Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-? in the Amyloid Pathology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
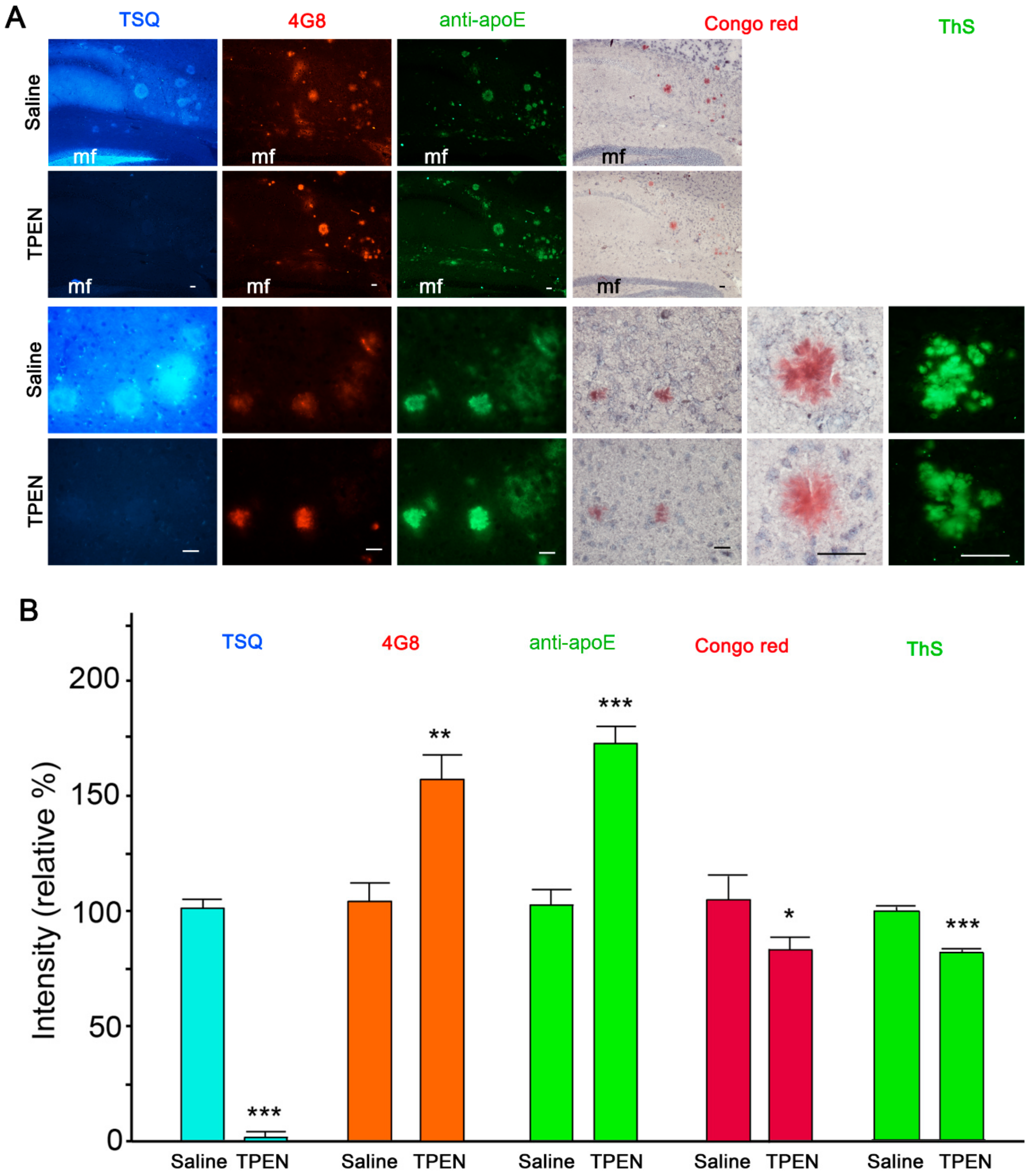
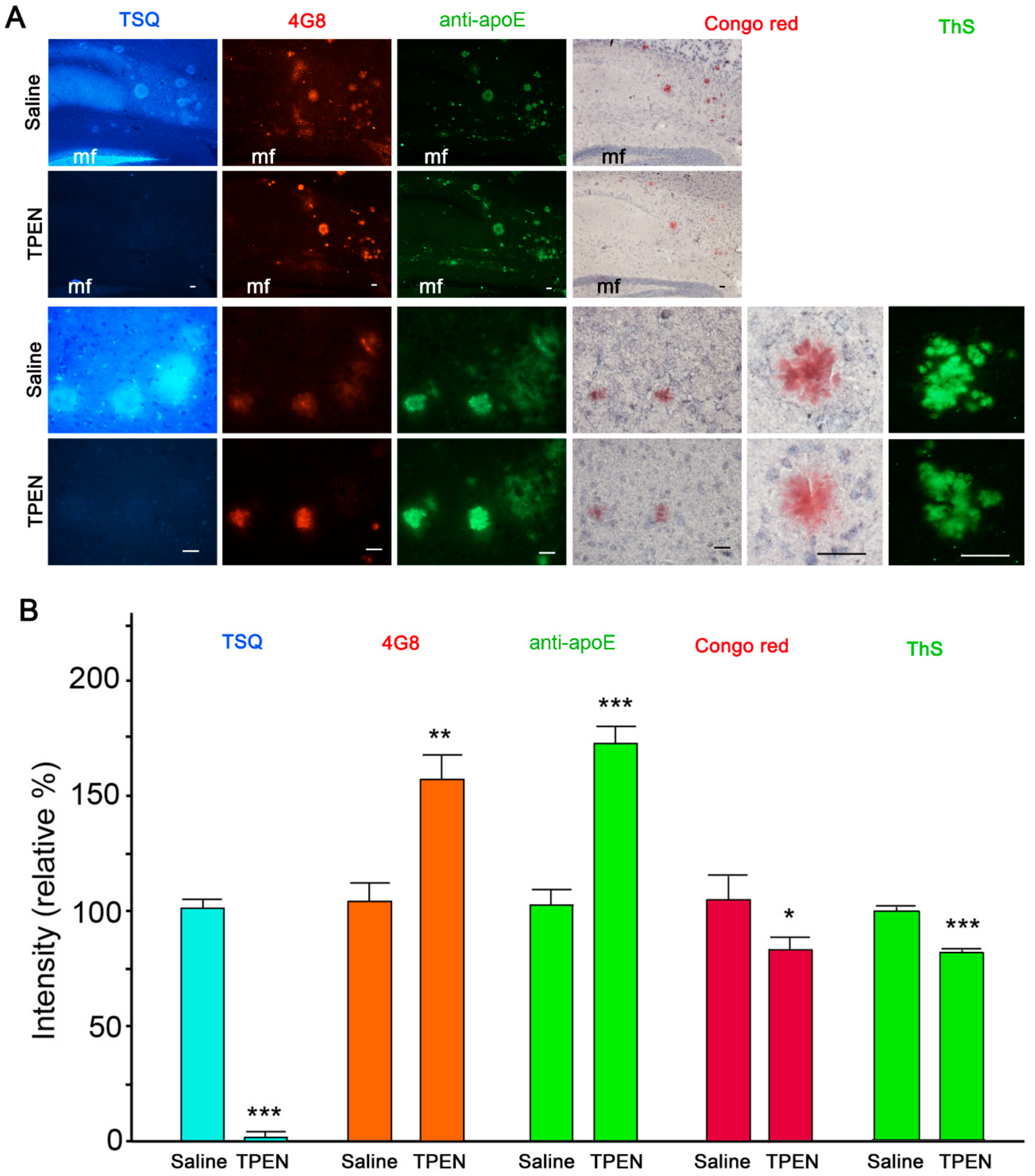
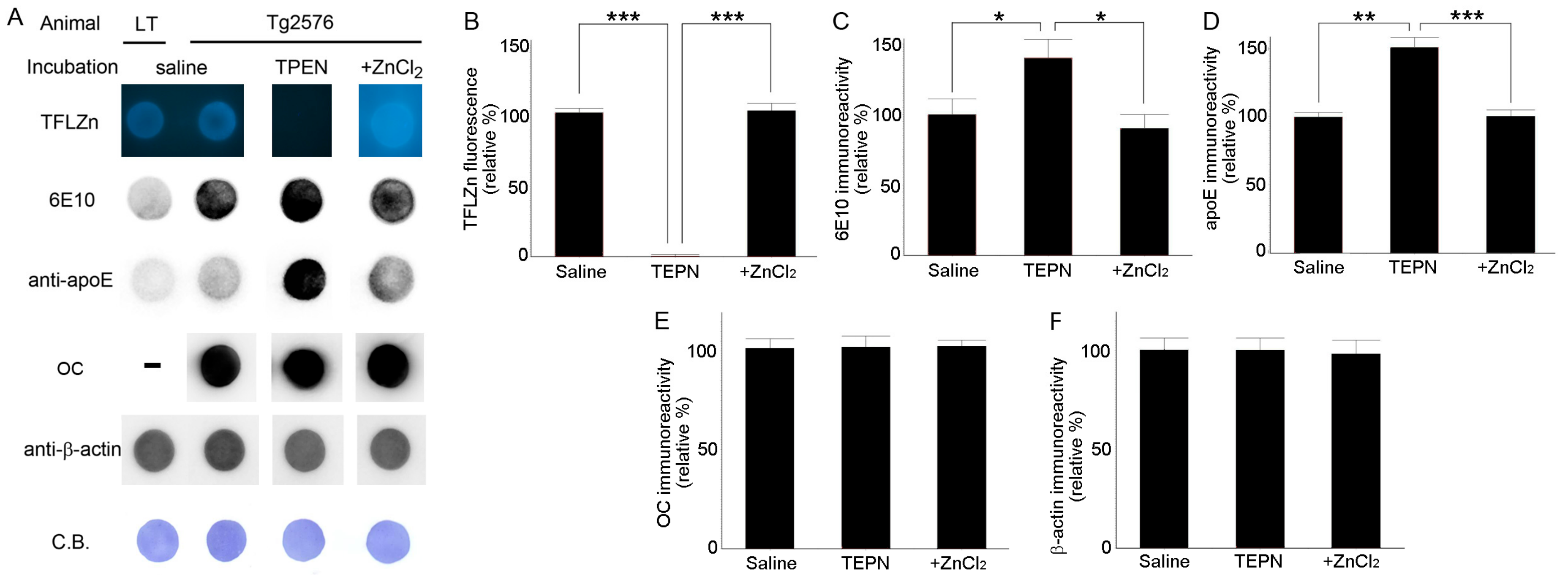
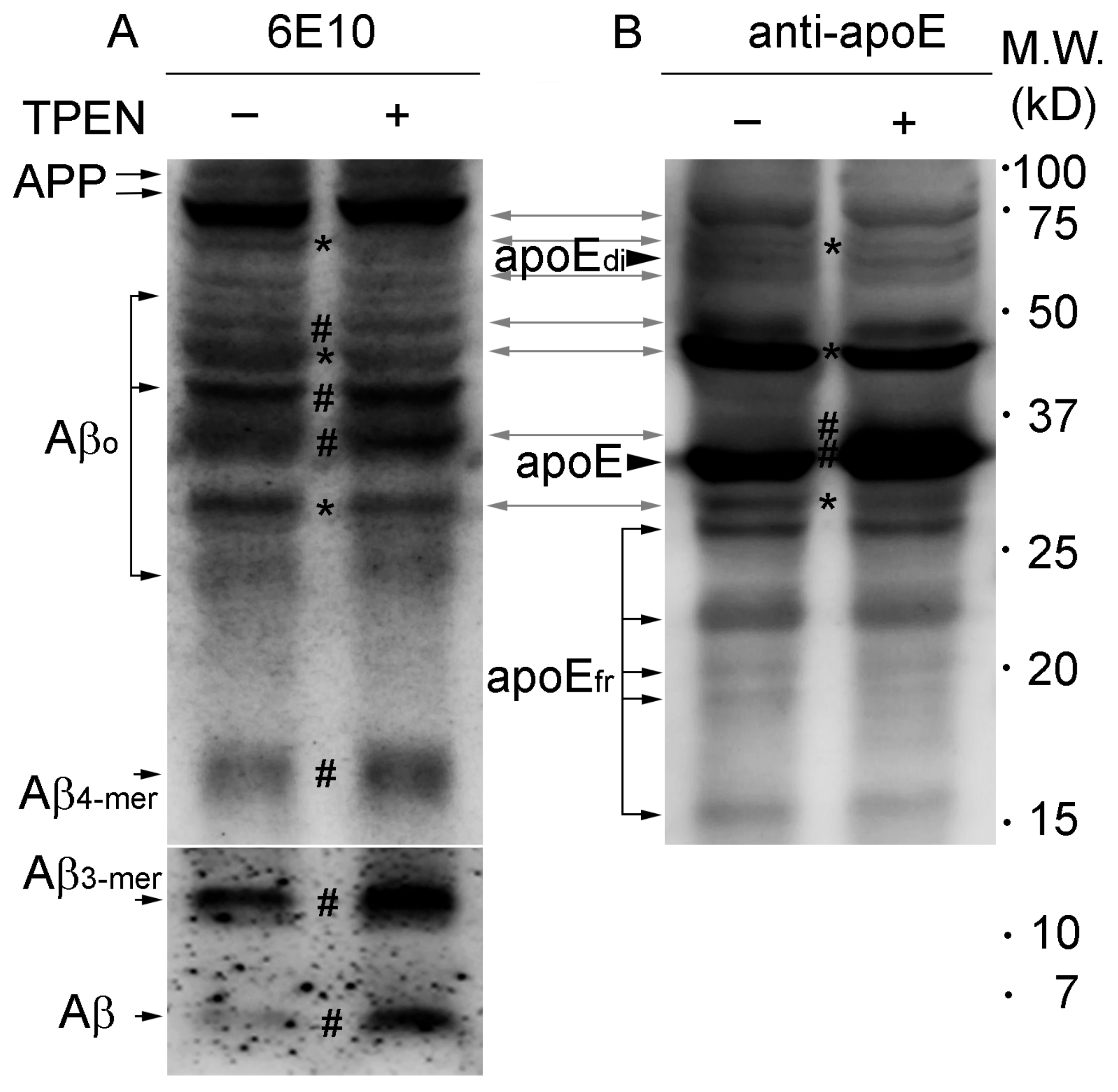
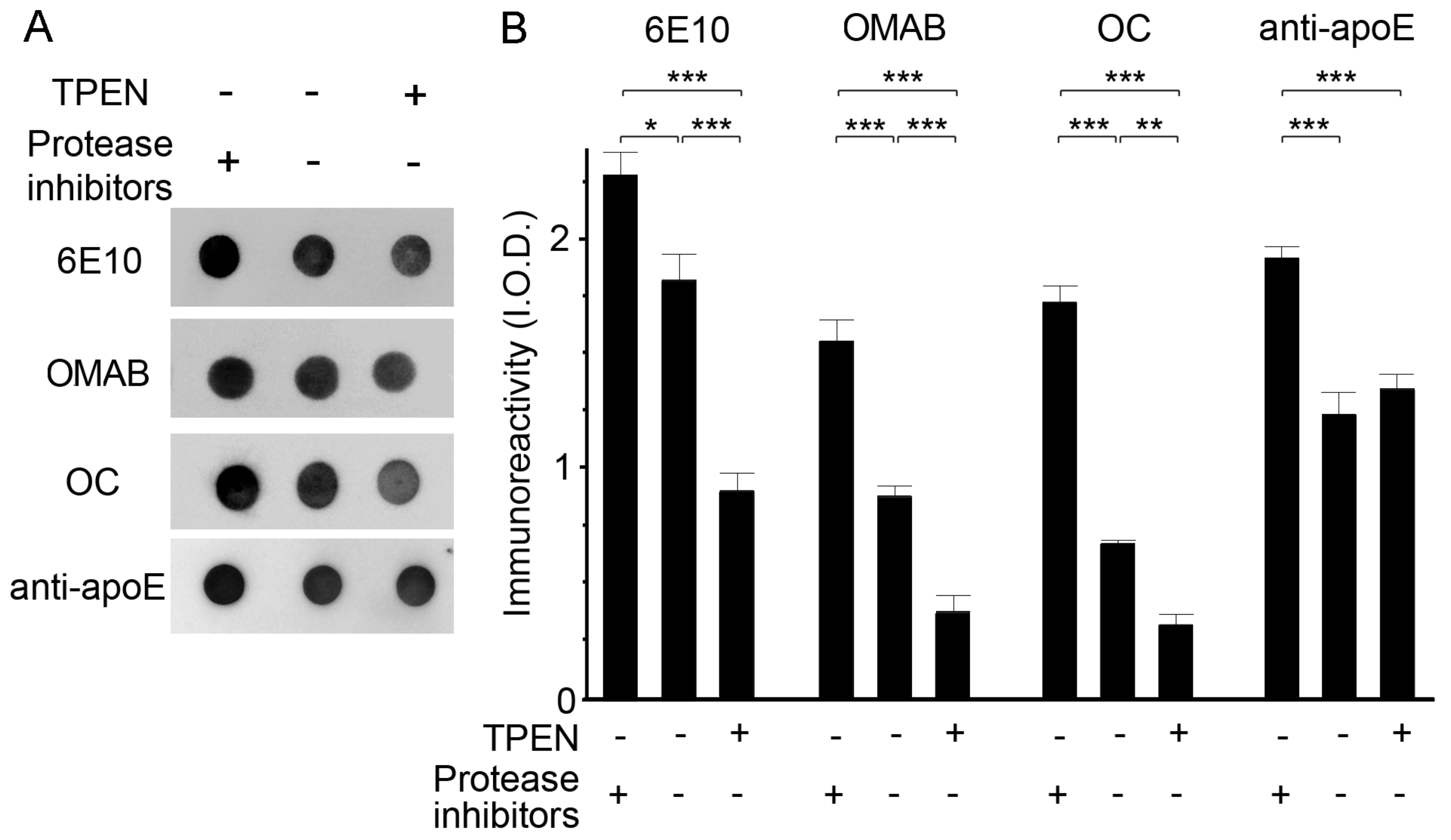
2.1. Zinc Chelation Enhances Aβ and apoE Immunoreactivities.
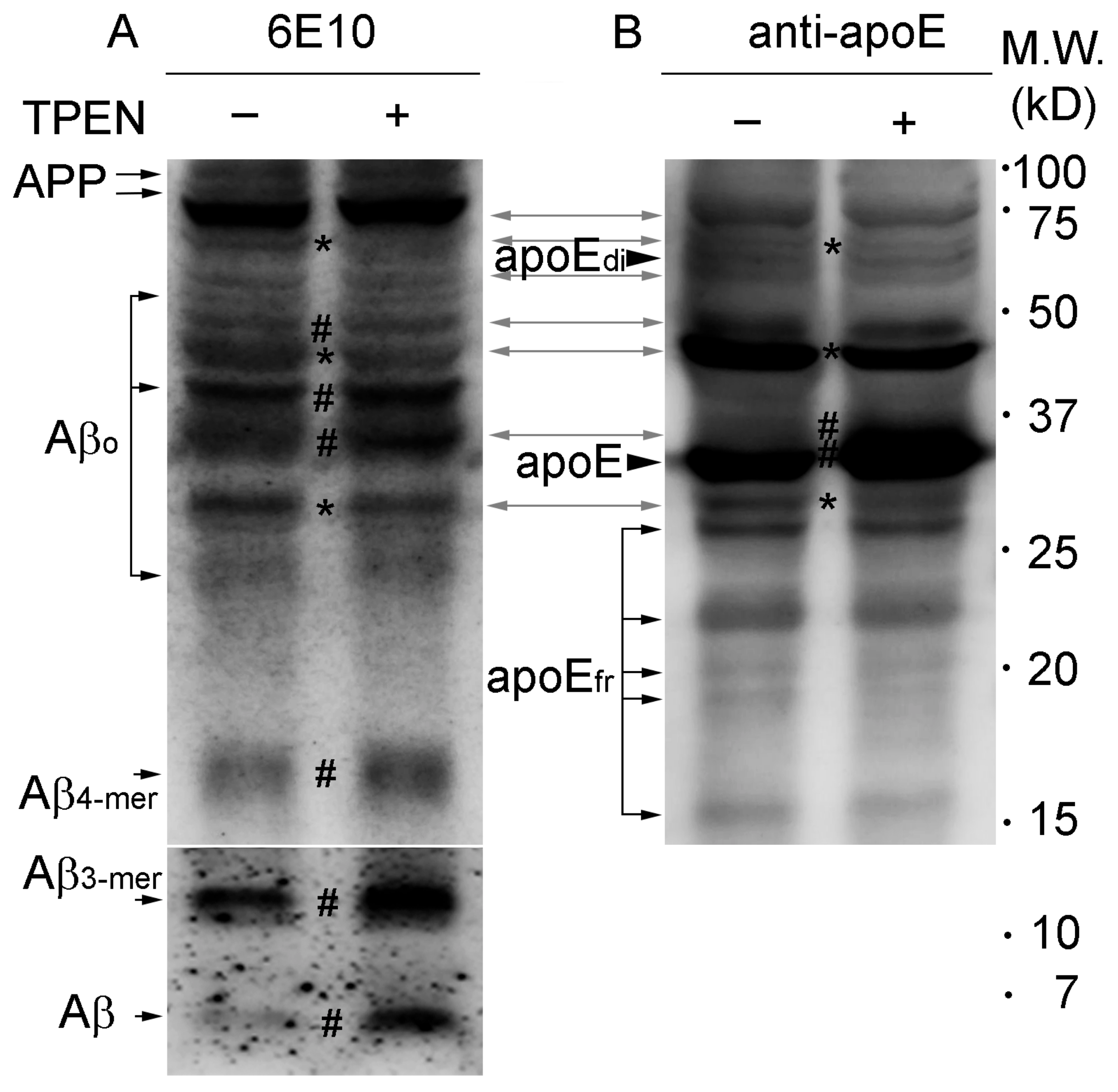
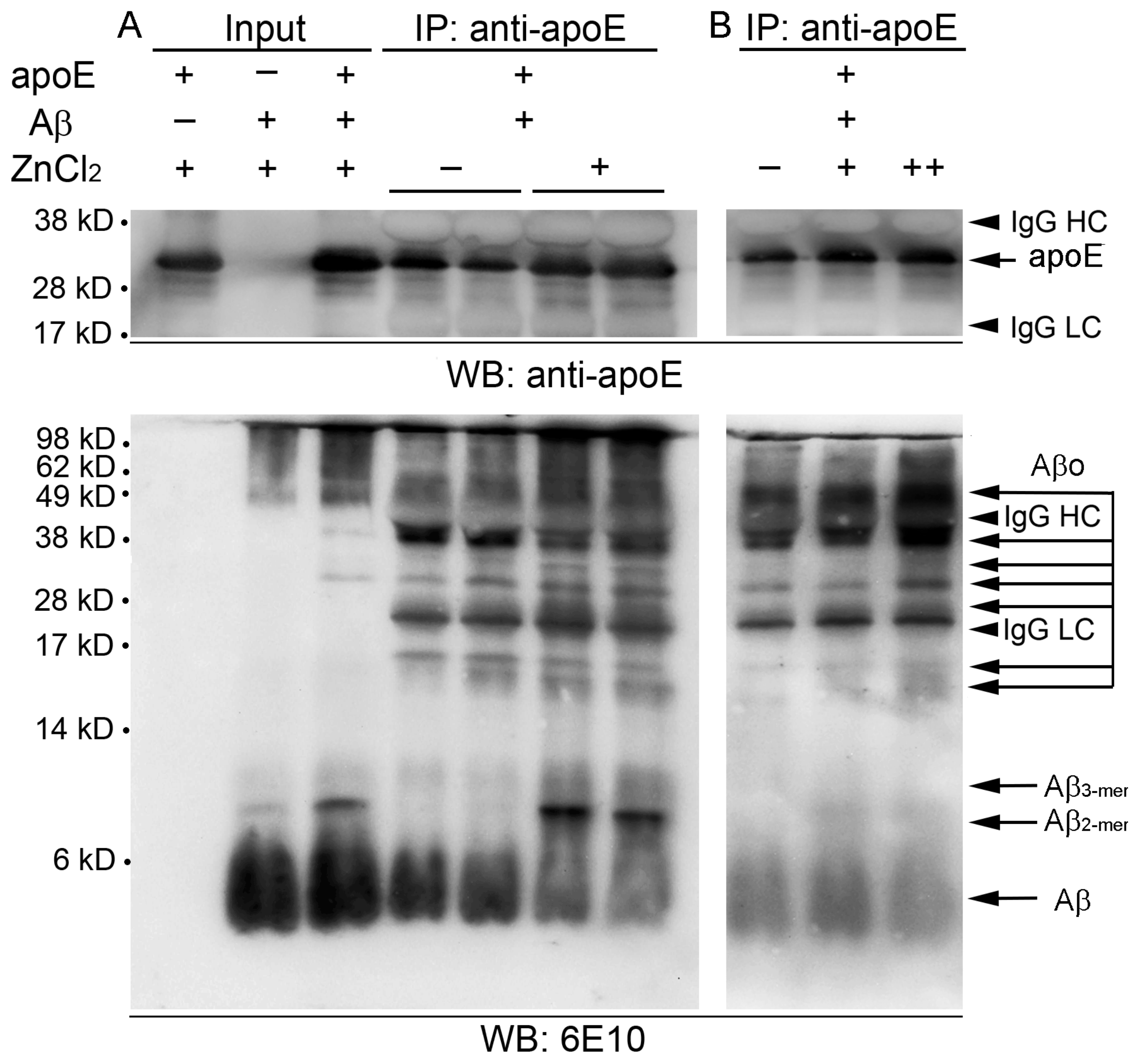
2.2. Zinc Promotes the Aggregations of apoE and/or Aβ Complexes
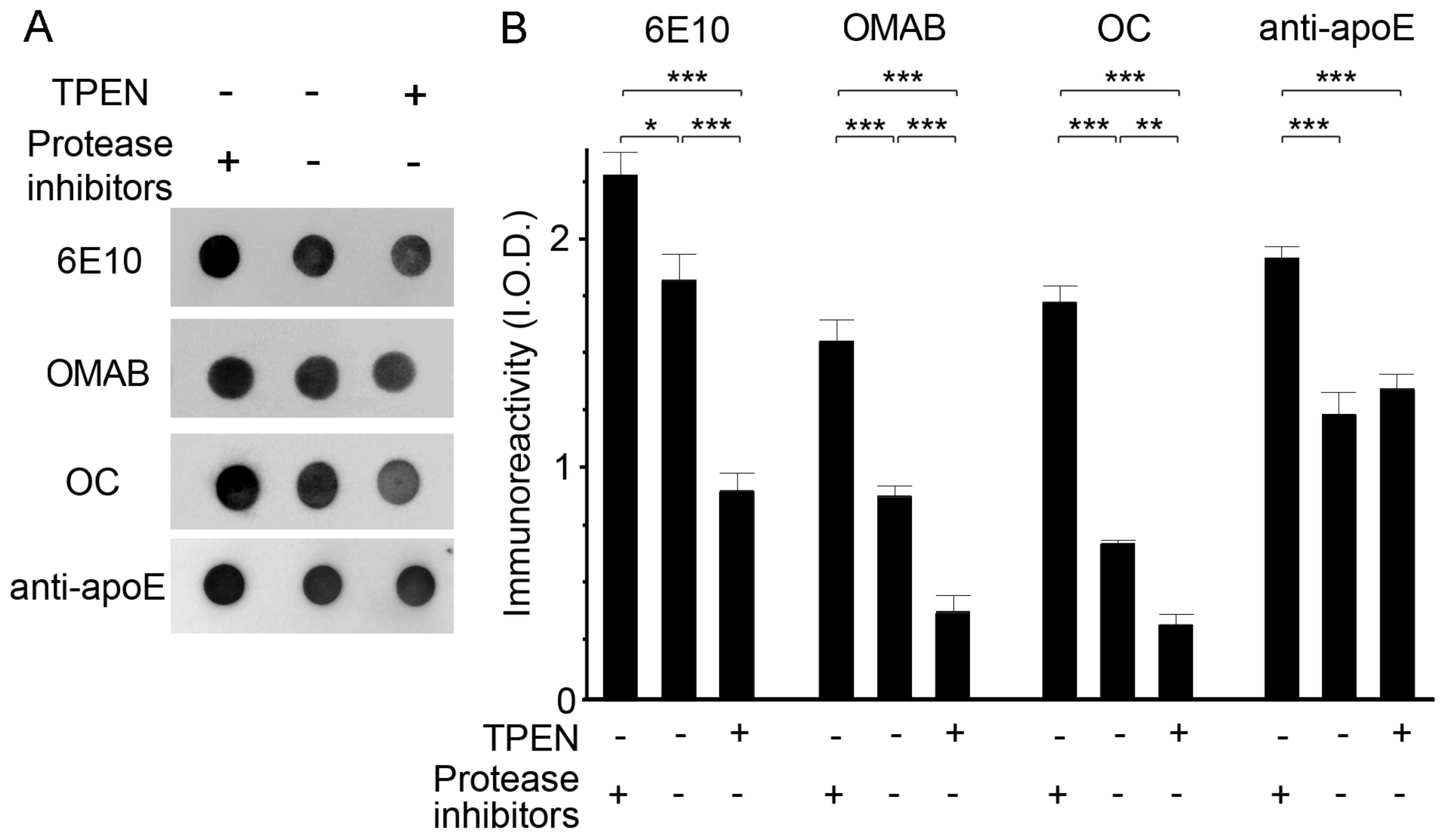
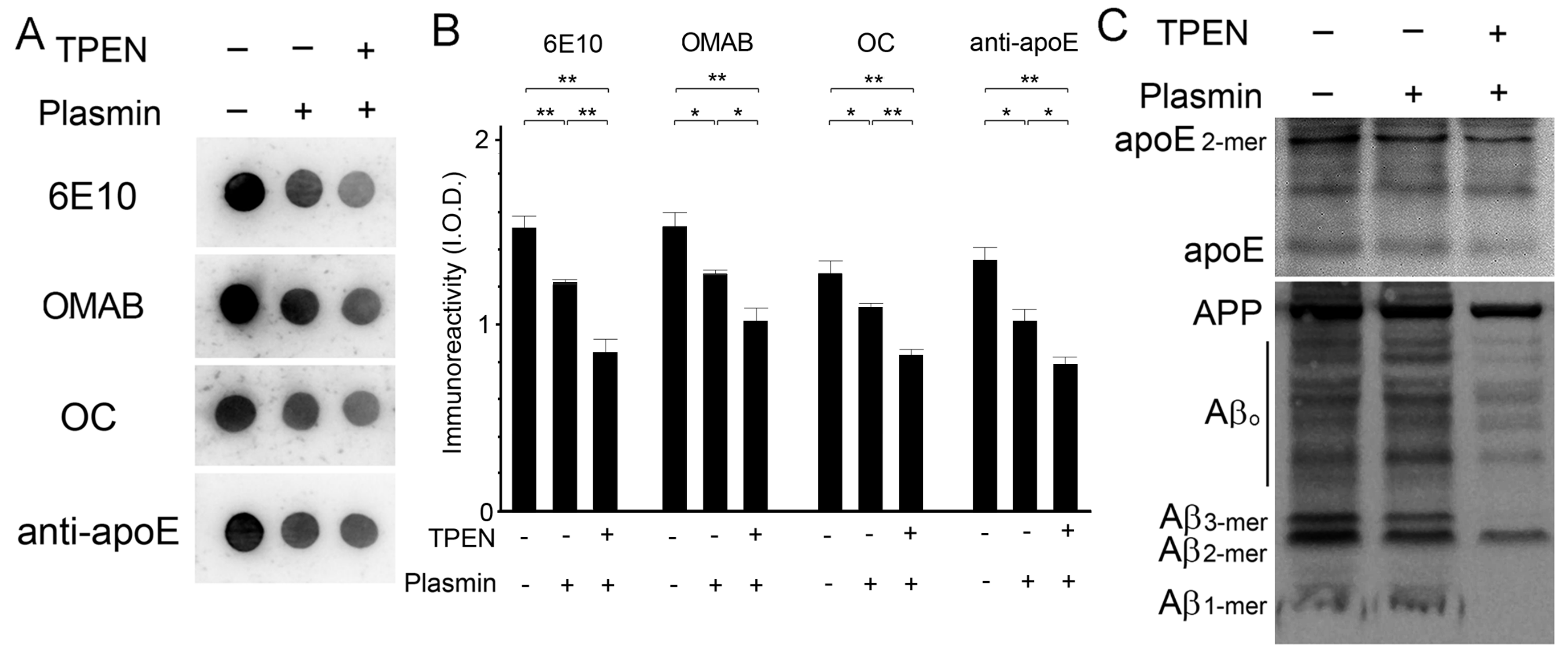
2.3. Zinc Depletion Encourages Aβ Degradation by Endogenous Proteinases
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.2. Tissue Preparations
4.3. Detection of Amyloid Plaques
4.4. Immunohistochemistry
4.5. Fluorescent Zinc Staining
4.6. Immunoblot Analysis
4.7. Co-immunoprecipitation
4.8. Proteolytic Degradation of apoE/Aβ Complexes by Endogenous Proteases or Plasmin
4.9. Statistics
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Normal and abnormal biology of the beta-amyloid precursor protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef]
- Lansbury, P.T., Jr. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA 1999, 96, 3342–3344. [Google Scholar] [CrossRef] [Green Version]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Patterson, J.C. Molecular dynamics study of Zn(abeta) and Zn(abeta)2. PLoS ONE 2013, 8, e70681. [Google Scholar] [CrossRef] [PubMed]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Segalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural changes of region 1-16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Cho, E.; Seo, J.W.; Hwang, J.J.; Koh, J.Y. Alteration of the cerebral zinc pool in a mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Mook-Jung, I.; Koh, J.Y. Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J. Neurosci. 1999, 19, RC10. [Google Scholar] [CrossRef]
- Cherny, R.A.; Legg, J.T.; McLean, C.A.; Fairlie, D.P.; Huang, X.; Atwood, C.S.; Beyreuther, K.; Tanzi, R.E.; Masters, C.L.; Bush, A.I. Aqueous dissolution of Alzheimer’s disease Abeta amyloid deposits by biometal depletion. J. Biol. Chem. 1999, 274, 23223–23228. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced Alzheimer’s Abeta1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasanthi, J.R.; Schrag, M.; Dasari, B.; Marwarha, G.; Dickson, A.; Kirsch, W.M.; Ghribi, O. Deferiprone reduces amyloid-beta and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J. Alzheimers Dis. 2012, 30, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Lee, J.Y.; Friedman, J.E.; Angel, I.; Kozak, A.; Koh, J.Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol. Aging 2004, 25, 1315–1321. [Google Scholar] [CrossRef]
- Friedlich, A.L.; Lee, J.Y.; van Groen, T.; Cherny, R.A.; Volitakis, I.; Cole, T.B.; Palmiter, R.D.; Koh, J.Y.; Bush, A.I. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3453–3459. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [Green Version]
- Budimir, A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm. 2011, 61, 1–14. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Yee, A.; Brewer, H.B., Jr.; Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Potter, H.; Wefes, I.M.; Nilsson, L.N. The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of Alzheimer amyloid formation. Neurobiol. Aging 2001, 22, 923–930. [Google Scholar] [CrossRef]
- Wisniewski, T.; Castano, E.M.; Golabek, A.; Vogel, T.; Frangione, B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am. J. Pathol. 1994, 145, 1030–1035. [Google Scholar] [PubMed]
- LaDu, M.J.; Falduto, M.T.; Manelli, A.M.; Reardon, C.A.; Getz, G.S.; Frail, D.E. Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 1994, 269, 23403–23406. [Google Scholar]
- Gearing, M.; Schneider, J.A.; Robbins, R.S.; Hollister, R.D.; Mori, H.; Games, D.; Hyman, B.T.; Mirra, S.S. Regional variation in the distribution of apolipoprotein E and A beta in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1995, 54, 833–841. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cho, E.; Kim, T.Y.; Kim, D.K.; Palmiter, R.D.; Volitakis, I.; Kim, J.S.; Bush, A.I.; Koh, J.Y. Apolipoprotein E ablation decreases synaptic vesicular zinc in the brain. Biometals 2010, 23, 1085–1095. [Google Scholar] [CrossRef]
- Sanan, D.A.; Weisgraber, K.H.; Russell, S.J.; Mahley, R.W.; Huang, D.; Saunders, A.; Schmechel, D.; Wisniewski, T.; Frangione, B.; Roses, A.D.; et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Investig. 1994, 94, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Bales, K.R.; Verina, T.; Cummins, D.J.; Du, Y.; Dodel, R.C.; Saura, J.; Fishman, C.E.; DeLong, C.A.; Piccardo, P.; Petegnief, V.; et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 15233–15238. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L. RING fingers and B-boxes: Zinc-binding protein-protein interaction domains. Biochem. Cell Biol. 1998, 76, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Klug, A.; Schwabe, J.W. Protein motifs 5. Zinc fingers. FASEB J. 1995, 9, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, M.E.; Wojcik, D.P.; Krone, C.A. Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimers Dis. 2003, 5, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moir, R.D.; Atwood, C.S.; Romano, D.M.; Laurans, M.H.; Huang, X.; Bush, A.I.; Smith, J.D.; Tanzi, R.E. Differential effects of apolipoprotein E isoforms on metal-induced aggregation of A beta using physiological concentrations. Biochemistry 1999, 38, 4595–4603. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, G.; Wenk, F. Pyridinderivate als Komplexbildner VIII Die Herstellung je eines neuen vier- und sechszähnigen Liganden. Helvetica Chimica Acta 1967, 50, 2330–2332. [Google Scholar] [CrossRef]
- Andrews, J.C.; Nolan, J.P.; Hammerstedt, R.H.; Bavister, B.D. Characterization of N-(6-methoxy-8-quinolyl)-p-toluenesulfonamide for the detection of zinc in living sperm cells. Cytometry 1995, 21, 153–159. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Naslund, J.; Thyberg, J.; Tjernberg, L.O.; Wernstedt, C.; Karlstrom, A.R.; Bogdanovic, N.; Gandy, S.E.; Lannfelt, L.; Terenius, L.; Nordstedt, C. Characterization of stable complexes involving apolipoprotein E and the amyloid beta peptide in Alzheimer’s disease brain. Neuron 1995, 15, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Wellnitz, S.; Friedlein, A.; Bonanni, C.; Anquez, V.; Goepfert, F.; Loetscher, H.; Adessi, C.; Czech, C. A 13 kDa carboxy-terminal fragment of ApoE stabilizes Abeta hexamers. J. Neurochem. 2005, 94, 1351–1360. [Google Scholar] [CrossRef]
- Lindhagen-Persson, M.; Brannstrom, K.; Vestling, M.; Steinitz, M.; Olofsson, A. Amyloid-beta oligomer specificity mediated by the IgM isotype--implications for a specific protective mechanism exerted by endogenous auto-antibodies. PLoS ONE 2010, 5, e13928. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, H.M.; Kihiko-Ehmann, M.; Wright, S.; Rydel, R.E.; Estus, S. Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J. Neurochem. 2000, 75, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, W.E.; Porter, M. Plasmin cleavage of the amyloid beta-protein: Alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 1999, 38, 11570–11576. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold. Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, N.; Zhou, T.; Hovmoller, S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics 2008, 24, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, G.W.; Roher, A.E.; Kuo, Y.M.; Gilligan, S.M.; Reardon, C.A.; Getz, G.S.; LaDu, M.J. SDS-stable complex formation between native apolipoprotein E3 and beta-amyloid peptides. Biochemistry 2000, 39, 16119–16124. [Google Scholar] [CrossRef]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Dibello, V.; Greco, A.; Daniele, A.; Seripa, D.; Logroscino, G.; Imbimbo, B.P. Are antibodies directed against amyloid-beta (Abeta) oligomers the last call for the Abeta hypothesis of Alzheimer’s disease? Immunotherapy 2019, 11, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, L.; Faustino, C. Therapeutic Strategies Targeting Amyloid-beta in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 418–452. [Google Scholar] [CrossRef]
- Van Dyck, C.H. Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.B.; Byun, C.J.; Yun, J.H.; Jo, D.G.; Carmeliet, P.; Koh, J.Y.; Lee, J.Y. Tissue plasminogen activator arrests Alzheimer’s disease pathogenesis. Neurobiol Aging 2014, 35, 511–519. [Google Scholar] [CrossRef]
- Grasso, G.; Giuffrida, M.L.; Rizzarelli, E. Metallostasis and amyloid beta-degrading enzymes. Metallomics 2012, 4, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Janc, J.W.; Clark, J.M.; Warne, R.L.; Elrod, K.C.; Katz, B.A.; Moore, W.R. A novel approach to serine protease inhibition: Kinetic characterization of inhibitors whose potencies and selectivities are dramatically enhanced by Zinc(II). Biochemistry 2000, 39, 4792–4800. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.A.; Clark, J.M.; Finer-Moore, J.S.; Jenkins, T.E.; Johnson, C.R.; Ross, M.J.; Luong, C.; Moore, W.R.; Stroud, R.M. Design of potent selective zinc-mediated serine protease inhibitors. Nature 1998, 391, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Bell, W.R.; Ebert, R.F.; Starksen, N.F. Protective effect of divalent cations in the plasmin degradation of fibrinogen. Arch. Biochem. Biophys. 1985, 238, 452–457. [Google Scholar] [CrossRef]
- Henderson, S.J.; Stafford, A.R.; Leslie, B.A.; Kim, P.Y.; Vaezzadeh, N.; Ni, R.; Fredenburgh, J.C.; Weitz, J.I. Zinc delays clot lysis by attenuating plasminogen activation and plasmin-mediated fibrin degradation. Thromb. Haemost 2015, 113, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Inhibitory zinc sites in enzymes. Biometals 2013, 26, 197–204. [Google Scholar] [CrossRef]
- Bush, A.I. Metals and neuroscience. Curr. Opin. Chem. Biol. 2000, 4, 184–191. [Google Scholar] [CrossRef]
- Price, K.A.; Crouch, P.J.; White, A.R. Therapeutic treatment of Alzheimer’s disease using metal complexing agents. Recent. Pat. CNS Drug Discov. 2007, 2, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Malm, T.M.; Iivonen, H.; Goldsteins, G.; Keksa-Goldsteine, V.; Ahtoniemi, T.; Kanninen, K.; Salminen, A.; Auriola, S.; Van Groen, T.; Tanila, H.; et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J. Neurosci. 2007, 27, 3712–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.W.; Oh, S.B.; Kerr, R.A.; Lee, H.J.; Kim, S.H.; Kim, S.; Jang, M.; Ruotolo, B.T.; Lee, J.Y.; Lim, M.H. A rationally designed small molecule for identifying an in vivo link between metal-amyloid-beta complexes and the pathogenesis of Alzheimer’s disease. Chem. Sci. 2015, 6, 1879–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.W.; Derrick, J.S.; Kerr, R.A.; Oh, S.B.; Cho, W.J.; Lee, S.J.; Ji, Y.; Han, J.; Tehrani, Z.A.; Suh, N.; et al. Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer’s disease. Nat. Commun. 2016, 7, 13115. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.B.; Kim, J.A.; Park, S.; Lee, J.-Y. Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-? in the Amyloid Pathology. Int. J. Mol. Sci. 2020, 21, 802. https://doi.org/10.3390/ijms21030802
Oh SB, Kim JA, Park S, Lee J-Y. Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-? in the Amyloid Pathology. International Journal of Molecular Sciences. 2020; 21(3):802. https://doi.org/10.3390/ijms21030802
Chicago/Turabian StyleOh, Shin Bi, Jung Ah Kim, SuJi Park, and Joo-Yong Lee. 2020. "Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-? in the Amyloid Pathology" International Journal of Molecular Sciences 21, no. 3: 802. https://doi.org/10.3390/ijms21030802
APA StyleOh, S. B., Kim, J. A., Park, S., & Lee, J.-Y. (2020). Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-? in the Amyloid Pathology. International Journal of Molecular Sciences, 21(3), 802. https://doi.org/10.3390/ijms21030802