Transgenic Mice Overexpressing Human STIM2 and ORAI1 in Neurons Exhibit Changes in Behavior and Calcium Homeostasis but Show No Signs of Neurodegeneration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
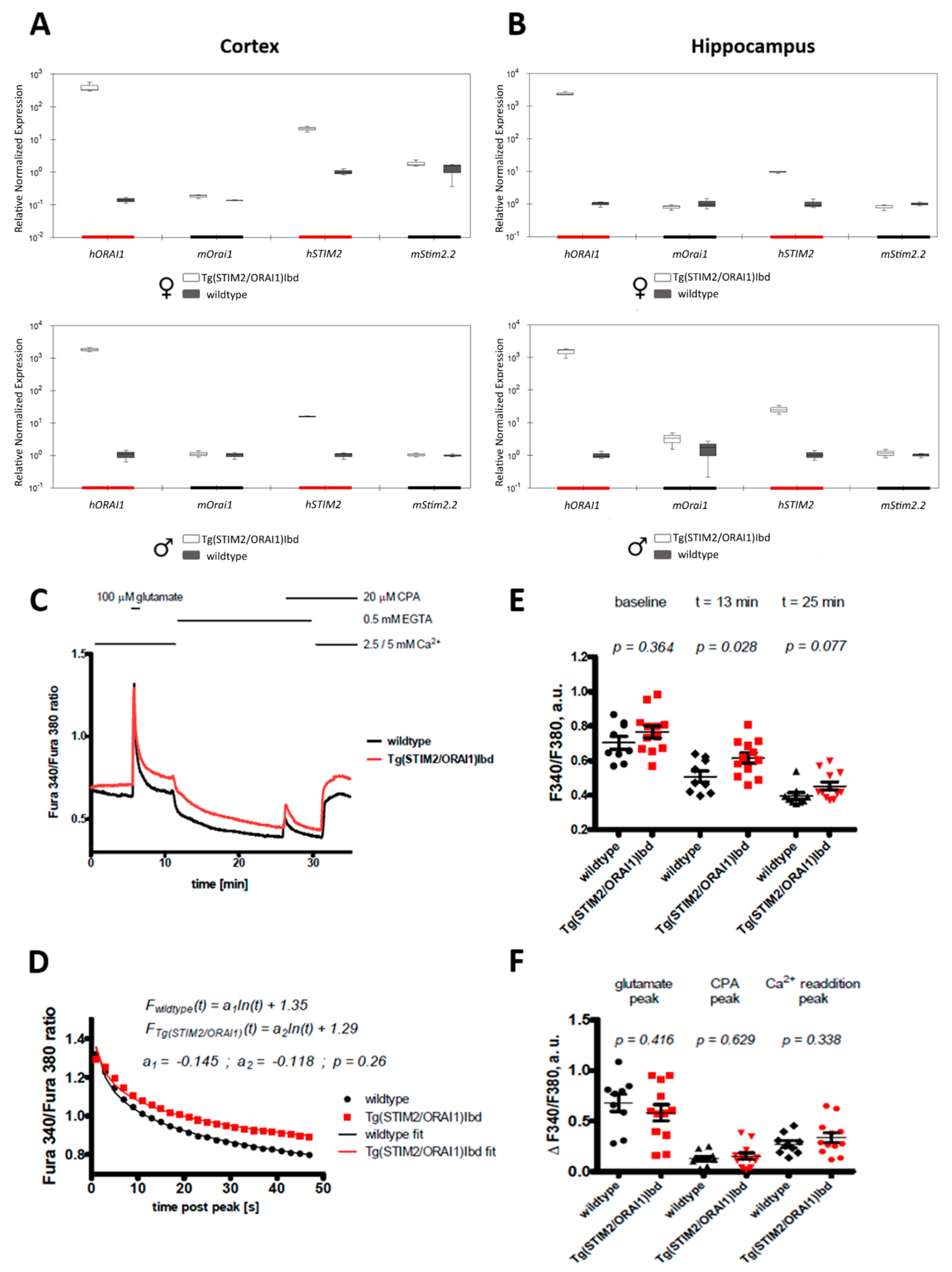
2. Results
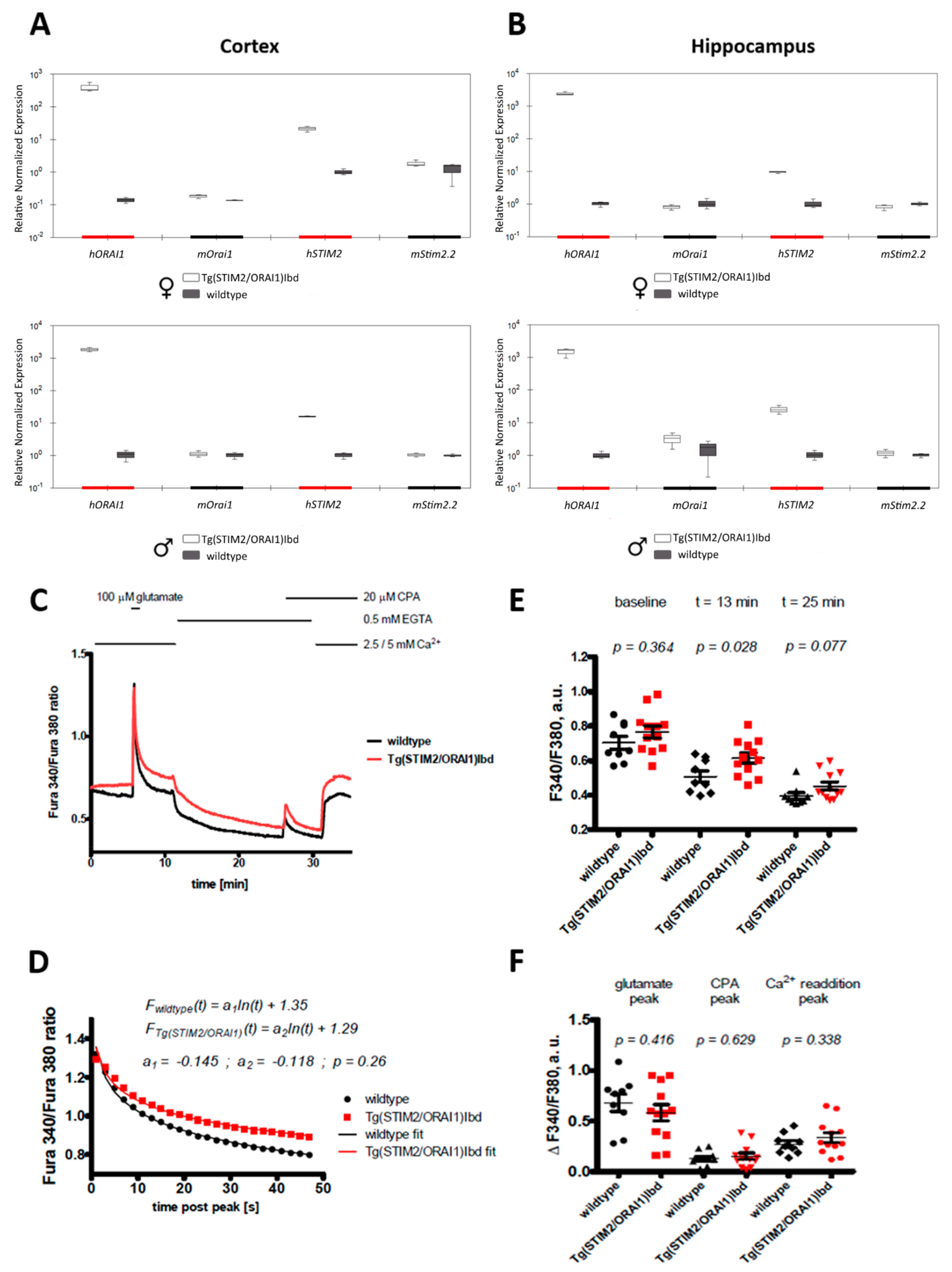
2.1. Overexpression of ORAI1 and STIM2 in Neurons Leads to Altered Ca2+ Response in a Modified Ca2+ Addback Assay in CA1 Hippocampal Region
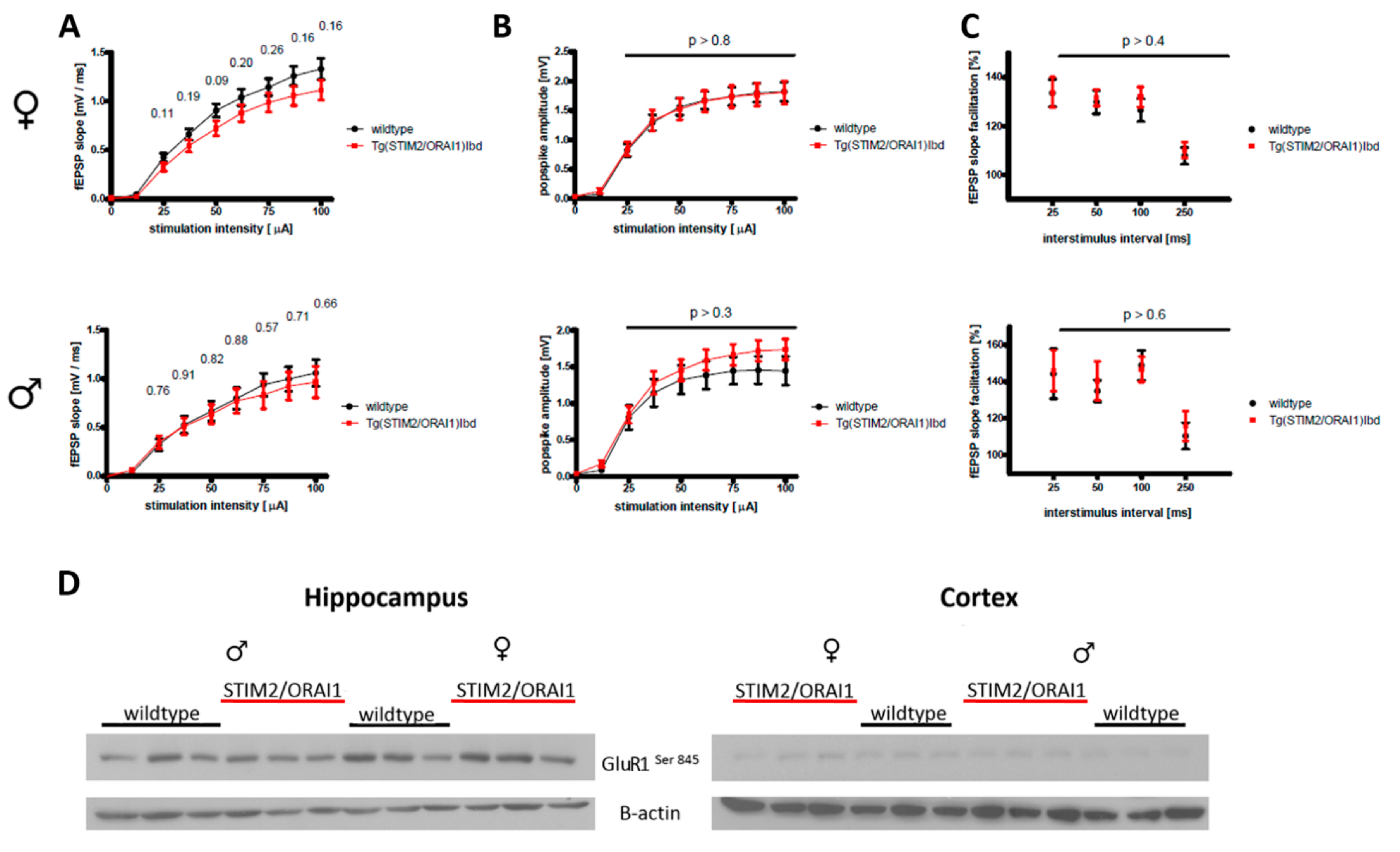
2.2. Impairment of Basal Synaptic Transmission in Adult Female Mice Overexpressing STIM2 and ORAI1
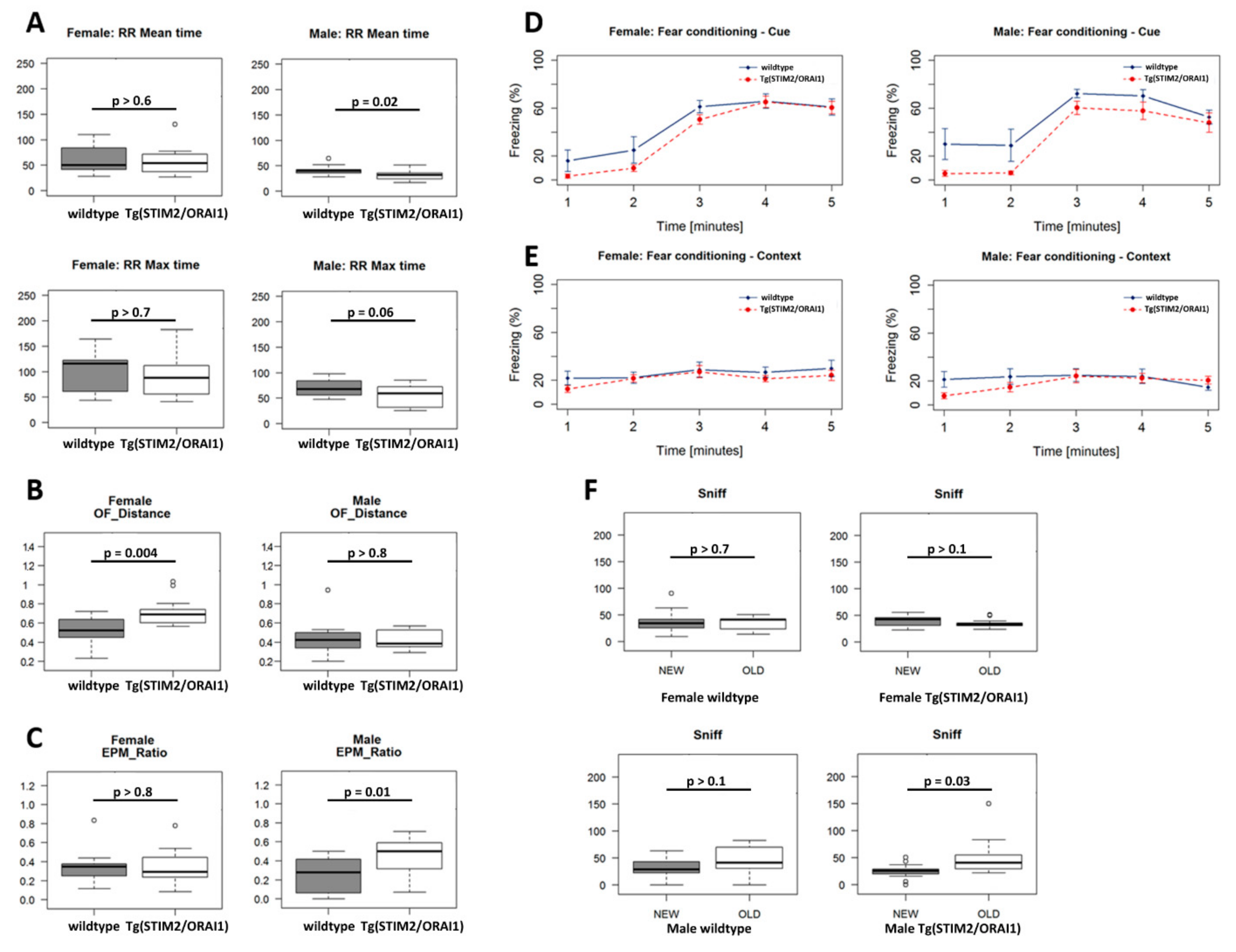
2.3. Modest Changes in Behavior of STIM2/ORAI1 Animals
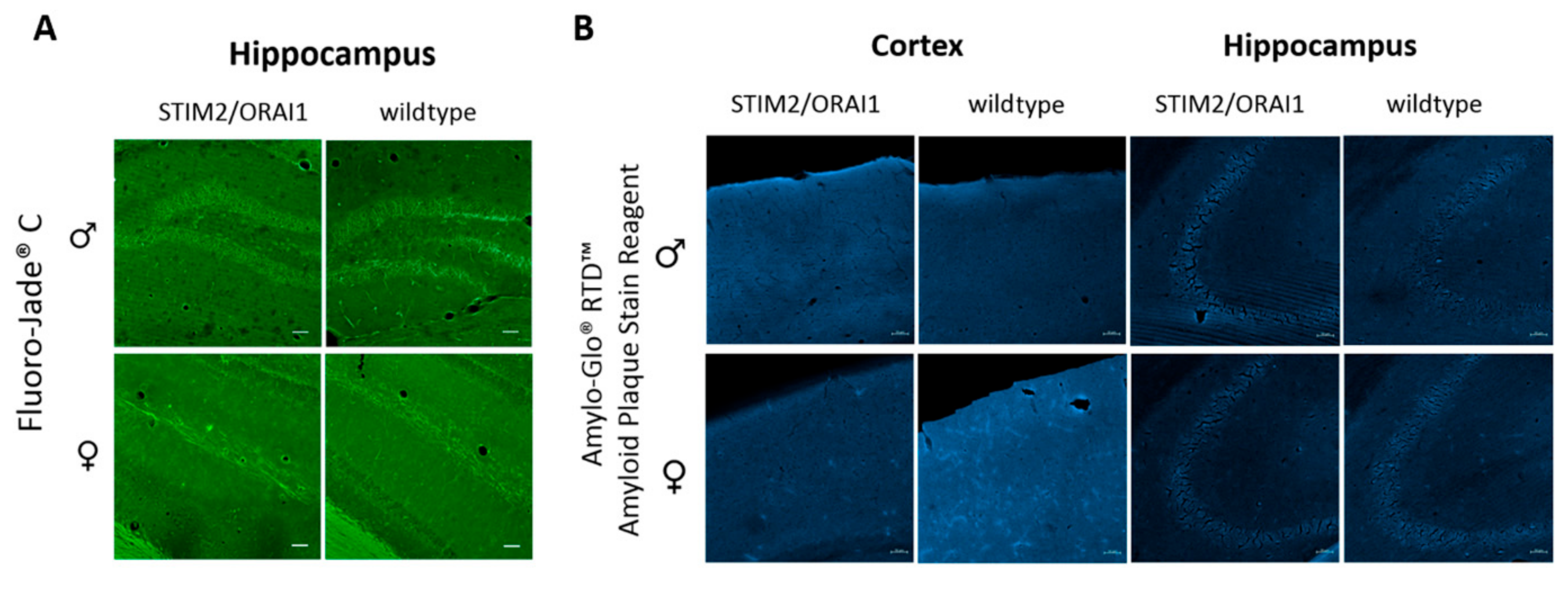
2.4. No Features of Neurodegeneration in Tg(STIM2/ORAI1)Ibd Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Animal Care
4.3. Generation of FVB/NJ–Tg(STIM2/ORAI1)Ibd Transgenic Mice
4.4. Real-Time PCR Analysis for Neuronal Confirmation of Expression of Both Transgenes
4.5. Protein Isolation and Immunoblotting
4.6. Perfusion of Mice and Brain Sectioning
4.7. Immunohistochemistry
4.8. Behavioral Analysis
4.9. Brain Slice Preparation, Ca2+ Imaging, and Electrophysiology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| aCSF | artificial cerebrospinal fluid |
| AD | Alzheimer’s Disease |
| AM | acetomethyl |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPK | adenosine monophosphate-activated protein kinase |
| APP | amyloid precursor protein |
| a.u. | arbitrary units |
| CPA | cyclopiazonic acid |
| DHPG | (S)-3,5-Dihydroxyphenylglycine |
| EDTA | ethylenediaminetetraacetic acid |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| EPM | elevated plus maze |
| ER | endoplasmic reticulum |
| fEPSP | field excitatory postsynaptic potentials |
| FRAP | fluorescence recovery after photobleaching |
| GluR1 | glutamate receptor 1 |
| NCLX | mitochondrial Na+/Ca2+ exchanger |
| NFAT | nuclear factor of activated T-cells |
| NOR | novel object recognition |
| nSOCE | neuronal store-operated calcium entry |
| OF | open field |
| PCR | polymerase chain reaction |
| PB | phosphate buffer |
| qPCR | quantitative polymerase chain reaction |
| RR | rotarod |
| sAD | sporadic Alzheimer’s disease |
| SDS | sodium dodecyl sulfate |
| SOCE | store operated calcium entry |
| SOCs | store-operated channels |
| STIM | stromal interaction molecule |
| Tg | transgenic |
| TX-100 | Polyethylene glycol p-(1,1,3,3-tetramethylbutyl)-phenyl ether |
References
- Klejman, M.E.; Gruszczynska-Biegala, J.; Skibinska-Kijek, A.; Wisniewska, M.B.; Misztal, K.; Blazejczyk, M.; Bojarski, L.; Kuznicki, J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 2009, 54, 49–55. [Google Scholar] [CrossRef]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Majewski, L.; Kuznicki, J. SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta 2015, 1853, 1940–1952. [Google Scholar] [CrossRef] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.N.; Krogh, M.; Toresson, H. Dendritic EGFP-STIM1 activation after type I metabotropic glutamate and muscarinic acetylcholine receptor stimulation in hippocampal neuron. J. Neurosci. Res. 2011, 89, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Kuznicki, J. Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J. Neurochem. 2013, 126, 727–738. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cardenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [Green Version]
- Wojda, U.; Kuznicki, J. Alzheimer’s disease modeling: Ups, downs, and perspectives for human induced pluripotent stem cells. J. Alzheimers Dis. 2013, 34, 563–588. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [Green Version]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182. [Google Scholar]
- Mattson, M.P.; Engle, M.G.; Rychlik, B. Effects of elevated intracellular calcium levels on the cytoskeleton and tau in cultured human cortical neurons. Mol. Chem. Neuropathol. 1991, 15, 117–142. [Google Scholar] [CrossRef]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.D.; Ruefli, A.A.; Parker, C.A.; Cypess, A.M.; Greengard, P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc. Natl. Acad. Sci. USA 1994, 91, 4489–4493. [Google Scholar] [CrossRef] [Green Version]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; Selkoe, D.J. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 1994, 33, 4550–4561. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Bezprozvanny, I. Role of endoplasmic reticulum Ca2+ signaling in the pathogenesis of Alzheimer disease. Front. Mol. Neurosci. 2013, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Olmedillas Del Moral, M.; Garaschuk, O. Intracellular Ca(2+) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, e1279–e1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazda, K.; Kuznicki, J.; Wegierski, T. Knockdown of amyloid precursor protein increases calcium levels in the endoplasmic reticulum. Sci. Rep. 2017, 7, 14512. [Google Scholar] [CrossRef] [Green Version]
- Mody, I.; MacDonald, J.F. NMDA receptor-dependent excitotoxicity: The role of intracellular Ca2+ release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, G.E. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist 2007, 13, 546–559. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria control store-operated Ca(2+) entry through Na(+) and redox signals. Embo J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L., 3rd; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging (Milano) 1989, 1, 17–34. [Google Scholar] [CrossRef]
- Majewski, L.; Wojtas, B.; Maciag, F.; Kuznicki, J. Changes in Calcium Homeostasis and Gene Expression Implicated in Epilepsy in Hippocampi of Mice Overexpressing ORAI1. Int. J. Mol. Sci. 2019, 20, E5539. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Maciag, F.; Majewski, L.; Boguszewski, P.M.; Gupta, R.K.; Wasilewska, I.; Wojtas, B.; Kuznicki, J. Behavioral and electrophysiological changes in female mice overexpressing ORAI1 in neurons. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1137–1150. [Google Scholar] [CrossRef]
- Majewski, L.; Maciag, F.; Boguszewski, P.M.; Wasilewska, I.; Wiera, G.; Wojtowicz, T.; Mozrzymas, J.; Kuznicki, J. Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1071–1087. [Google Scholar] [CrossRef]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.; Adelsberger, H.; Brill, M.S.; Ruhlmann, C.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca(2)(+) signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alvarez, G.; Lu, B.; Yap, K.A.; Wong, L.C.; Thevathasan, J.V.; Lim, L.; Ji, F.; Tan, K.W.; Mancuso, J.J.; Tang, W.; et al. STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol. Biol. Cell 2015, 26, 1141–1159. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.; Jang, D.C.; Jung, D.; Kim, Y.G.; Shim, H.G.; Ryu, H.H.; Lee, Y.S.; Linden, D.J.; Worley, P.F.; Kim, S.J. STIM1 Regulates Somatic Ca(2+) Signals and Intrinsic Firing Properties of Cerebellar Purkinje Neurons. J. Neurosci. 2017, 37, 8876–8894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, M.; Korkotian, E. Roles of Calcium Stores and Store-Operated Channels in Plasticity of Dendritic Spines. Neuroscientist 2016, 22, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Sladowska, M.; Kuznicki, J. AMPA Receptors Are Involved in Store-Operated Calcium Entry and Interact with STIM Proteins in Rat Primary Cortical Neurons. Front. Cell Neurosci. 2016, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Kreiner, G. Compensatory mechanisms in genetic models of neurodegeneration: Are the mice better than humans? Front. Cell Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Chen-Engerer, H.J.; Hartmann, J.; Karl, R.M.; Yang, J.; Feske, S.; Konnerth, A. Two types of functionally distinct Ca(2+) stores in hippocampal neurons. Nat. Commun. 2019, 10, 3223. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Fon Tacer, K.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [Green Version]
- Sanati, M.; Khodagholi, F.; Aminyavari, S.; Ghasemi, F.; Gholami, M.; Kebriaeezadeh, A.; Sabzevari, O.; Hajipour, M.J.; Imani, M.; Mahmoudi, M.; et al. Impact of Gold Nanoparticles on Amyloid beta-Induced Alzheimer’s Disease in a Rat Animal Model: Involvement of STIM Proteins. ACS Chem. Neurosci. 2019, 10, 2299–2309. [Google Scholar] [CrossRef]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Song, J.; Wu, S. Autophagic degradation of stromal interaction molecule 2 mediates disruption of neuronal dendrites by endoplasmic reticulum stress. J. Neurochem. 2019, 151, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.C.; Wan, B.Y.; Zeng, Y. STIM2 knockdown protects against ischemia/reperfusion injury through reducing mitochondrial calcium overload and preserving mitochondrial function. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Oni-Biton, E.; Segal, M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 2017, 595, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, M.; Morris, R.; Grosveld, F.; Spanopoulou, E. Tissue-specific control elements of the Thy-1 gene. Embo J. 1990, 9, 833–840. [Google Scholar] [CrossRef]
- Caroni, P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J. Neurosci. Methods 1997, 71, 3–9. [Google Scholar] [CrossRef]
- Murphy, D.; Carter, D.A. An overview of transgenic mouse production. Methods Mol. Biol. 1993, 18, 111–114. [Google Scholar]
- Kholodar-Smith, D.B.; Boguszewski, P.; Brown, T.H. Auditory trace fear conditioning requires perirhinal cortex. Neurobiol. Learn Mem. 2008, 90, 537–543. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majewski, L.; Maciąg, F.; Boguszewski, P.M.; Kuznicki, J. Transgenic Mice Overexpressing Human STIM2 and ORAI1 in Neurons Exhibit Changes in Behavior and Calcium Homeostasis but Show No Signs of Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 842. https://doi.org/10.3390/ijms21030842
Majewski L, Maciąg F, Boguszewski PM, Kuznicki J. Transgenic Mice Overexpressing Human STIM2 and ORAI1 in Neurons Exhibit Changes in Behavior and Calcium Homeostasis but Show No Signs of Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(3):842. https://doi.org/10.3390/ijms21030842
Chicago/Turabian StyleMajewski, Lukasz, Filip Maciąg, Pawel M. Boguszewski, and Jacek Kuznicki. 2020. "Transgenic Mice Overexpressing Human STIM2 and ORAI1 in Neurons Exhibit Changes in Behavior and Calcium Homeostasis but Show No Signs of Neurodegeneration" International Journal of Molecular Sciences 21, no. 3: 842. https://doi.org/10.3390/ijms21030842
APA StyleMajewski, L., Maciąg, F., Boguszewski, P. M., & Kuznicki, J. (2020). Transgenic Mice Overexpressing Human STIM2 and ORAI1 in Neurons Exhibit Changes in Behavior and Calcium Homeostasis but Show No Signs of Neurodegeneration. International Journal of Molecular Sciences, 21(3), 842. https://doi.org/10.3390/ijms21030842