Bumetanide Suppression of Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy



Abstract
:1. Introduction
2. Results
2.1. Effect of Bumetanide on Eye Opening
2.2. Effect of Bumetanide on Growth
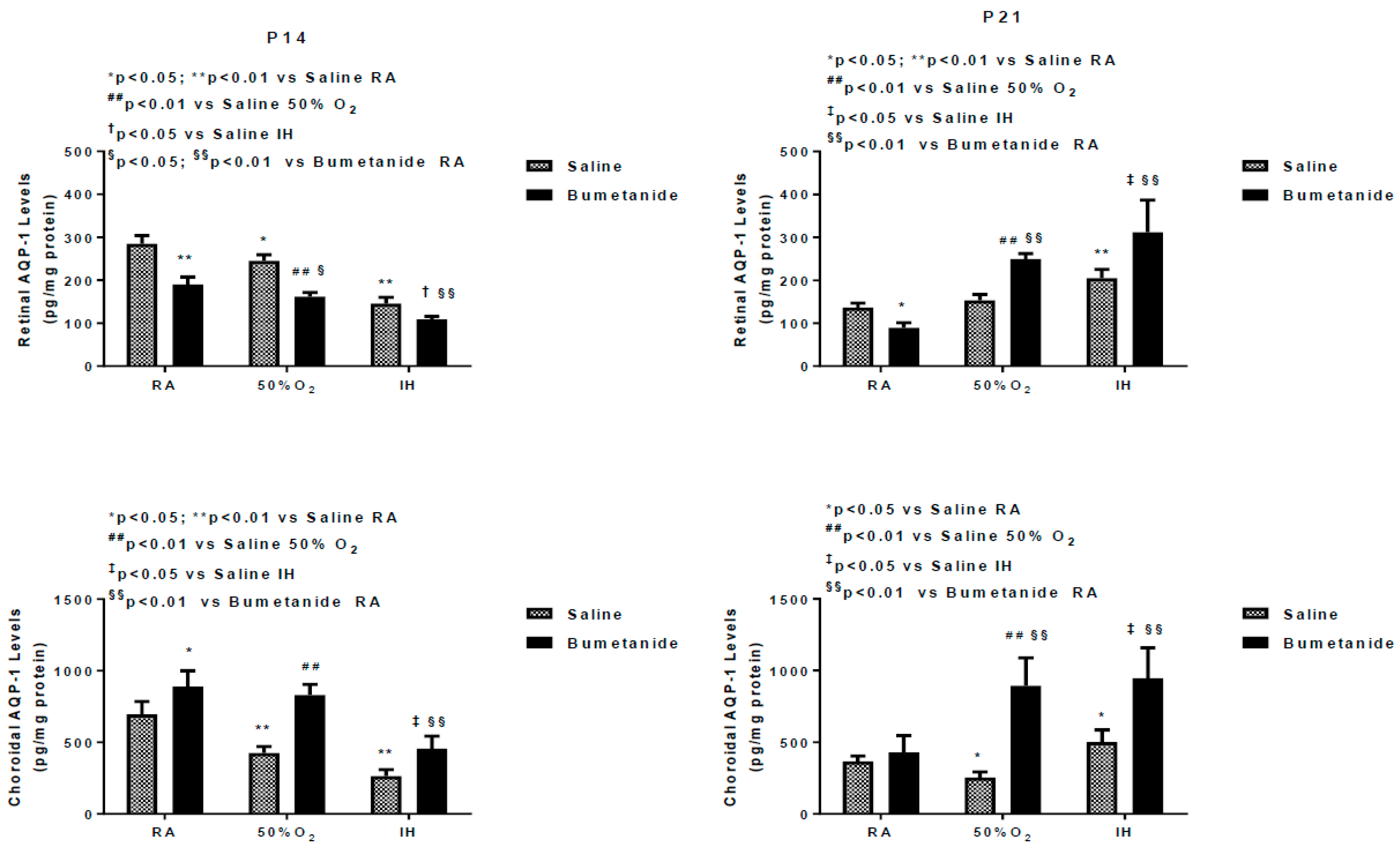
2.3. Effect of Bumetanide on AQPs
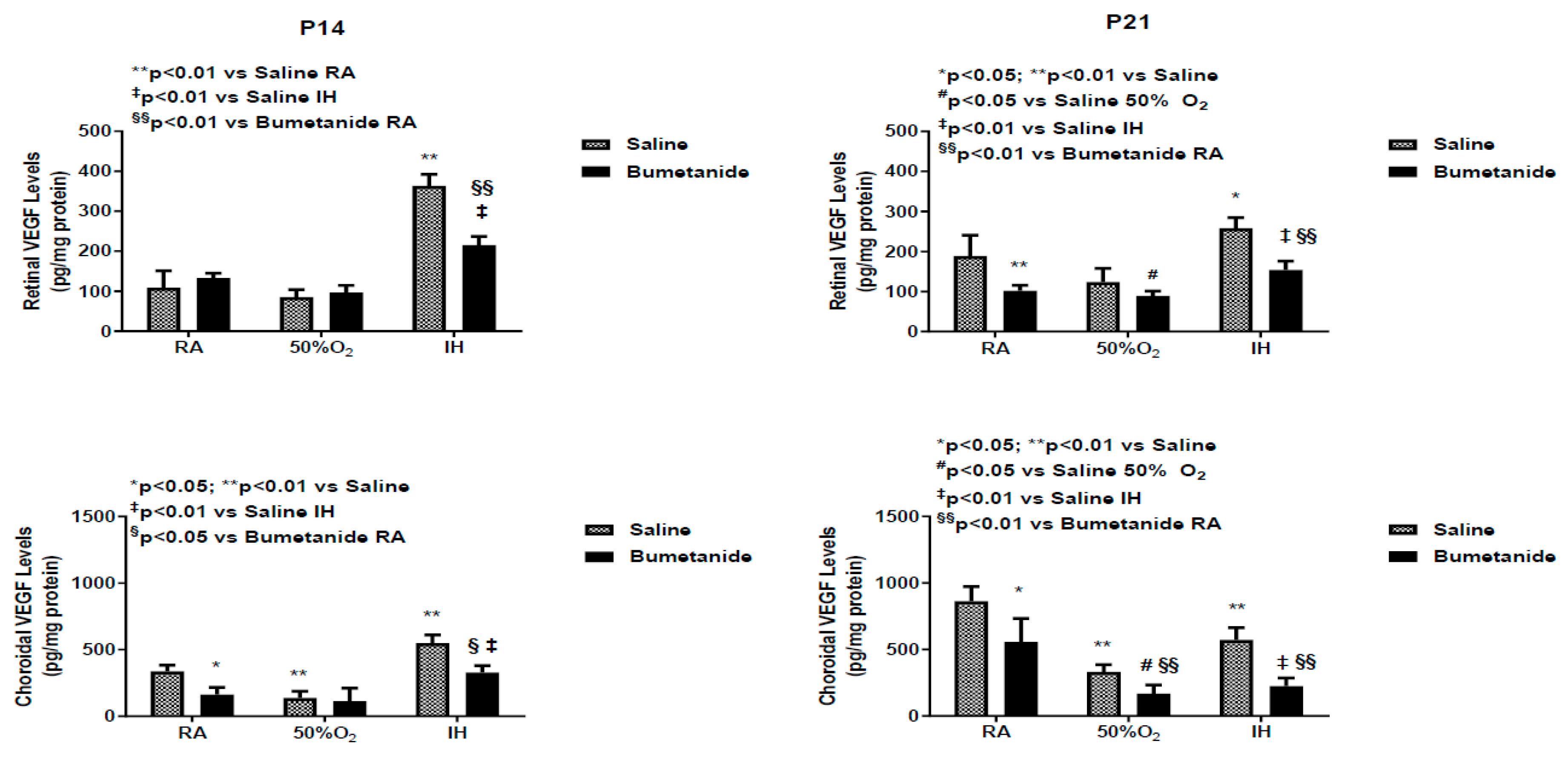
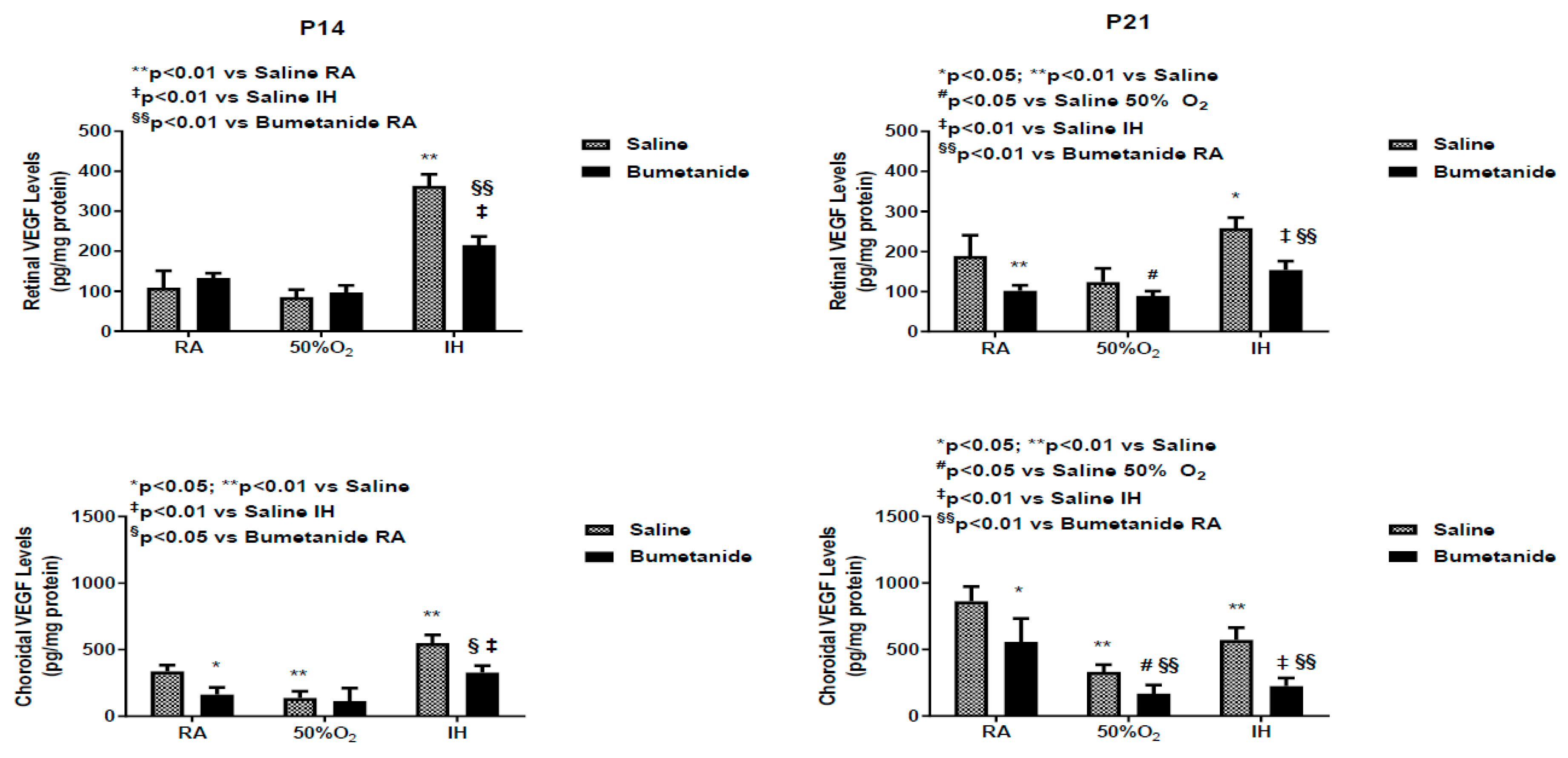
2.4. Effect of Bumetanide on VEGF
2.5. Effect of Bumetanide on sVEGFR-1
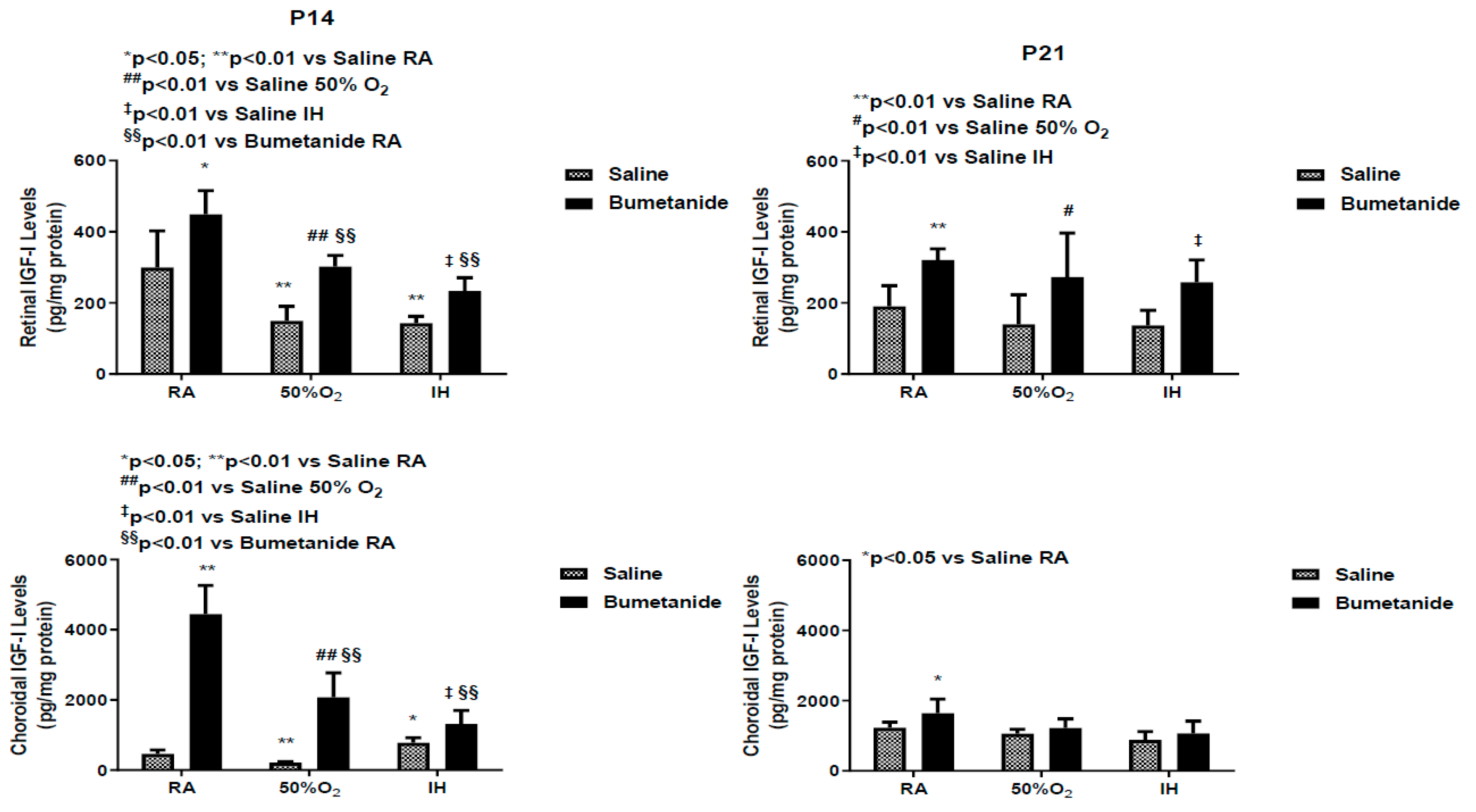
2.6. Effect of Bumetanide on IGF-1 Levels
2.7. Effect of Bumetanide on Retinal Vasculature
2.8. Effect of Bumetanide on Retinal Layers
2.9. Effect of Bumetanide on Retinal Morphometry
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Hyperoxia or IH Cycling
4.3. Sample Collection and Processing
4.4. Aquaporin Assays
4.5. Assay of Angiogenesis Biomarkers
4.6. Total Cellular Protein Levels
4.7. ADPase Staining of Retinal Flatmounts
4.8. Retinal Morphometric Analyses
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADPase | Adenosine diphosphatase |
| ANOVA | Analysis of variance |
| AQPs | Aquaporins |
| ARVO ELISA GCL | Association for research in vision and ophthalmology Enzyme-linked immunosorbent assay Ganglion cell layer |
| GFAP | Glial fibrillary acidic protein |
| H&E IGF-I | Hematoxylin and eosin Insulin-like growth factor-1 |
| IH | Intermittent hypoxia |
| INL | Inner nuclear layer |
| IP | Intraperitoneal |
| IPL | Inner plexiform layer |
| KCC2 | K+Cl2 cotransporter isoform 2 |
| NFL | Nerve fiber layer |
| NKCC1 | Na+/K+/Cl− cotransporter |
| O2 OIR | Oxygen Oxygen-induced retinopathy |
| ONL | Outer nuclear layer |
| OPL | Outer plexiform layer |
| P14 | Postnatal day 14 |
| P21 | Postnatal day 21 |
| P0 RA R/C | Postnatal age at birth Room air Rods and cones |
| ROP | Retinopathy of prematurity |
| SEM | Standard error of the mean |
| SPSS sVEGFR-1 | Statistical package for social sciences Soluble vascular endothelial growth factor receptor-1 |
| VEGF | Vascular endothelial growth factor |
References
- Gilbert, C. Retinopathy of prematurity: A global perspective of the epidemics, population of babies at risk and implications for control. Early Hum. Dev. 2008, 84, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Fry, M.; Al-Samarraie, M.; Gilbert, C.; Steinkuller, P.G. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J. AAPOS 2012, 16, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Group TS-RMS. Supplemental Therapeutic Oxygen for Prethreshold Retinopathy of Prematurity (STOP-ROP), a randomized, controlled trial. I: Primary outcomes. Pediatrics 2000, 105, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E.; Lane, R.H. Effects of oxygen on the development and severity of retinopathy of prematurity. J. AAPOS 2013, 17, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Whyte, R.K.; Asztalos, E.V.; Moddemann, D.; Poets, C.; Rabi, Y.; Solimano, A.; Roberts, R.S.; Canadian Oxygen Trial (COT) Group. Canadian Oxygen Trial (COT) Group. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: A randomized clinical trial. JAMA 2013, 309, 2111–2120. [Google Scholar] [CrossRef]
- Chen, J.; Smith, L.E. Retinopathy of prematurity. Angiogenesis 2007, 10, 133–140. [Google Scholar] [CrossRef] [Green Version]
- York, J.R.; Landers, S.; Kirby, R.S.; Arbogast, P.G.; Penn, J.S. Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birthweight infants. J. Perinatol. 2004, 24, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, J.M.; Kaffashi, F.; Loparo, K.; Sattar, A.; Schluchter, M.; Foglyano, R.; Martin, R.J.; Wilson, C.G. The relationship between the patterns of intermittent hypoxia and retinopathy of prematurity. Pediatr. Res. 2012, 72, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.J.; Di Fiore, J.M.; Macfarlane, P.M.; Wilson, C.G. Physiologic basis for intermittent hypoxic episodes in preterm infants. Adv. Exp. Med. Biol. 2012, 758, 351–358. [Google Scholar]
- Hellstrom, A.; Hard, A.L.; Engstrom, E.; Niklasson, A.; Andersson, E.; Smith, L.; Lofqvist, C. Early weight gain predicts retinopathy in preterm infants: New, simple, efficient approach to screening. Pediatrics 2009, 123, e638–e645. [Google Scholar] [CrossRef] [Green Version]
- Penn, J.S. Oxygen-induced retinopathy in the rat: Possible contribution of peroxidation reactions. Doc. Ophthalmol. 1990, 74, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Phelps, D.L. Vitamin E and retinopathy of prematurity: The clinical investigator’s perspective on antioxidant therapy: Side effects and balancing risks and benefits. Birth Defects Orig. Artic. Ser. 1988, 24, 209–218. [Google Scholar] [PubMed]
- Raju, T.N.K.; Langenberg, P.; Bhutani, V.; Quinn, G.E. Vitamin E prophylaxis to reduce retinopathy of prematurity: A reappraisal of published trials. J. Pediatrics 1997, 131, 844–850. [Google Scholar] [CrossRef]
- Early Treatment for Retinopathy of Prematurity Cooperative Group. Revised indications for the treatment of retinopathy of prematurity: Results of the early treatment for retinopathy of prematurity randomized trial. Arch. Ophthalmol. 2003, 121, 1684–1694. [Google Scholar] [CrossRef] [Green Version]
- Cryotherapy for Retinopathy of Prematurity Cooperative Group. Multicenter trial of cryotherapy for retinopathy of prematurity: Snellen visual acuity and structural outcome at 51/2 years after randomization. Arch. Ophthalmol. 1996, 114, 417–424. [Google Scholar] [CrossRef]
- Hartnett, M.E. Vascular endothelial growth factor antagonist therapy for ROP. Clin. Perinatol. 2014, 41, 925–943. [Google Scholar] [CrossRef] [Green Version]
- Morin, J.; Luu, T.M.; Superstein, R.; Ospina, L.H.; Lefebvre, F.; Simard, M.N.; Shah, V.; Shah, P.S.; Kelly, E.N.; Canadian Neonatal Network and the Canadian Neonatal Follow-Up Network Investigators. Neurodevelopmental outcomes following bevacizumab injections for retinopathy of prematurity. Pediatrics 2016, 137, e20153218. [Google Scholar] [CrossRef] [Green Version]
- Schey, K.L.; Wang, Z.L.; Wenke, J.; Qi, Y. Aquaporins in the eye: Expression, function, and roles in ocular disease. Biochim. Biophys. Acta 2014, 1840, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and Cell Migration. Pflugers Arch. 2008, 456, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Tenckhoff, S.; Hollborn, M.; Kohen, L.; Wolf, S.; Wiedemann, P.; Bringmann, A. Diversity of aquaporin mRNA expressed by rat and human retinas. Neuroreport 2005, 16, 53–56. [Google Scholar] [CrossRef]
- Patil, R.V.; Saito, I.; Yang, X.; Wax, M.B. Expression of aquaporins in the rat ocular tissue. Exp. Eye Res. 1997, 64, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Ruiz-Ederra, J.; Levin, M.H. Functions of Aquaporins in the Eye. Prog. Retin Eye Res. 2008, 27, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.J.; Tsujita, M.; Nakada, T. Aquaporins in drug discovery and pharmacotherapy. Mol. Aspects Med. 2012, 33, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Jullien, V.; Pressler, R.M.; Boylan, G.; Blennow, M.; Marlow, N.; Chiron, C.; Pons, G.; NEMO Consortium Neonatal Seizure Treatment With Medication Off-Patent. Pilot evaluation of the population pharmacokinetics of bumetanide in the term newborn infants with seizure. J. Clin. Pharmacol. 2016, 56, 284–290. [Google Scholar] [CrossRef]
- Glass, H.C. Bumetanide for treatment of seizures in neonates. Lancet Neurol. 2015, 14, 456–457. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef]
- Zhang, L.L.; Pathak, H.R.; Coulterm, D.A.; Freed, M.A.; Vardi, N. Shift of intracellular chloride concentration in ganglion and amacrine cells of developing mouse retina. J. Neurophysiol. 2006, 95, 2404–2416. [Google Scholar] [CrossRef] [Green Version]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Liu, J.; Wang, X.; Li, W.; Chen, J.; Sun, H. Pretreatment with AQP4 and NKCC1 inhibitors concurrently attenuated spinal cord edema and tissue damage after spinal cord injury in rats. Front. Physiol. 2018, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.J.; Beharry, K.D.; Brock, R.; Abad-Santos, P.; Abad-Santos, M.; Modanlou, H.D. Effects of brief, clustered versus dispersed hypoxic episodes on systemic and ocular growth factors in a rat model of oxygen-induced retinopathy. Pediatr. Res. 2008, 64, 1–6. [Google Scholar] [CrossRef]
- Aranda, J.V.; Cai, C.L.; Ahmad, T.; Bronshtein, V.; Sadeh, J.; Valencia, G.B.; Lazzaro, D.R.; Beharry, K.D. Pharmacologic synergism of ocular ketorolac and systemic caffeine citrate in rat oxygen-induced retinopathy. Pediatr. Res. 2016, 80, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Beharry, K.D.; Cai, C.L.; Sharma, P.; Bronshtein, V.; Valencia, G.B.; Lazzaro, D.R.; Aranda, J.V. Hydrogen peroxide accumulation in the choroid during intermittent hypoxia increases risk of severe oxygen-induced retinopathy in neonatal rats. Invest. Ophthalmol. Vis. Sci. 2013, 54, 7644–7657. [Google Scholar] [CrossRef] [Green Version]
- Beharry, K.D.; Cai, C.L.; Ahmad, T.; Guzel, S.; Valencia, G.B.; Aranda, J.V. Impact of chronic neonatal intermittent hypoxia on severity of retinal damage in a rat model of oxygen-induced retinopathy. J. Nat. Sci. 2018, 4, e488. [Google Scholar]
- Beharry, K.D.; Cai, C.L.; Skelton, J.; Siddiqui, F. Christina D’Agrosa,1 Johanna Calo,1 Gloria B. Valencia,1 and Jacob V. Aranda Oxygen-induced retinopathy from recurrent intermittent hypoxia is not dependent on resolution with room air or oxygen, in neonatal rats. Int. J. Mol. Sci. 2018, 19, 1337. [Google Scholar] [CrossRef] [Green Version]
- Modanlou, H.D.; Gharraee, Z.; Hasan, J.; Waltzman, J.; Nageotte, S.; Beharry, K.D. Ontogeny of VEGF, IGF-I, and GH in neonatal rat serum, vitreous fluid, and retina from birth to weaning. Invest. Ophthalmol. Vis. Sci. 2006, 47, 738–744. [Google Scholar] [CrossRef]
- Jensen, A.K.; Ying, G.S.; Huang, J.; Quinn, G.E.; Binenbaum, G. Postnatal serum insulin-like growth factor I and retinopathy of prematurity. Retina 2017, 37, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Hellström, A.; Engström, E.; Hård, A.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Löfqvist, C.; Svensson, E.; Holm, S.; Ewald, U.; et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics 2003, 112, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Pressler, R.M.; Boylan, G.B.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; de Vries, L.S.; Hallberg, B.; Hellström-Westas, L.; Jullien, V.; et al. Neonatal seizure treatment with Medication Off-patent (NEMO) consortium. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): An open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef]
- Pisani, F.; Cammalleri, M.; Dal Monte, M.; Locri, F.; Mola, M.G.; Nicchia, G.P.; Frigeri, A.; Bagnoli, P.; Svelto, M. Potential role of the methylation of VEGF gene promoter in response to hypoxia in oxygen-induced retinopathy: Beneficial effect of the absence of AQP4. J. Cell. Mol. Med. 2018, 22, 613–627. [Google Scholar] [CrossRef]
- Kida, T.; Oku, H.; Horie, T.; Fukumoto, M.; Okuda, Y.; Morishita, S.; Ikeda, T. Implication of VEGF and aquaporin 4 mediating Müller cell swelling to diabetic retinal edema. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1149–1157. [Google Scholar] [CrossRef]
- Da, T.; Verkman, A.S. Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest. Ophthalmol. Vis. Sci. 2004, 45, 4477–4483. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Y.T.; Chen, S.N. Foveal microvascular anomalies on optical coherence tomography angiography and the correlation with foveal thickness and visual acuity in retinopathy of prematurity. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 23–30. [Google Scholar] [CrossRef]
- Nambu, H.; Umeda, N.; Kachi, S.; Oshima, Y.; Akiyama, H.; Nambu, R.; Campochiaro, P.A. Angiopoietin 1 prevents retinal detachment in an aggressive model of proliferative retinopathy, but has no effect on established neovascularization. J. Cell. Physiol. 2005, 204, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Ruizz-Ederra, J.; Verkman, A.S. Aquaporin-1 independent microvessel proliferation in a neonatal mouse model of oxygen-induced retinopathy. Invest. Ophthalmol. Vis. Sci. 2007, 48, 4802–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzani, E.; Bazzotti, R.; Perego, C.; La Porta, C.A. AQP1 is not only a water channel, it contributes to cell migration through Lin7/B catenin. PLoS ONE 2009, 4, e6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iandiev, I.; Pannicke, T.; Biedermann, B.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Ischemia–reperfusion alters the immunolocalization of glial aquaporins in rat retina. Neurosci. Lett. 2006, 408, 108–112. [Google Scholar] [CrossRef]
- Bringmann, A.; Uckermann, O.; Pannicke, T.; Iandiev, I.; Reichenbach, A.; Wiedemann, P. Neuronal versus glial cell swelling in the ischemic retina. Acta Ophthalmol. Scand. 2005, 83, 528–538. [Google Scholar] [CrossRef]
- Zou, Y.Y.; Kan, E.M.; Lu, J.; Ng, K.C.; Tan, M.H.; Yao, L.; Ling, E.-A. Primary blast injury-induced lesions in the retina of adult rats. J. Neuroinflammation. 2013, 10, 792013. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [Green Version]
- Lip, P.L.; Belgore, F.; Blann, A.D.; Hope-Ross, M.W.; Gibson, J.M.; Lip, G.Y.H. Plasma VEGF and soluble VEGF receptor FLT-1 in proliferative retinopathy: Relationship to endothelial dysfunction and laser treatment. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2115–2119. [Google Scholar]
- Panet, R.; Atlan, H. Stimulation of bumetanide-sensitive Na+/K+/Cl- cotransport by different mitogens in synchronized human skin fibroblasts is essential for cell proliferation. J. Cell Biol. 1991, 114, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, L.; Cenni, M.C.; Melani, R.; Deidda, G.; Landi, S.; Narducci, R.; Cancedda, L.; Maffei, L.; Berardi, N. Early IGF1 primes visual cortex maturation and accelerates developmental switch between NKCC1 and KCC2 chloride transporters in enriched animals. Neuropharmacology 2017, 113, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, A.L.; Joly, S.; Hardy, P.; Chemtob, S.; Lachapelle, P. The effect of oxygen and light on the structure and function of the neonatal rat retina. Doc. Ophthalmol. 2009, 118, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Binns, K.E.; Salt, T.E. Post eye-opening maturation of visual receptive field diameters in superior colliculus of normal- and dark-reared rats. Dev. Brain Res. 1997, 99, 263–266. [Google Scholar] [CrossRef]
- Zieske, J.D. Corneal Development associated with eyelid opening. Int. J. Dev. Biol. 2004, 48, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Löfqvist, C.; Hansen-Pupp, I.; Andersson, E.; Holm, K.; Smith, L.E.; Ley, D.; Hellström, A. Validation of a new retinopathy of prematurity screening method monitoring longitudinal postnatal weight and insulin like growth factor I. Arch. Ophthalmol. 2009, 127, 622–627. [Google Scholar] [CrossRef]
- Madan, A.; Penn, J.S. Animal models of oxygen-induced retinopathy. Front. Biosci. 2003, 8, d1030–d1043. [Google Scholar]
- Penn, J.S.; Henry, M.M.; Tolman, B.L. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr. Res. 1994, 36, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, J.M.; Bloom, J.N.; Orge, F.; Schutt, A.; Schluchter, M.; Cheruvu, V.K.; Walsh, M.; Finer, N.; Martin, R.J. A higher incidence of intermittent hypoxia/hypoxemic episodes is associated with severe retinopathy of prematurity. J Pediatr. 2010, 157, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Nakano, A.; Nakahara, T.; Mori, A.; Ushikubo, H.; Sakamoto, K.; Ishii, K. Short-term treatment with VEGF receptor inhibitors induces retinopathy of prematurity-like abnormal vascular growth in neonatal rats. Exp. Eye Res. 2016, 143, 120–131. [Google Scholar] [CrossRef]
- Beharry, K.D.; Cai, C.L.; Siddiqui, F.; Chowdhury, S.; D’Agrosa, C.; Valencia, G.B.; Aranda, J.V. Comparative Effects of Coenzyme Q10 or n-3 Polyunsaturated Fatty Acid Supplementation on Retinal Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy. Antioxidants (Basel) 2018, 7, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, A.; Wong, T.Y.; Khachigian, L.M. Emerging therapeutic approaches in the management of retinal angiogenesis and edema. J. Mol. Med. 2011, 89, 343–361. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
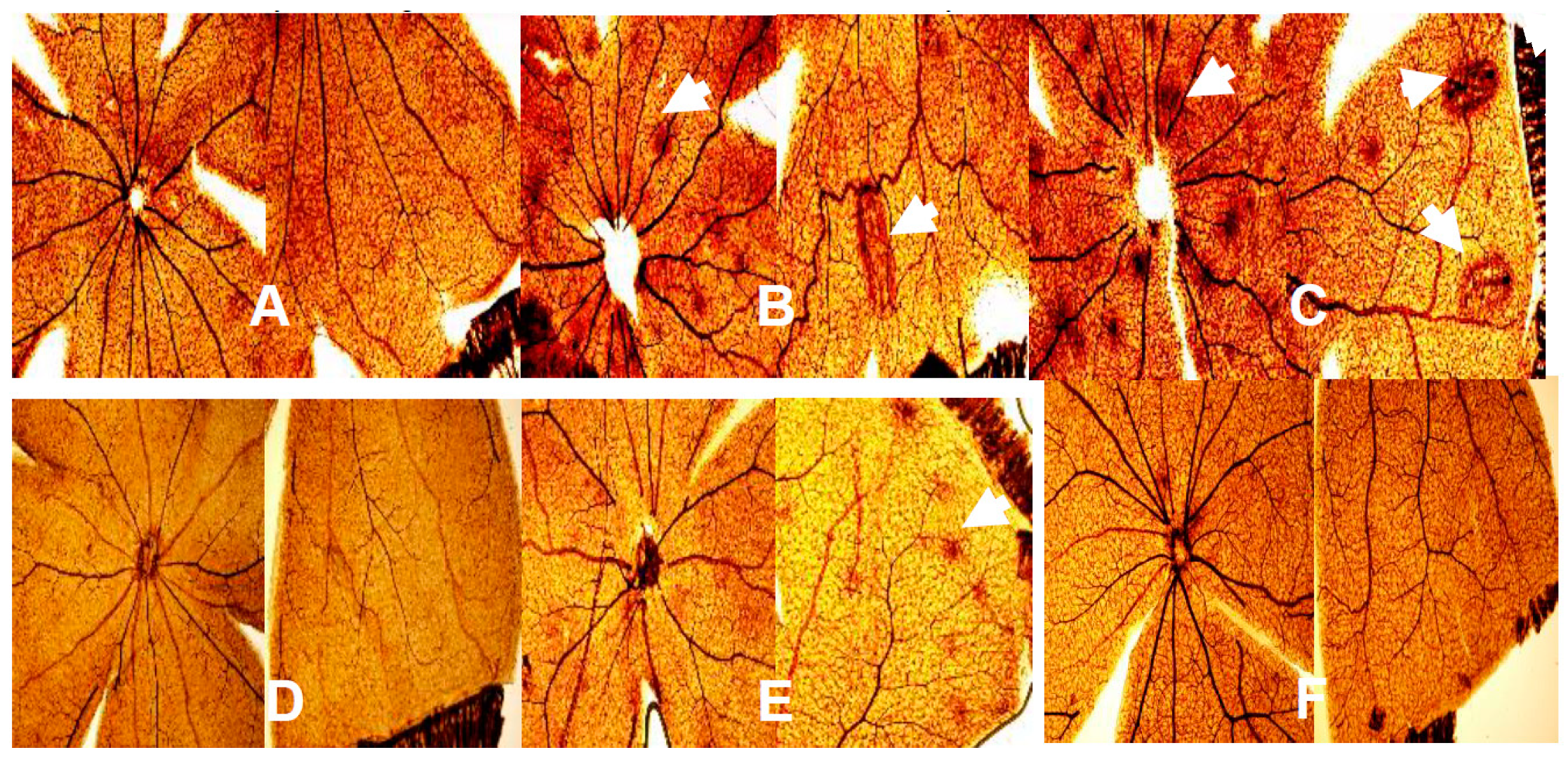{kind=link}
{kind=link}
| Eye | Saline RA | Bumetanide RA | Saline 50% O2 | Bumetanide 50% O2 | Saline IH | Bumetanide IH |
|---|---|---|---|---|---|---|
| Left Eye | 21 (58%) | 30 (83%) * | 22 (61%) | 23 (64%) | 13 (36%) | 31 (86%) ** |
| Right Eye | 22 (61%) | 33 (92%) ** | 24 (67%) | 23 (64%) | 11 (31%) | 28 (78%) ** |
| Both Eyes | 20 (56%) | 29 (81%) * | 23 (64%) | 22 (61%) | 4 (11%) | 27 (75%) ** |
| % Change from P0 | Saline RA | Bumetanide RA | Saline 50% O2 | Bumetanide 50% O2 | Saline IH | Bumetanide IH |
|---|---|---|---|---|---|---|
| P14 (n = 18/group): | ||||||
| Weight (g) | 229.5 ± 12.2 | 216.7 ± 8.2 | 306.4 ± 8.7 ** | 255.6 ± 11.5 §§ | 263.6 ± 11.4 | 232.1 ± 6.0 † |
| Length (cm) | 61.3 ± 2.2 | 62.0 ± 3.6 | 40.3 ± 1.5 ** | 55.0 ± 2.4 ## | 43.8 ± 1.6 | 54.1 ± 2.2 ‡ |
| P21 (n = 18/group): | ||||||
| Weight (g) | 444.7 ± 13.4 | 474.5 ± 19.7 | 456.9 ± 13.4 | 268.1 ± 10.1 ##,§§ | 376.0 ± 17.6 ** | 233.0 ± 7.6 §§,‡ |
| Length (cm) | 86.3 ± 4.2 | 100.9 ± 3.1 ** | 100.5 ± 1.2 ** | 64.1 ± 2.6 ##,§§ | 89.6 ± 2.7 | 59.6 ± 2.1 §§,‡ |
| Morphometric Parameters | Saline RA | Bumetanide RA | Saline 50% O2 | Bumetanide 50% O2 | Saline IH | Bumetanide IH |
|---|---|---|---|---|---|---|
| Tortuosity Index | 1.00 ± 0.01 | 1.02 ± 0.03 | 1.15 ± 0.01 ** | 1.04 ± 0.04 ‡ | 1.2 ± 0.01 ** | 1.04 ± 0.04 §§ |
| Diameter of Arteries | 36.6 ± 0.79 | 31.5 ± 3.3 | 39.7 ± 1.0 * | 32.4 ± 0.80 ‡ | 27.5 ± 0.88 ** | 28.1 ± 0.85 |
| Diameter of Veins (µm) | 45.2 ± 0.99 | 52.10 ± 4.5 | 23.1 ± 0.76 ** | 42.1 ± 1.6 #,‡ | 43.8 ± 1.2 | 38.6 ± 1.3 ##,§§ |
| Number of Cells in NFL/GCL (µm) | 210.0 ± 13.6 | 330.1 ± 5.4 ** | 424.8 ± 47.4 * | 344.4 ± 19.6 ‡ | 994.7 ± 93.4 ** | 571.5 ± 49.3 ##,§§ |
| Total Retinal Thickness (µm) | 263.5 ± 4.3 | 321.3 ± 7.0 ** | 364.0 ± 3.8 ** | 326.0 ± 8.7 ‡ | 441.8 ± 14.6 ** | 366.9 ± 7.9 ##,§§ |
| NFL/GCL Thickness (µm) | 42.4 ± 1.4 | 55.8 ± 2.2 ** | 61.8 ± 2.9 * | 57.3 ± 2.0 | 117.1 ± 8.8 ** | 64.4 ± 1.9 ##,§§ |
| IPL Thickness (µm) | 56.3 ± 0.88 | 77.2 ± 1.4 ** | 73.4 ± 3.3 ** | 64.7 ± 1.9 † | 63.8 ± 3.2 * | 63.4 ± 2.4 ## |
| INL Thickness (µm) | 48.4 ± 1.1 | 70.5 ± 2.0 ** | 70.5 ± 2.0 ** | 65.7 ± 1.4 ‡ | 78.1 ± 3.3 ** | 65.6 ± 1.9 ##,§§ |
| ONL Thickness (µm) | 67.5 ± 2.8 | 83.1 ± 1.8 ** | 102.6 ± 3.7 ** | 62.6 ± 1.7 ‡ | 110.8 ± 1.5 ** | 100.3 ± 1.7 ##,§§ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzel, S.; Cai, C.L.; Ahmad, T.; Quan, M.; Valencia, G.B.; Aranda, J.V.; Beharry, K.D. Bumetanide Suppression of Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy. Int. J. Mol. Sci. 2020, 21, 987. https://doi.org/10.3390/ijms21030987
Guzel S, Cai CL, Ahmad T, Quan M, Valencia GB, Aranda JV, Beharry KD. Bumetanide Suppression of Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy. International Journal of Molecular Sciences. 2020; 21(3):987. https://doi.org/10.3390/ijms21030987
Chicago/Turabian StyleGuzel, Sibel, Charles L. Cai, Taimur Ahmad, Michelle Quan, Gloria B. Valencia, Jacob V. Aranda, and Kay D. Beharry. 2020. "Bumetanide Suppression of Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy" International Journal of Molecular Sciences 21, no. 3: 987. https://doi.org/10.3390/ijms21030987
APA StyleGuzel, S., Cai, C. L., Ahmad, T., Quan, M., Valencia, G. B., Aranda, J. V., & Beharry, K. D. (2020). Bumetanide Suppression of Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy. International Journal of Molecular Sciences, 21(3), 987. https://doi.org/10.3390/ijms21030987