DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia

 ,
, 
Abstract
:1. Introduction
2. Results
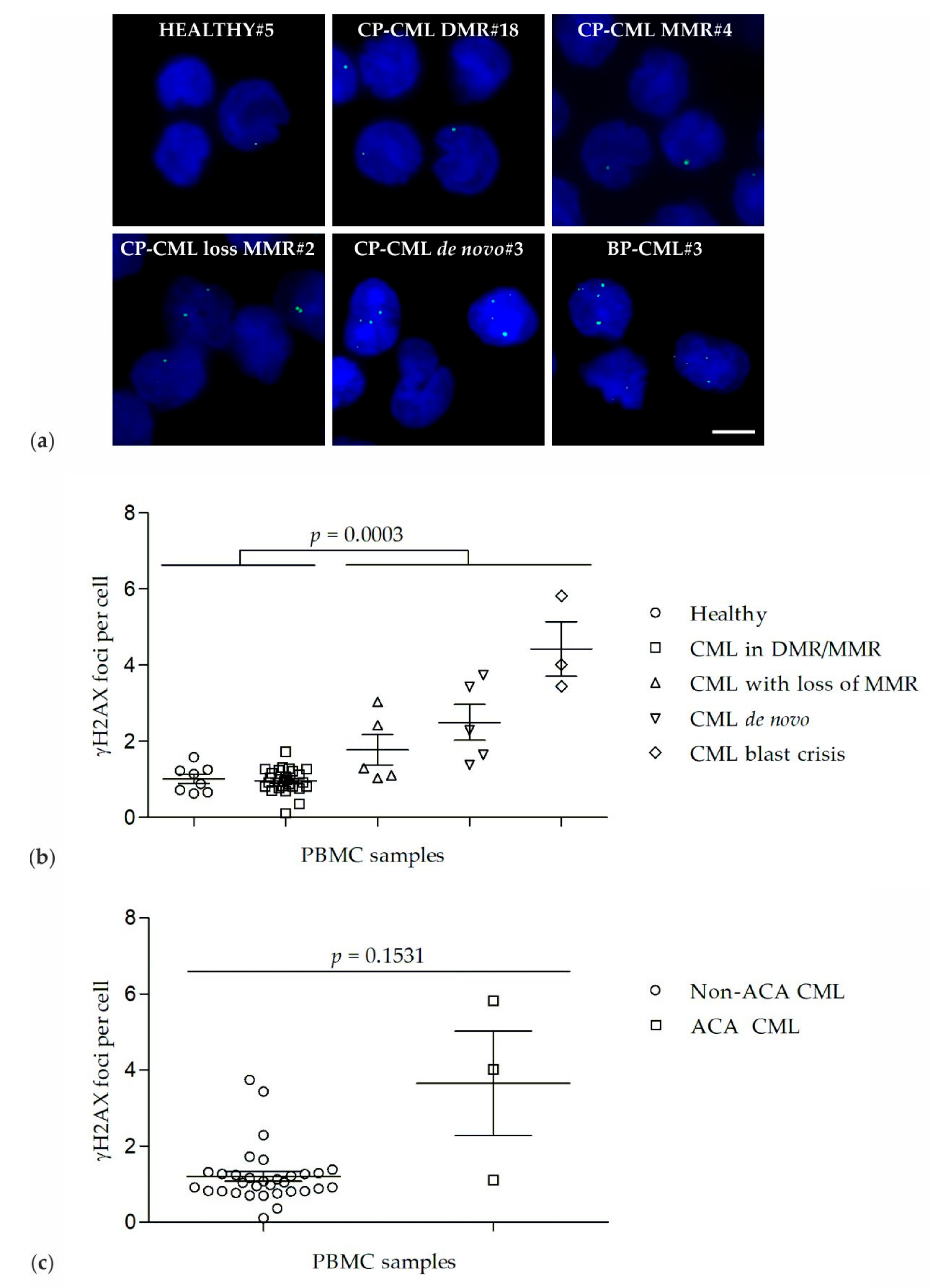
2.1. γH2AX Foci in PBMCs of Healthy Donors and CML Patients
2.2. Co-Localization of γH2AX and 53BP1 Foci
2.3. DNA Damage Response
3. Discussion
4. Materials and Methods
4.1. Blood Samples
4.2. Cytology, Cytogenetics, and Molecular Analyses
4.3. Immunofluorescence Staining of γH2AX and 53BP1
4.4. Western Blotting
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Accelerated phase |
| ACA | Additional chromosomal aberrations |
| BP | Blast phase |
| CML | Chronic myeloid leukemia |
| CP | Chronic phase |
| DASA | Dasatinib |
| DDR | DNA damage response |
| DMR | Deep molecular response |
| DSB | DNA double-strand breaks |
| IMA | Imatinib |
| MMEJ | Microhomology mediated end-joining |
| MMR | Major molecular response |
| MR | Molecular response |
| NHEJ | Non-homologous end joining |
| NILO | Nilotinib |
| PBMC | Peripheral blood mononuclear cells |
| PONA | Ponatinib |
| ROS | Reactive oxygen species |
| TKI | Tyrosine kinase inhibitor |
References
- Vardiman, J.W.; Melo, J.V.; Baccarani, M.; Radich, J.P.; Kvasnicka, H.M. Chronic myeloid leukaemia, BCR-ABL1-positive. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; pp. 30–36. [Google Scholar]
- NCCN. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. Available online: https://jnccn.org/view/journals/jnccn/16/9/article-p1108.xml (accessed on 05 November 2019).
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.; Nagaria, P.; Rassool, F.V. Genomic instability in chronic myeloid leukemia: Targets for therapy? Curr. Hematol. Malig. Rep. 2012, 7, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef] [Green Version]
- Stoklosa, T.; Poplawski, T.; Koptyra, M.; Nieborowska-Skorska, M.; Basak, G.; Slupianek, A.; Rayevskaya, M.; Seferynska, I.; Herrera, L.; Blasiak, J.; et al. BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res. 2008, 68, 2576–2580. [Google Scholar] [CrossRef] [Green Version]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Patel, A.B.; O’Hare, T.; Deininger, M.W. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol. Oncol. Clin. North. Am. 2017, 31, 589–612. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, J.; Tauchi, H.; Sakamoto, S.; Nakamura, A.; Morishima, K.; Matsuura, S.; Kobayashi, T.; Tamai, K.; Tanimoto, K.; Komatsu, K. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002, 12, 1846–1851. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Ward, I.M.; Minn, K.; Jorda, K.G.; Chen, J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 2003, 278, 19579–19582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Difilippantonio, S.; Gapud, E.; Wong, N.; Huang, C.Y.; Mahowald, G.; Chen, H.T.; Kruhlak, M.J.; Callen, E.; Livak, F.; Nussenzweig, M.C.; et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008, 456, 529–533. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Callen, E.; Di Virgilio, M.; Kruhlak, M.J.; Nieto-Soler, M.; Wong, N.; Chen, H.T.; Faryabi, R.B.; Polato, F.; Santos, M.; Starnes, L.M.; et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013, 153, 1266–1280. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F. The cytogenetic scenario of chronic myeloid leukemia. Leuk Lymphoma 1993, 11, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Leitner, A.; Hochhaus, A.; Muller, M.C.; Hanfstein, B.; Haferlach, C.; Gohring, G.; Schlegelberger, B.; Jotterand, M.; Reiter, A.; et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011, 118, 6760–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2013, 32, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Löffler, H.; Rastetter, J.; Haferlach, T. Light microscopic procedures. In Atlas of Clinical Hematology, 6th ed.; Springer: Heidelberg, Germany, 2005; p. 8. [Google Scholar]
- MLL. Request for Testing Form. Available online: https://www.mll.com/en.html (accessed on 05 November 2019).
- Gisselsson, D. Cytogenetic methods. In Cancer Cytogenetics, 3rd ed.; Heim, S., Mitelman, F., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; pp. 9–16. [Google Scholar]
- UMM. Request for Testing Form. Available online: https://www.umm.de/iii-medizinische-klinik/wissenschaftliches-labor/ (accessed on 05 November 2019).
- Emig, M.; Saussele, S.; Wittor, H.; Weisser, A.; Reiter, A.; Willer, A.; Berger, U.; Hehlmann, R.; Cross, N.C.; Hochhaus, A. Accurate and rapid analysis of residual disease in patients with CML using specific fluorescent hybridization probes for real time quantitative RT-PCR. Leukemia 1999, 13, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Spiess, B.; Rinaldetti, S.; Naumann, N.; Galuschek, N.; Kossak-Roth, U.; Wuchter, P.; Tarnopolscaia, I.; Rose, D.; Voskanyan, A.; Fabarius, A.; et al. Diagnostic performance of the molecular BCR-ABL1 monitoring system may impact on inclusion of CML patients in stopping trials. PLoS ONE 2019, 14, e0214305. [Google Scholar] [CrossRef] [PubMed]
- Popp, H.D.; Brendel, S.; Hofmann, W.K.; Fabarius, A. Immunofluorescence Microscopy of gammaH2AX and 53BP1 for Analyzing the Formation and Repair of DNA Double-strand Breaks. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Popp, H.D.; Naumann, N.; Brendel, S.; Henzler, T.; Weiss, C.; Hofmann, W.K.; Fabarius, A. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk Res. 2017, 57, 112–118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gp | Pt | Age/Sex | Disease | TKI | γH2AX Foci Per Nucleus ± SEM | BCR/ABL MR | BCR/ABL Transcript | Cytogenetics/FISH |
|---|---|---|---|---|---|---|---|---|
| Group 1 | HEALTHY#1 | 49/♂ | Healthy | - | 0.6 ± 0.1 | - | - | - |
| HEALTHY#2 | 62/♀ | Healthy | - | 0.7 ± 0.1 | - | - | - | |
| HEALTHY#3 | 43/♂ | Healthy | - | 0.7 ± 0.1 | - | - | - | |
| HEALTHY#4 | NA | Healthy | - | 0.9 ± 0.2 | - | - | - | |
| HEALTHY#5 | 54/♀ | Healthy | - | 1.2 ± 0.2 | - | - | - | |
| HEALTHY#6 | 36/♀ | Healthy | - | 1.2 ± 0.2 | - | - | - | |
| HEALTHY#7 | 54/♀ | Healthy | - | 1.3 ± 0.1 | - | - | - | |
| HEALTHY#8 | 53/♀ | Healthy | - | 1.6 ± 0.2 | - | - | - | |
| Group 2 | CP-CML DMR#1 | 61/♀ | CP-CML | DASA | 0.1 ± 0.1 | MR4.5 | e13a2 (b2a2) | 46,XX,t(9;22)(q34;q11) [25] |
| CP-CML DMR#2 | 64/♀ | CP-CML | Stop | 0.4 ± 0.1 | MR4.5 | e13a2 (b2a2) | NA | |
| CP-CML DMR#3 | 74/♀ | CP-CML | Stop | 0.7 ± 0.2 | MR4.5 | e13a2/e14a2 (b2a2/b3a2) | NA | |
| CP-CML DMR#4 | 48/♂ | CP-CML | Stop | 0.7 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#5 | 63/♀ | CP-CML | Stop | 0.8 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#6 | 55/♂ | CP-CML | DASA | 0.8 ± 0.2 | MR4 | e14a2 (b3a2) | NA | |
| CP-CML DMR#7 | 61/♀ | CP-CML | Stop | 0.8 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#8 | 33/♀ | CP-CML | DASA | 0.8 ± 0.2 | MR4 | e13a2 (b2a2) | NA | |
| CP-CML DMR#9 | 58/♂ | CP-CML | Stop | 0.8 ± 0.2 | MR5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#10 | 72/♂ | CP-CML | Stop | 0.9 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#11 | 34/♂ | CP-CML | DASA | 0.9 ± 0.1 | MR4 | e13a2 (b2a2) | NA | |
| CP-CML DMR#12 | 68/♀ | CP-CML | DASA | 0.9 ± 0.2 | MR4 | e13a2 (b2a2) | NA | |
| CP-CML DMR#13 | 78/♀ | CP-CML | NILO | 1.0 ± 0.2 | MR4.5 | e13a2/e14a2 (b2a2/b3a2) | NA | |
| CP-CML DMR#14 | 59/♀ | CP-CML | IMA | 1.0 ± 0.2 | MR4 | e13a2 (b2a2) | NA | |
| CP-CML DMR#15 | 60/♀ | CP-CML | DASA | 1.1 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#16 | 67/♀ | CP-CML | Stop | 1.1 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#17 | 76/♀ | CP-CML | Stop | 1.1 ± 0.2 | MR4.5 | e13a2 (b2a2) | NA | |
| CP-CML DMR#18 | 77/♂ | CP-CML | Stop | 1.2 ± 0.2 | MR4.5 | e13a2/e14a2 (b2a2/b3a2) | NA | |
| CP-CML DMR#19 | 84/♀ | CP-CML | IMA | 1.3 ± 0.2 | MR4 | e13a2 (b2a2) | NA | |
| CP-CML DMR#20 | 71/♀ | CP-CML | Stop | 1.3 ± 0.3 | MR5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#21 | 63/♂ | CP-CML | Stop | 1.3 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML DMR#22 | 62/♂ | CP-CML | DASA | 1.7 ± 0.2 | MR4.5 | e14a2 (b3a2) | NA | |
| CP-CML MMR#1 | 70/♀ | CP-CML | IMA | 0.8 ± 0.2 | 0.04% | e13a2 (b2a2) | NA | |
| CP-CML MMR#2 | 59/♀ | CP-CML | Stop | 1.2 ± 0.2 | 0.07% | e13a2/e14a2 (b2a2/b3a2) | NA | |
| CP-CML MMR#3 | 77/♀ | CP-CML | DASA | 1.2 ± 0.2 | 0.09% | e14a2 (b3a2) | NA | |
| CP-CML MMR#4 | 54/♂ | CP-CML | DASA | 1.3 ± 0.2 | 0.02% | e14a2 (b3a2) | NA | |
| Group 3 | CP-CML loss MMR#1 | 54/♀ | CP-CML | NILO | 1.0 ± 0.2 | 0.18% | e1a2 | 46,XX,t(9;22)(q34;q11) [25] |
| CP-CML loss MMR#2 | 46/♂ | CP-CML | DASA | 1.1 ± 0.2 | 45% | e14a2 (b3a2) | 51,XY,+6,+8,+8,+8, t(9;22)(q34;q11),+19 [25] | |
| CP-CML loss MMR#3 | 81/♂ | CP-CML | Stop | 1.3 ± 0.2 | 0.35% | e13a2/e14a2 (b2a2/b3a2) | NA | |
| CP-CML loss MMR#4 * | 83/♀ | CP-CML | Stop | 2.4 ± 0.2 | 1.00% | e14a2 (b3a2) | NA | |
| CP-CML loss MMR#5 | 83/♀ | CP-CML | Stop | 3.0 ± 0.2 | 30% | e13a2/e14a2 (b2a2/b3a2) | NA | |
| Group 4 | CP-CML de novo#1 | 19/♂ | CP-CML | - | 1.4 ± 0.2 | 60% | e13a2 (b2a2) | 46,XY,t(9;22)(q34;q11) [25] |
| CP-CML de novo#2 | 79/♀ | CP-CML | - | 1.6 ± 0.3 | 63% | e13a2 (b2a2) | 46,XX,t(9;22)(q34;q11) [25] | |
| CP-CML de novo#3 | 18/♀ | CP-CML | - | 2.3 ± 0.3 | 68% | e13a2 (b2a2) | 46,XX,t(9;22)(q34;q11) [25] | |
| CP-CML de novo#4 | 66/♀ | CP-CML | - | 3.4 ± 0.2 | 29% | e14a2 (b3a2) | 46,XX,t(9;22)(q34;q11) [25] | |
| CP-CML de novo#5 | 53/♀ | CP-CML | - | 3.7 ± 0.3 | 68% | e13a2 (b2a2) | 46,XX,t(9;22)(q34;q11) [25] | |
| Group 5 | BP-CML#1 ** | 76/♀ | BP-CML | Stop | 3.4 ± 0.4 | 32% | e13a2 (b2a2) | NA (rejected) |
| BP-CML#2 | 63/♂ | BP-CML | DASA | 4.0 ± 0.4 | 63% | e14a2 (b3a2) | 46,XY,inv(3)(q21;q26), t(9;22)(q34;q11) [25] | |
| BP-CML#3 *** | 38/♀ | BP-CML | PONA | 5.8 ± 0.4 | 256% | e13a2 (b2a2) | 46,XX,t(9;22)(q34;q11) [1] 44,XX,der(3)t(3;9)(p11;q11) t(9;22)(q34;q11),−7,−9, der(13)t(7;13)(q22;q34),der(22) t(9;22)(q34;q11) [5] 46,XX [14] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.-K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. https://doi.org/10.3390/ijms21041177
Popp HD, Kohl V, Naumann N, Flach J, Brendel S, Kleiner H, Weiss C, Seifarth W, Saussele S, Hofmann W-K, et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2020; 21(4):1177. https://doi.org/10.3390/ijms21041177
Chicago/Turabian StylePopp, Henning D., Vanessa Kohl, Nicole Naumann, Johanna Flach, Susanne Brendel, Helga Kleiner, Christel Weiss, Wolfgang Seifarth, Susanne Saussele, Wolf-Karsten Hofmann, and et al. 2020. "DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia" International Journal of Molecular Sciences 21, no. 4: 1177. https://doi.org/10.3390/ijms21041177