Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance Through the CXCL12/CXCR4/mTOR/TGFβ Signaling Pathway in Breast Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Chemotherapy in DPP-4-Deficient Breast Cancer Cells Facilitated the Expression of ABC Transporters
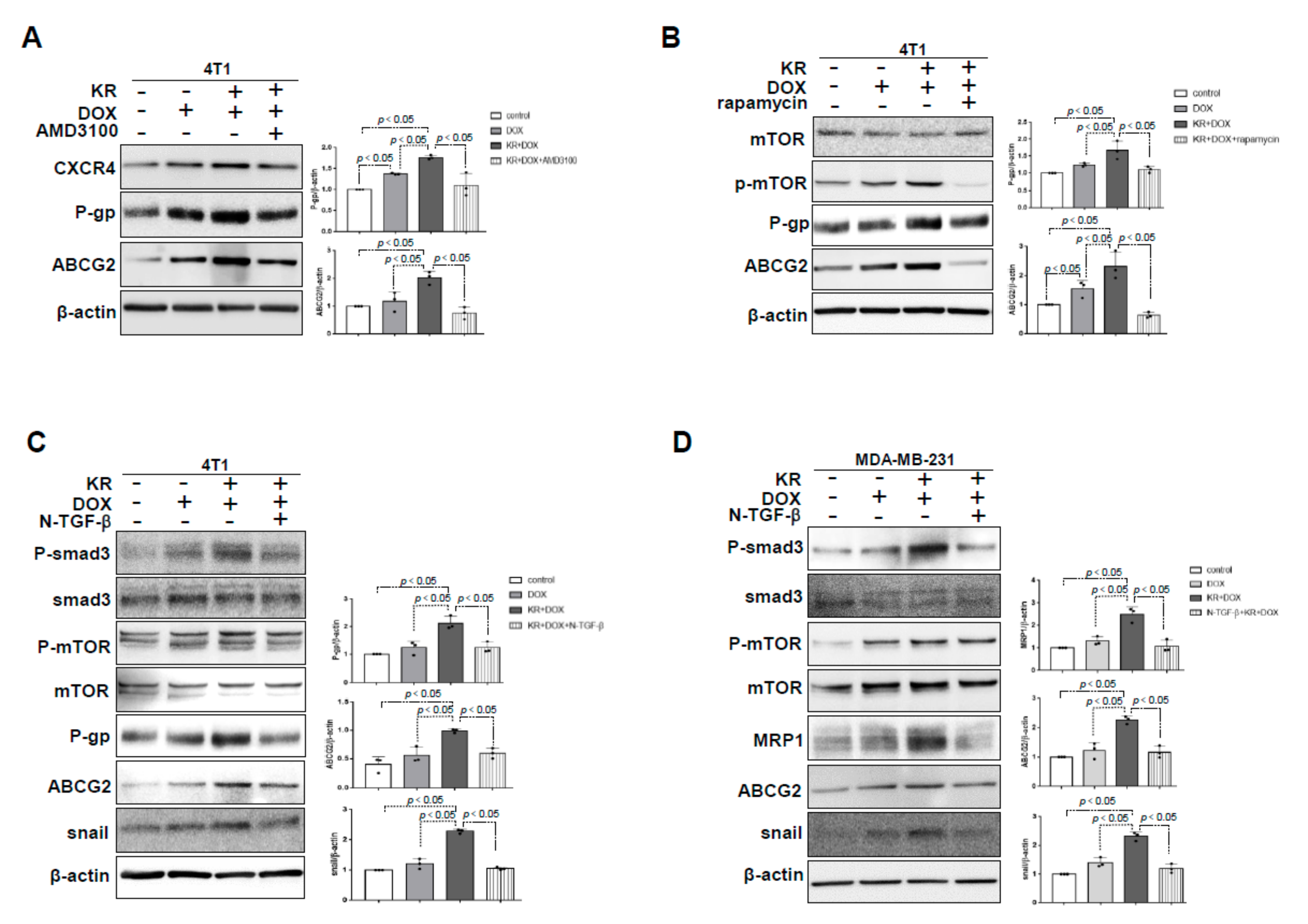
2.2. DPP-4 Deficiency Induced ABC Transporters via CXCL12/CXCR4/mTOR and the TGF-β Signaling Axis in 4T1 Cells
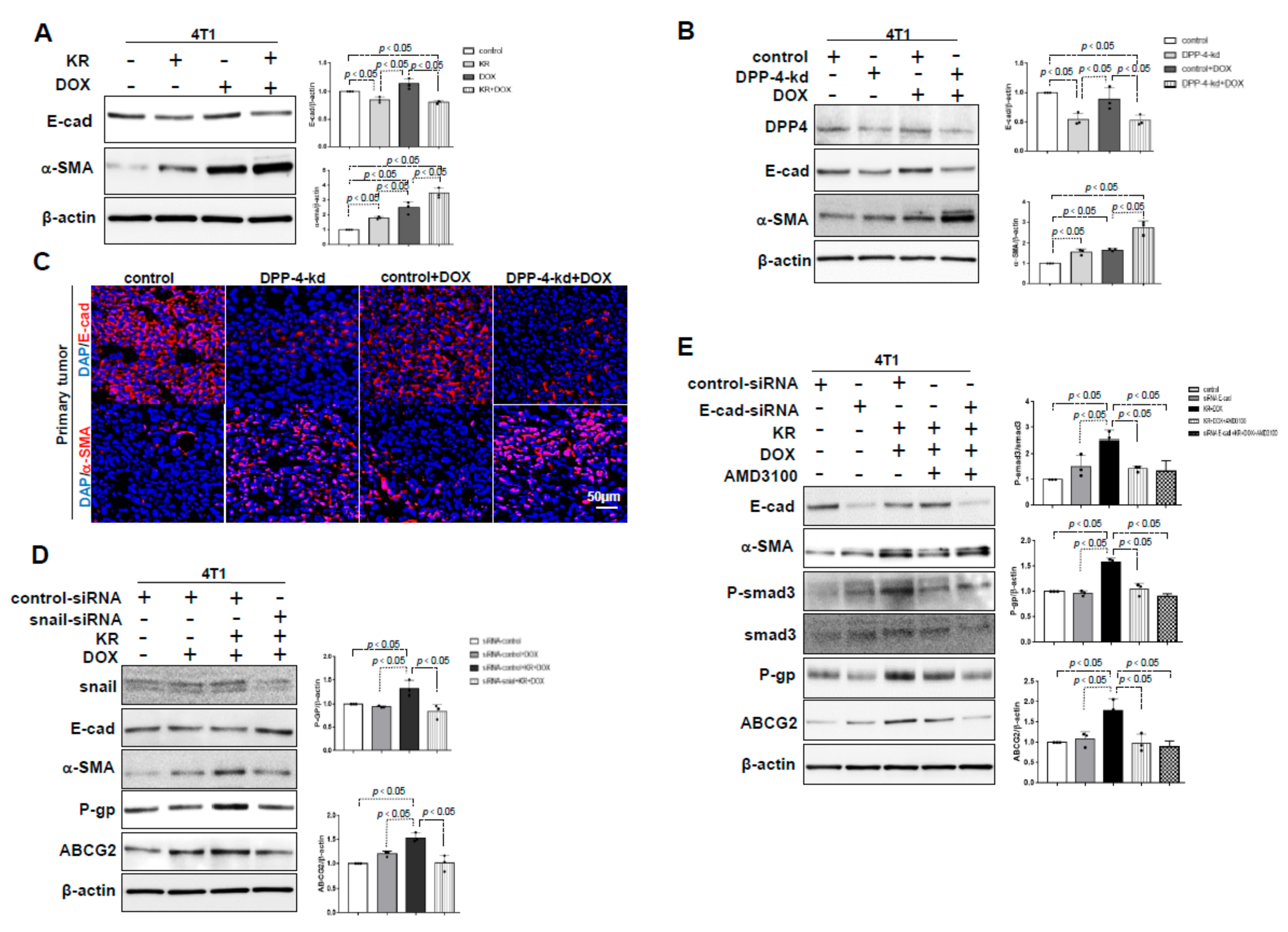
2.3. The Role of EMT in DPP-4 Deficiency-Induced ABC Transporters
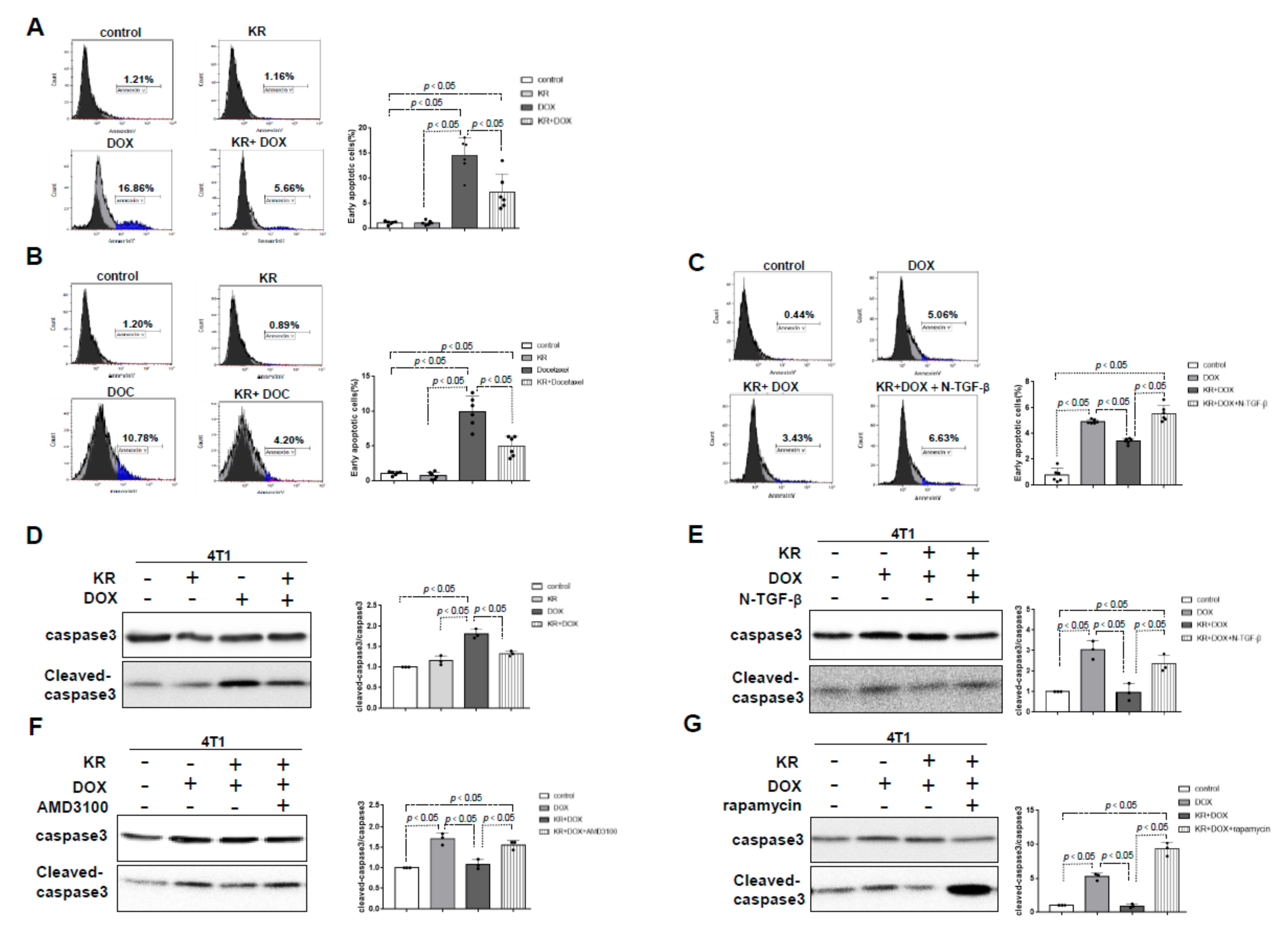
2.4. The Effects of DPP-4 Deficiency on Chemotherapy-Induced Apoptosis in Breast Cancer Cells
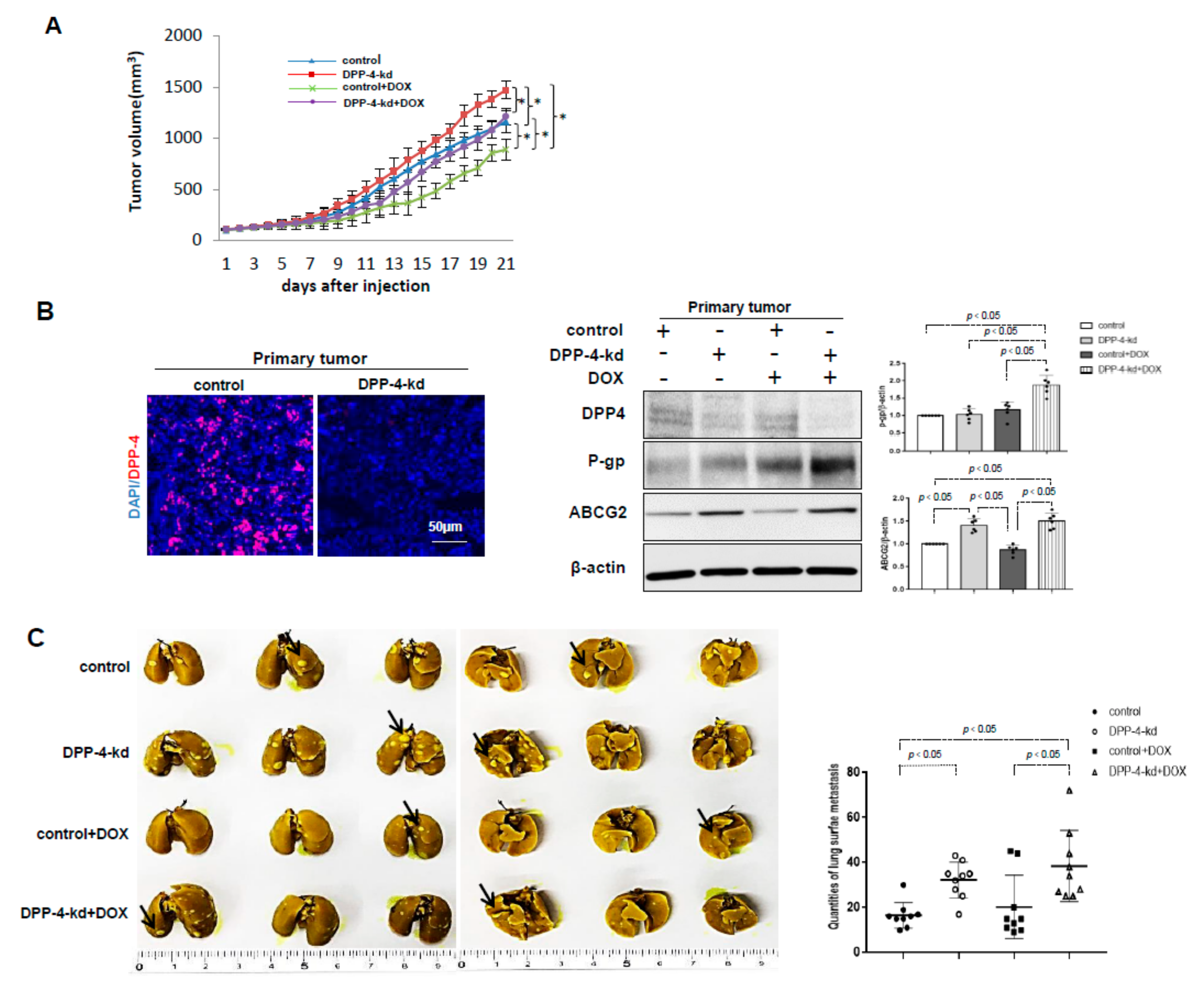
2.5. DPP-4 Deficiency Induced the Expression of ABC Transporters and Was Associated With Chemoresistance in the Allograft Breast Cancer Model
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Treatment
4.3. Transfection Experiments
4.4. Allograft Breast Cancer Mouse Model
4.5. Ethics Statement
4.6. Western Blot Analysis
4.7. Flow Cytometry
4.8. Immunofluorescence in Mouse Tissue
4.9. Bouin Buffer Staining
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.; He, Y.; Koya, D.; Kanasaki, K. Cancer biology in diabetes. J. Diabetes invest. 2014, 5, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J. A Increased Cancer-Related Mortality for Patients With Type 2 Diabetes Who Use Sulfonylureas or Insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanasaki, K. The role of renal dipeptidyl peptidase-4 in kidney disease: renal effects of dipeptidyl peptidase-4 inhibitors with a focus on linagliptin. Clin. Sci. 2018, 132, 489–507. [Google Scholar] [CrossRef] [Green Version]
- Busek, P.; Sedo, A. Dipeptidyl Peptidase-IV and Related Proteases in Brain Tumors. In Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications; InTech: London, UK, 2013; pp. 235–269. [Google Scholar]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4—A potential biomarker and target for cancer therapy. Pharmacol. Ther. 2019, 198, 135–159. [Google Scholar] [CrossRef]
- Abrahami, D.; Douros, A.; Yin, H.; Yu, O.H.; Faillie, J.-L.; Montastruc, F.; Platt, R.W.; Bouganim, N.; Azoulay, L. Incretin based drugs and risk of cholangiocarcinoma among patients with type 2 diabetes: Population based cohort study. BMJ 2018, 363, k4880. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Vilsbøll, T.; Gallwitz, B.; Garber, A.; Madsbad, S. Incretin-based therapies: Viewpoints on the way to consensus. Diabetes Care 2009, 32, S223–S231. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.W.; Gao, C.; Bhasin, S.S.; Voznesensky, O.S.; Calagua, C.; Arai, S.; Nelson, P.S.; Montgomery, B.; Mostaghel, E.A.; Corey, E.; et al. Downregulation of Dipeptidyl Peptidase 4 Accelerates Progression to Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 6354–6362. [Google Scholar] [CrossRef] [Green Version]
- Wesley, U.V. A Role for Dipeptidyl Peptidase IV in Suppressing the Malignant Phenotype of Melanocytic Cells. J. Exp. Med. 1999, 190, 311–322. [Google Scholar] [CrossRef]
- Wesley, U.V.; Tiwari, S.; Houghton, A.N. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int. J. Cancer 2004, 109, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arscott, W.T.; LaBauve, A.E.; May, V.; Wesley, U.V. Suppression of neuroblastoma growth by dipeptidyl peptidase IV: relevance of chemokine regulation and caspase activation. Oncogene 2009, 28, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumour Biol. 2014, 35, 7765–7773. [Google Scholar] [CrossRef] [Green Version]
- Kajiyama, H.; Shibata, K.; Ino, K.; Mizutani, S.; Nawa, A.; Kikkawa, F. The expression of dipeptidyl peptidase IV (DPPIV/CD26) is associated with enhanced chemosensitivity to paclitaxel in epithelial ovarian carcinoma cells. Cancer Sci. 2010, 101, 347–354. [Google Scholar] [CrossRef]
- Yang, F.; Takagaki, Y.; Yoshitomi, Y.; Ikeda, T.; Li, J.; Kitada, M.; Kumagai, A.; Kawakita, E.; Shi, S.; Kanasaki, K.; et al. Inhibition of Dipeptidyl Peptidase-4 Accelerates Epithelial-Mesenchymal Transition and Breast Cancer Metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019, 79, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.Q.; Buhrer, E.D.; Berezowska, S.; Marti, T.M.; Xu, D.; Froment, L.; Yang, H.; Hall, S.R.R.; Vassella, E.; Yang, Z.; et al. mTOR mediates a mechanism of resistance to chemotherapy and defines a rational combination strategy to treat KRAS-mutant lung cancer. Oncogene 2019, 38, 622–636. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Geng, L.; Yi, H.; Huo, W.; Talmon, G.; Kim, Y.C.; Wang, S.M.; Wang, J. Transforming Growth Factor beta Mediates Drug Resistance by Regulating the Expression of Pyruvate Dehydrogenase Kinase 4 in Colorectal Cancer. J. Biolog. Chem. 2016, 291, 17405–17416. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.A.; Cronin, K.A.; Plevritis, S.K.; Fryback, D.G.; Clarke, L.; Zelen, M.; Mandelblatt, J.S.; Yakovlev, A.Y.; Habbema, J.D.F.; Feuer, E.J. Effect of screening and adjuvant therapy on mortality from breast cancer. N. Engl. J. Med. 2005, 353, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Yan, M. Over-expression of FSIP1 promotes breast cancer progression and confers resistance to docetaxel via MRP1 stabilization. Cell Death Dis. 2019, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavi, O.; Gottesman, M.M.; Levy, D. The dynamics of drug resistance: a mathematical perspective. Drug Resist. Updat. 2012, 15, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genom. Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: more than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Zhang, J.T. Use of arrays to investigate the contribution of ATP-binding cassette transporters to drug resistance in cancer chemotherapy and prediction of chemosensitivity. Cell Res. 2007, 17, 311–323. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Arumugam, A.; Subramani, R.; Nandy, S.B.; Terreros, D.; Dwivedi, A.K.; Saltzstein, E.; Lakshmanaswamy, R. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 2019, 51, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.; Xu, Z.D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.S.; Sun, Y.Z.; Wang, S.M.; Ruan, J.S. Epithelial-mesenchymal transition: Potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, S.; Lehmann, R. An ABC transporter controls export of a Drosophila germ cell attractant. Science 2009, 323, 943–946. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, H.; Li, Z.; You, J.; Wu, Q.W.; Zhao, C.; Tzeng, C.M.; Zhang, Z.M. Interaction of WBP2 with ERalpha increases doxorubicin resistance of breast cancer cells by modulating MDR1 transcription. Br. J. Cancer 2018, 182–192. [Google Scholar] [CrossRef]
- Chen, W.; Wang, H.; Tang, Y.; Liu, C.; Li, H.; Li, W. Multidrug resistance in breast cancer cells during epithelialmesenchymal transition is modulated by breast cancer resistant protein. Chin. J. Cancer 2010, 29, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, C.; Tang, Y.; Li, H.; Zhou, F.; Lv, S. Overexpression of Snail accelerates adriamycin induction of multidrug resistance in breast cancer cells. Asian Pac. J. Cancer Prev. 12, 2575–2580. [PubMed]
- Ma, S.Y.; Park, J.H.; Jung, H.; Ha, S.M.; Kim, Y.; Park, D.H.; Lee, D.H.; Lee, S.; Chu, I.H.; Jung, S.Y.; et al. Snail maintains metastatic potential, cancer stem-like properties, and chemoresistance in mesenchymal mouse breast cancer TUBOP2J cells. Oncol. Rep. 2017, 38, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Fan, Y.; Kumagai, A.; Kawakita, E.; Kitada, M.; Kanasaki, K.; Koya, D. Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance Through the CXCL12/CXCR4/mTOR/TGFβ Signaling Pathway in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 805. https://doi.org/10.3390/ijms21030805
Li S, Fan Y, Kumagai A, Kawakita E, Kitada M, Kanasaki K, Koya D. Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance Through the CXCL12/CXCR4/mTOR/TGFβ Signaling Pathway in Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(3):805. https://doi.org/10.3390/ijms21030805
Chicago/Turabian StyleLi, Shaolan, Yang Fan, Asako Kumagai, Emi Kawakita, Munehiro Kitada, Keizo Kanasaki, and Daisuke Koya. 2020. "Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance Through the CXCL12/CXCR4/mTOR/TGFβ Signaling Pathway in Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 3: 805. https://doi.org/10.3390/ijms21030805
APA StyleLi, S., Fan, Y., Kumagai, A., Kawakita, E., Kitada, M., Kanasaki, K., & Koya, D. (2020). Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance Through the CXCL12/CXCR4/mTOR/TGFβ Signaling Pathway in Breast Cancer Cells. International Journal of Molecular Sciences, 21(3), 805. https://doi.org/10.3390/ijms21030805