The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone



{kind=link}
{kind=link}
Abstract
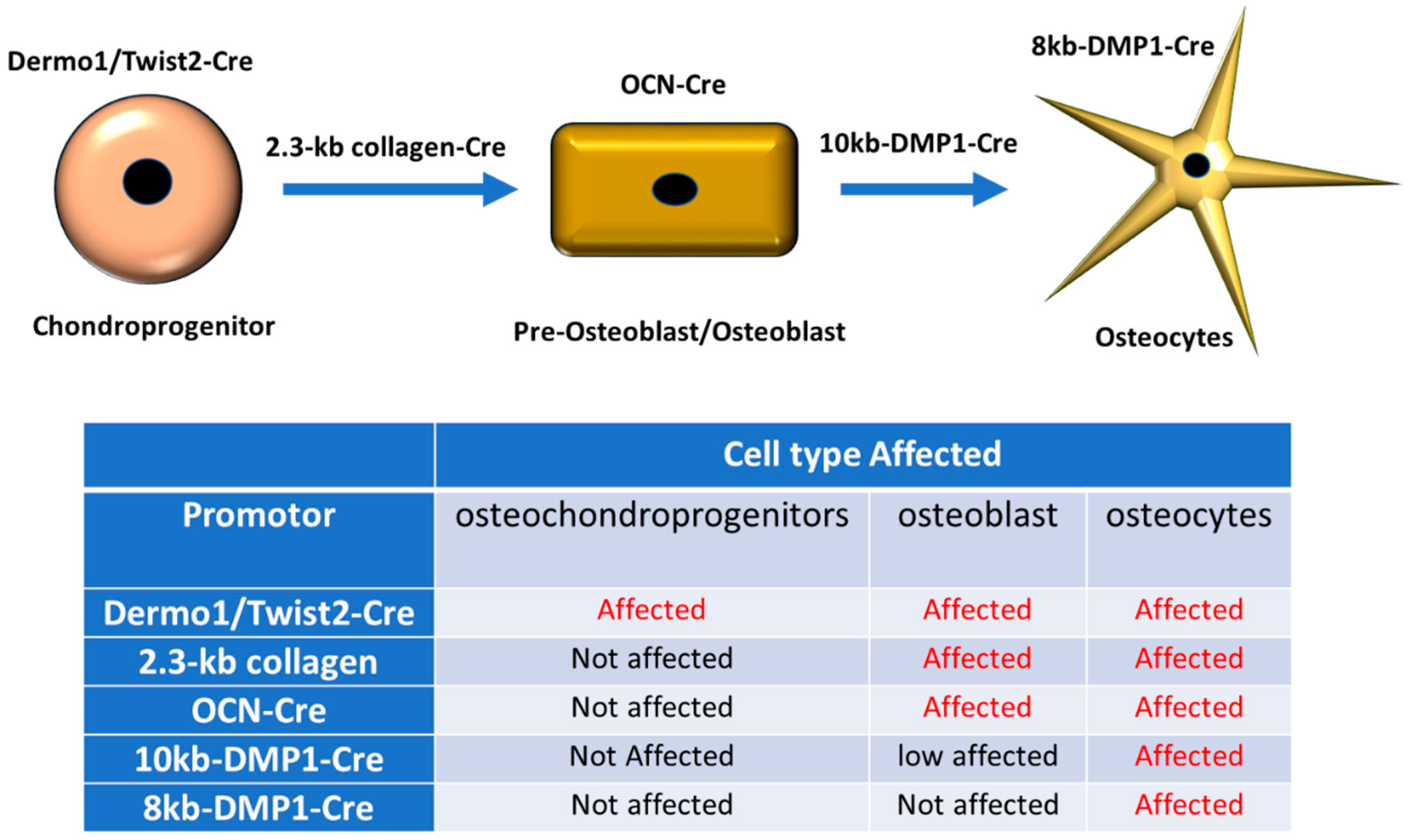
1. Connexin 43 Gap Junctions, Hemichannels and Bone Homeostasis
2. Mechanical Loading Signaling in the Bone and Involvement of Connexins and Pannexins
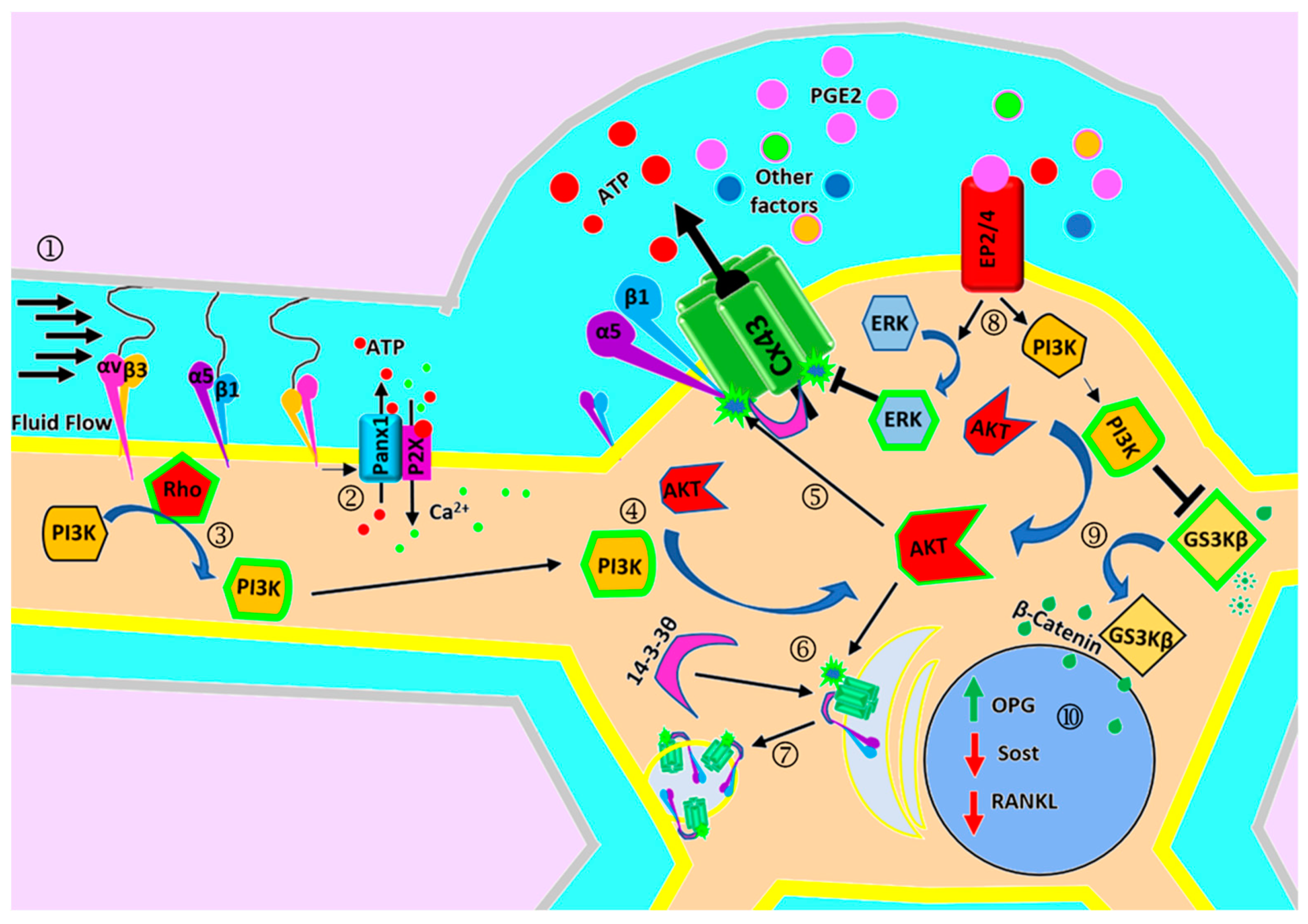
3. The Role of Cx43 and Cx43 Channels in Mechanotransduction
4. Regulation of Cx43 and Cx43 Channels by Mechanical Loading
5. Role of Cx43 in Response to Mechanical Unloading/Disuse
6. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Goodenough, D.A.; Paul, D.L. Beyond the gap: Functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 2003, 4, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Riquelme, M.A.; Stehberg, J.; Alcayaga, J. Connexin43 Hemichannels in Satellite Glial Cells, Can They Influence Sensory Neuron Activity? Front. Mol. Neurosci. 2017, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Buo, A.M.; Stains, J.P. Gap junctional regulation of signal transduction in bone cells. FEBS Lett. 2014, 588, 1315–1321. [Google Scholar] [CrossRef]
- Pacheco-Costa, R.; Hassan, I.; Reginato, R.D.; Davis, H.M.; Bruzzaniti, A.; Allen, M.R.; Plotkin, L.I. High bone mass in mice lacking Cx37 because of defective osteoclast differentiation. J. Biol. Chem. 2014, 289, 8508–8520. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Stains, J.P. Connexins and pannexins in the skeleton: Gap junctions, hemichannels and more. Cell. Mol. Life Sci. CMLS 2015, 72, 2853–2867. [Google Scholar] [CrossRef]
- Reaume, A.G.; De Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Lecanda, F.; Warlow, P.M.; Sheikh, S.; Furlan, F.; Steinberg, T.H.; Civitelli, R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 2000, 151, 931–943. [Google Scholar] [CrossRef]
- Watkins, M.; Grimston, S.K.; Norris, J.Y.; Guillotin, B.; Shaw, A.; Beniash, E.; Civitelli, R. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol. Biol. Cell 2011, 22, 1240–1251. [Google Scholar] [CrossRef]
- Chung, D.J.; Castro, C.H.; Watkins, M.; Stains, J.P.; Chung, M.Y.; Szejnfeld, V.L.; Willecke, K.; Theis, M.; Civitelli, R. Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin. J. Cell Sci. 2006, 119 Pt 20, 4187–4198. [Google Scholar] [CrossRef]
- Bivi, N.; Pacheco-Costa, R.; Brun, L.R.; Murphy, T.R.; Farlow, N.R.; Robling, A.G.; Bellido, T.; Plotkin, L.I. Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J. Orthop. Res. 2013, 31, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Paul, E.M.; Sathyendra, V.; Davison, A.; Sharkey, N.; Bronson, S.; Srinivasan, S.; Gross, T.S.; Donahue, H.J. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE 2011, 6, e23516. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.A.; Lewis, G.S.; Zhang, Y.; Paul, E.M.; Donahue, H.J. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J. Bone Miner. Res. 2012, 27, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Bivi, N.; Nelson, M.T.; Faillace, M.E.; Li, J.; Miller, L.M.; Plotkin, L.I. Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif. Tissue Int. 2012, 91, 215–224. [Google Scholar] [CrossRef]
- Lloyd, S.A.; Loiselle, A.E.; Zhang, Y.; Donahue, H.J. Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone 2013, 57, 76–83. [Google Scholar] [CrossRef]
- Moorer, M.C.; Hebert, C.; Tomlinson, R.E.; Iyer, S.R.; Chason, M.; Stains, J.P. Defective signaling, osteoblastogenesis and bone remodeling in a mouse model of connexin 43 C-terminal truncation. J. Cell Sci. 2017, 130, 531–540. [Google Scholar] [CrossRef]
- Davis, H.M.; Aref, M.W.; Aguilar-Perez, A.; Pacheco-Costa, R.; Allen, K.; Valdez, S.; Herrera, C.; Atkinson, E.G.; Mohammad, A.; Lopez, D.; et al. Cx43 overexpression in osteocytes prevents osteocyte apoptosis and preserves cortical bone quality in aging mice. JBMR Plus 2018, 2, 206–216. [Google Scholar] [CrossRef]
- Xu, H.; Gu, S.; Riquelme, M.A.; Burra, S.; Callaway, D.; Cheng, H.; Guda, T.; Schmitz, J.; Fajardo, R.J.; Werner, S.L.; et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J. Bone Miner. Res. 2015, 30, 436–448. [Google Scholar] [CrossRef]
- Laird, D.W. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014, 588, 1339–1348. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, I.P.; de Almeida, S.; Tiziani, V.; Do Amaral, C.M.; Gowrishankar, K.; Passos-Bueno, M.R.; Reichenberger, E.J. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS ONE 2013, 8, e73576. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Sasse, P.; Schrickel, J.W.; Watkins, M.; Kim, J.S.; Rackauskas, M.; Troatz, C.; Ghanem, A.; Tiemann, K.; Degen, J.; et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 2008, 17, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Zappitelli, T.; Chen, F.; Moreno, L.; Zirngibl, R.A.; Grynpas, M.; Henderson, J.E.; Aubin, J.E. The G60S connexin 43 mutation activates the osteoblast lineage and results in a resorption-stimulating bone matrix and abrogation of old-age-related bone loss. J. Bone Miner. Res. 2013, 28, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Flenniken, A.M.; Osborne, L.R.; Anderson, N.; Ciliberti, N.; Fleming, C.; Gittens, J.E.; Gong, X.Q.; Kelsey, L.B.; Lounsbury, C.; Moreno, L.; et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 2005, 132, 4375–4386. [Google Scholar] [CrossRef] [PubMed]
- Zappitelli, T.; Chen, F.; Aubin, J.E. Up-regulation of BMP2/4 signaling increases both osteoblast-specific marker expression and bone marrow adipogenesis in Gja1Jrt/+ stromal cell cultures. Mol. Biol. Cell 2015, 26, 832–842. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Sommershof, A.; Willecke, K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 2007, 219, 9–17. [Google Scholar] [CrossRef]
- Jacobs, C.R.; Temiyasathit, S.; Castillo, A.B. Osteocyte mechanobiology and pericellular mechanics. Annu. Rev. Biomed. Eng. 2010, 12, 369–400. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Cowin, S.C.; Zeng, Y. A model for the excitation of osteocytes by mechanical loading induced bone fluid shear stresses. J. Biomech. 1994, 27, 339–360. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Burra, S.; Nicolella, D.P.; Francis, W.L.; Freitas, C.J.; Mueschke, N.J.; Poole, K.; Jiang, J.X. Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc. Natl. Acad. Sci. USA 2010, 107, 13648–13653. [Google Scholar] [CrossRef]
- Miyauchi, A.; Gotoh, M.; Kamioka, H.; Notoya, K.; Sekiya, H.; Takagi, Y.; Yoshimoto, Y.; Ishikawa, H.; Chihara, K.; Takano-Yamamoto, T.; et al. AlphaVbeta3 integrin ligands enhance volume-sensitive calcium influx in mechanically stretched osteocytes. J. Bone Miner. Metab. 2006, 24, 498–504. [Google Scholar] [CrossRef]
- Thi, M.M.; Suadicani, S.O.; Schaffler, M.B.; Weinbaum, S.; Spray, D.C. Mechanosensory responses of osteocytes to physiological forces occur along processes and not cell body and require alphaVbeta3 integrin. Proc. Natl. Acad. Sci. USA 2013, 110, 21012–21017. [Google Scholar] [CrossRef] [PubMed]
- Yavropoulou, M.P.; Yovos, J.G. The molecular basis of bone mechanotransduction. J. Musculoskelet. Neuronal Interact. 2016, 16, 221–236. [Google Scholar] [PubMed]
- Wu, X.T.; Sun, L.W.; Yang, X.; Ding, D.; Han, D.; Fan, Y.B. The potential role of spectrin network in the mechanotransduction of MLO-Y4 osteocytes. Sci. Rep. 2017, 7, 40940. [Google Scholar] [CrossRef] [PubMed]
- Grimston, S.K.; Watkins, M.P.; Brodt, M.D.; Silva, M.J.; Civitelli, R. Enhanced periosteal and endocortical responses to axial tibial compression loading in conditional connexin43 deficient mice. PLoS ONE 2012, 7, e44222. [Google Scholar] [CrossRef]
- Joeng, K.S.; Lee, Y.C.; Lim, J.; Chen, Y.; Jiang, M.M.; Munivez, E.; Ambrose, C.; Lee, B.H. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J. Clin. Investig. 2017, 127, 2678–2688. [Google Scholar] [CrossRef]
- Sawakami, K.; Robling, A.G.; Ai, M.; Pitner, N.D.; Liu, D.; Warden, S.J.; Li, J.; Maye, P.; Rowe, D.W.; Duncan, R.L.; et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J. Biol. Chem. 2006, 281, 23698–23711. [Google Scholar] [CrossRef]
- Robinson, J.A.; Chatterjee-Kishore, M.; Yaworsky, P.J.; Cullen, D.M.; Zhao, W.; Li, C.; Kharode, Y.; Sauter, L.; Babij, P.; Brown, E.L.; et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J. Biol. Chem. 2006, 281, 31720–31728. [Google Scholar] [CrossRef]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef]
- Spagnol, G.; Trease, A.J.; Zheng, L.; Gutierrez, M.; Basu, I.; Sarmiento, C.; Moore, G.; Cervantes, M.; Sorgen, P.L. Connexin43 Carboxyl-Terminal Domain Directly Interacts with beta-Catenin. Int. J. Mol. Sci. 2018, 19, 1562. [Google Scholar] [CrossRef]
- Ai, Z.; Fischer, A.; Spray, D.C.; Brown, A.M.; Fishman, G.I. Wnt-1 regulation of connexin43 in cardiac myocytes. J. Clin. Investig. 2000, 105, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Spencer, G.J.; Utting, J.C.; Etheridge, S.L.; Arnett, T.R.; Genever, P.G. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J. Cell Sci. 2006, 119 Pt 7, 1283–1296. [Google Scholar] [CrossRef]
- Lloyd, S.A.; Loiselle, A.E.; Zhang, Y.; Donahue, H.J. Shifting paradigms on the role of connexin43 in the skeletal response to mechanical load. J. Bone Miner. Res. 2014, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, B.; Stern, A.R.; Lara, N.; Dallas, M.; Zhao, H.; Liu, Y.; Bonewald, L.F.; Johnson, M.L. Deletion of a single beta-catenin allele in osteocytes abolishes the bone anabolic response to loading. J. Bone Miner. Res. 2014, 29, 705–715. [Google Scholar] [CrossRef]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 2012, 27, 374–389. [Google Scholar] [CrossRef]
- Grimston, S.K.; Fontana, F.; Watkins, M.; Civitelli, R. Heterozygous deletion of both sclerostin (Sost) and connexin43 (Gja1) genes in mice is not sufficient to impair cortical bone modeling. PLoS ONE 2017, 12, e0187980. [Google Scholar] [CrossRef]
- Cheung, W.Y.; Fritton, J.C.; Morgan, S.A.; Seref-Ferlengez, Z.; Basta-Pljakic, J.; Thi, M.M.; Suadicani, S.O.; Spray, D.C.; Majeska, R.J.; Schaffler, M.B. Pannexin-1 and P2X7-Receptor Are Required for Apoptotic Osteocytes in Fatigued Bone to Trigger RANKL Production in Neighboring Bystander Osteocytes. J. Bone Miner. Res. 2016, 31, 890–899. [Google Scholar] [CrossRef]
- Morvan, F.; Boulukos, K.; Clement-Lacroix, P.; Roman Roman, S.; Suc-Royer, I.; Vayssiere, B.; Ammann, P.; Martin, P.; Pinho, S.; Pognonec, P.; et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J. Bone Miner. Res. 2006, 21, 934–945. [Google Scholar] [CrossRef]
- Guo, J.; Liu, M.; Yang, D.; Bouxsein, M.L.; Saito, H.; Galvin, R.J.; Kuhstoss, S.A.; Thomas, C.C.; Schipani, E.; Baron, R.; et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010, 11, 161–171. [Google Scholar] [CrossRef]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front. Biosci. (Landmark Ed.) 2011, 16, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Bakker, A.D.; Zandieh-Doulabi, B.; Semeins, C.M.; Klein-Nulend, J. Pulsating fluid flow modulates gene expression of proteins involved in Wnt signaling pathways in osteocytes. J. Orthop. Res. 2009, 27, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, P.; Wu, J.; Feng, X.; Cai, J.; Zhai, M.; Li, J.; Liu, X.; Jiang, M.; Luo, E.; et al. Fluid shear stress improves morphology, cytoskeleton architecture, viability, and regulates cytokine expression in a time-dependent manner in MLO-Y4 cells. Cell Biol. Int. 2018, 42, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Huo, B.; Park, M.; Guo, X.E. Calcium response in osteocytic networks under steady and oscillatory fluid flow. Bone 2012, 51, 466–473. [Google Scholar] [CrossRef]
- Batra, N.; Burra, S.; Siller-Jackson, A.J.; Gu, S.; Xia, X.; Weber, G.F.; DeSimone, D.; Bonewald, L.F.; Lafer, E.M.; Sprague, E.; et al. Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 2012, 109, 3359–3364. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Burra, S.; Kar, R.; Lampe, P.D.; Jiang, J.X. Mitogen-activated Protein Kinase (MAPK) Activated by Prostaglandin E2 Phosphorylates Connexin 43 and Closes Osteocytic Hemichannels in Response to Continuous Flow Shear Stress. J. Biol. Chem. 2015, 290, 28321–28328. [Google Scholar] [CrossRef]
- Batra, N.; Riquelme, M.A.; Burra, S.; Kar, R.; Gu, S.; Jiang, J.X. Direct regulation of osteocytic connexin 43 hemichannels through AKT kinase activated by mechanical stimulation. J. Biol. Chem. 2014, 289, 10582–10591. [Google Scholar] [CrossRef]
- Batra, N.; Riquelme, M.A.; Burra, S.; Jiang, J.X. 14-3-3 theta facilitates plasma membrane delivery and function of mechanosensitive connexin 43 hemichannels. J. Cell Sci. 2014, 127, 137–146. [Google Scholar] [CrossRef]
- Cherian, P.P.; Siller-Jackson, A.J.; Gu, S.; Wang, X.; Bonewald, L.F.; Sprague, E.; Jiang, J.X. Mechanical strain opens connexin 43 hemichannels in osteocytes: A novel mechanism for the release of prostaglandin. Mol. Biol. Cell 2005, 16, 3100–3106. [Google Scholar] [CrossRef]
- Genetos, D.C.; Kephart, C.J.; Zhang, Y.; Yellowley, C.E.; Donahue, H.J. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J. Cell Physiol. 2007, 212, 207–214. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Mathov, I.; Aguirre, J.I.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Mechanical stimulation prevents osteocyte apoptosis: Requirement of integrins, Src kinases, and ERKs. Am. J. Physiol. Cell Physiol. 2005, 289, C633–C643. [Google Scholar] [CrossRef] [PubMed]
- Kitase, Y.; Barragan, L.; Qing, H.; Kondoh, S.; Jiang, J.X.; Johnson, M.L.; Bonewald, L.F. Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J. Bone Miner. Res. 2010, 25, 2657–2668. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Manolagas, S.C.; Bellido, T. Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 2002, 277, 8648–8657. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Aguirre, J.I.; Kousteni, S.; Manolagas, S.C.; Bellido, T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal-regulated kinase activation. J. Biol. Chem. 2005, 280, 7317–7325. [Google Scholar] [CrossRef] [PubMed]
- Thi, M.M.; Islam, S.; Suadicani, S.O.; Spray, D.C. Connexin43 and pannexin1 channels in osteoblasts: Who is the “hemichannel”? J. Membr. Biol. 2012, 245, 401–409. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keyak, J.H.; Koyama, A.K.; LeBlanc, A.; Lu, Y.; Lang, T.F. Reduction in proximal femoral strength due to long-duration spaceflight. Bone 2009, 44, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Spector, E.R.; Smith, S.M.; Sibonga, J.D. Skeletal effects of long-duration head-down bed rest. Aviat. Space Environ. Med. 2009, 80 (Suppl. S5), A23–A28. [Google Scholar] [CrossRef] [PubMed]
- Maimoun, L.; Fattal, C.; Micallef, J.P.; Peruchon, E.; Rabischong, P. Bone loss in spinal cord-injured patients: From physiopathology to therapy. Spinal Cord 2006, 44, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Globus, R.K.; Bikle, D.D.; Morey-Holton, E. The temporal response of bone to unloading. Endocrinology 1986, 118, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Kurata, K.; Fukunaga, T.; Higaki, H. Bone marrow cell differentiation regulated by gel-embedded osteocyte under multi-dimensional gravity. J. Biomech. 2006, 39, S469. [Google Scholar] [CrossRef]
- Di, S.; Qian, A.; Qu, L.; Zhang, W.; Wang, Z.; Ding, C.; Li, Y.; Ren, H.; Shang, P. Graviresponses of osteocytes under altered gravity. Adv. Space Res. 2011, 48, 1161–1166. [Google Scholar] [CrossRef]
- Xu, H.; Liu, R.; Ning, D.; Zhang, J.; Yang, R.; Riquelme, M.A.; Li, J.; Jiang, J.X.; Shang, P. Biological responses of osteocytic connexin 43 hemichannels to simulated microgravity. J. Orthop. Res. 2017, 35, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Grimston, S.K.; Goldberg, D.B.; Watkins, M.; Brodt, M.D.; Silva, M.J.; Civitelli, R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J. Bone Miner. Res. 2011, 26, 2151–2160. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riquelme, M.A.; Cardenas, E.R.; Xu, H.; Jiang, J.X. The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone. Int. J. Mol. Sci. 2020, 21, 1146. https://doi.org/10.3390/ijms21031146
Riquelme MA, Cardenas ER, Xu H, Jiang JX. The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone. International Journal of Molecular Sciences. 2020; 21(3):1146. https://doi.org/10.3390/ijms21031146
Chicago/Turabian StyleRiquelme, Manuel A., Eduardo R. Cardenas, Huiyun Xu, and Jean X. Jiang. 2020. "The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone" International Journal of Molecular Sciences 21, no. 3: 1146. https://doi.org/10.3390/ijms21031146
APA StyleRiquelme, M. A., Cardenas, E. R., Xu, H., & Jiang, J. X. (2020). The Role of Connexin Channels in the Response of Mechanical Loading and Unloading of Bone. International Journal of Molecular Sciences, 21(3), 1146. https://doi.org/10.3390/ijms21031146