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: Focus on Rho GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014, 307, C2–C13. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Safety and Efficacy Study of EXC 001 to Improve the Appearance of Scars from Prior Breast Surgery. Available online: https://ClinicalTrials.gov/show/NCT01346969 (accessed on 20 November 2019).
- ClinicalTrials.gov. Fat Grafting in Skin-Grafted Deep Burn Scars. Available online: https://ClinicalTrials.gov/show/NCT03627650 (accessed on 20 November 2019).
- ClinicalTrials.gov. Therapy of Scars and Cutis Laxa With Autologous Adipose Derived Mesenchymal Stem Cells. Available online: https://ClinicalTrials.gov/show/NCT03887208 (accessed on 20 November 2019).
- ClinicalTrials.gov. Safety and Efficacy Study of PF-06473871 to Reduce Hypertrophic Scars from Recurring Post-Revision Surgery. Available online: https://ClinicalTrials.gov/show/NCT01730339 (accessed on 20 November 2019).
- ClinicalTrials.gov. Evaluation of Safety, PK, and Preliminary Effects on Scar Formation of Multiple Intradermal Administrations of RXI-109. Available online: https://ClinicalTrials.gov/show/NCT01780077 (accessed on 20 November 2019).
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-Beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, D.; Khaw, P.T.; King, A.J.; Donaldson, M.L.; Overton, B.M.; Migdal, C.; Cordeiro, M.F. Human antitransforming growth factor beta(2) monoclonal antibody—A new modulator of wound healing in trabeculectomy: A randomized placebo controlled clinical study. Ophthalmology 2002, 109, 427–431. [Google Scholar] [CrossRef]
- Denton, C.P.; Merkel, P.A.; Furst, D.E.; Khanna, D.; Emery, P.; Hsu, V.M.; Silliman, N.; Streisand, J.; Powell, J.; Akesson, A.; et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007, 56, 323–333. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Measurement and Comparison of the Occlusive and Hydrating Capacity of Three Silicone Gels and One Gel Cream. Available online: https://ClinicalTrials.gov/show/NCT01413789 (accessed on 20 November 2019).
- Finnerty, C.C.; Jeschke, M.G.; Branski, L.K.; Barret, J.P.; Dziewulski, P.; Herndon, D.N. Hypertrophic scarring: The greatest unmet challenge after burn injury. Lancet 2016, 388, 1427–1436. [Google Scholar] [CrossRef]
- Chae, J.K.; Kim, J.H.; Kim, E.J.; Park, K. Values of a patient and observer scar assessment scale to evaluate the facial skin graft scar. Ann. Dermatol. 2016, 28, 615–623. [Google Scholar] [CrossRef]
- Zhang, N.; Shi, K.; Hong, L.; Zhao, J.; Yu, J. Antera 3D camera: A novel method for evaluating the therapeutic efficacy of fractional CO2 laser for surgical incision scars. J. Cosmet. Dermatol. 2018, 17, 1041–1045. [Google Scholar] [CrossRef]
- Peake, M.; Pan, K.; Rotatori, R.M.; Powell, H.; Fowler, L.; James, L.; Dale, E. Incorporation of 3D stereophotogrammetry as a reliable method for assessing scar volume in standard clinical practice. Burns 2019, 45, 1614–1620. [Google Scholar] [CrossRef]
- Ngaage, M.; Agius, M. The psychology of scars: A mini-review. Psychiatr. Danub. 2018, 30, 633–638. [Google Scholar]
- Robert, R.; Meyer, W.; Bishop, S.; Rosenberg, L.; Murphy, L.; Blakeney, P. Disfiguring burn scars and adolescent self-esteem. Burns 1999, 25, 581–585. [Google Scholar] [CrossRef]
- Ai, J.W.; Liu, J.T.; Pei, S.D.; Liu, Y.; Li, D.S.; Lin, H.M.; Pei, B. The effectiveness of pressure therapy (15–25 mmHg) for hypertrophic burn scars: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 40185. [Google Scholar] [CrossRef]
- Clayton, N.A.; Haertsch, P.A.; Maitz, P.K.; Issler-Fisher, A.C. Ablative fractional resurfacing in acute care management of facial burns: A new approach to minimize the need for acute surgical reconstruction. J. Burn Care Res. 2019, 40, 368–372. [Google Scholar] [CrossRef]
- Issler-Fisher, A.C.; Fisher, O.M.; Haertsch, P.; Li, Z.; Maitz, P.K.M. Ablative fractional resurfacing with laser-facilitated steroid delivery for burn scar management: Does the depth of laser penetration matter? Lasers Surg. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shavit, R.; Dierickx, C. A new method for percutaneous drug delivery by thermo-mechanical fractional injury. Lasers Surg. Med. 2019. [Google Scholar] [CrossRef]
- He, X.; Zhang, J.; Bai, X.; Shi, J.; Hu, D. New progress of adipose-derived stem cells in the therapy of hypertrophic scars. Curr. Stem. Cell Res. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Doersch, K.M.; DelloStritto, D.J.; Newell-Rogers, M.K. The contribution of interleukin-2 to effective wound healing. Exp. Biol. Med. (Maywood) 2017, 242, 384–396. [Google Scholar] [CrossRef]
- Tabola, R.; Augoff, K.; Grabowski, K.; Cirocchi, R. Role of hyaluronidase in the treatment of hypertrophic scars. Dermatol. Surg. 2018, 44, 1155–1157. [Google Scholar] [CrossRef]
- Klinger, M.; Caviggioli, F.; Klinger, F.M.; Giannasi, S.; Bandi, V.; Banzatti, B.; Forcellini, D.; Maione, L.; Catania, B.; Vinci, V. Autologous fat graft in scar treatment. J. Craniofac. Surg. 2013, 24, 1610–1615. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).