Fluorescent Analogues of Human α-Calcitonin Gene-Related Peptide with Potent Vasodilator Activity

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Fluorescent CGRP Derivatives
2.2. Biological Activity of Three Fluorescently Tagged CGRP Analogues
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Animals
3.3. Human Subcutaneous Arteries
3.4. Data Analysis and Statistics
3.5. CD Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| Ahx | 6-aminohexanoic acid (Ahx) |
| Boc | tert-butyloxycarbonyl |
| CD | circular dichroism |
| CF | 5(6)-carboxyfluorescein |
| CFSE | carboxyfluorescein-N-succinimidylester |
| CLR | calcitonin receptor-like receptor |
| DCM | dichloromethane |
| DIEA | diisopropylamine |
| DMF | dimethylformamide |
| FITC | fluorescein isothiocyanate |
| Fmoc | 9-fluorenylmethyloxycarbonyl |
| h-α-CGRP | human α-calcitonin gene-related peptide |
| HATU | 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| HOAt | 1-hydroxy-7-azabenzotriazole |
| HPLC | high-performance liquid chromatography |
| ivDde | 1-(4,4-Dimethyl-2,6-dioxo-cyclohexylidene)-3-methyl-butyl |
| MALDI-TOF-MS | matrix-assisted linear desorption ionization time-of-flight mass spectrometry |
| PyAOP | (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate) |
| Mtt | 4-methyltrityl |
| SPPS | solid-phase peptide synthesis |
| TIS | triisopropylsilane |
| Trt | trityl |
References
- Poyner, D.R. Calcitonin gene-related peptide: Multiple actions, multiple receptors. Pharmacol. Ther. 1992, 56, 23–51. [Google Scholar] [CrossRef]
- Russell, F.A.; King, R.; Smillie, S.J.; Kodji, X.; Brain, S.D. Calcitonin gene-related peptide: Physiology and pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingell, J.J.; Hendrikse, E.R.; Hay, D.L. New Insights into the Regulation of CGRP-Family Receptors. Trends Pharmacol. Sci. 2019, 40, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.A.; Rathbone, D.L.; Barwell, J.; Hay, D.L.; Poyner, D.R. Structure-activity relationships for alpha-calcitonin gene-related peptide. Br. J. Pharmacol. 2013, 170, 1308–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urits, I.; Jones, M.R.; Gress, K.; Charipova, K.; Fiocchi, J.; Kaye, A.D.; Viswanath, O. CGRP Antagonists for the Treatment of Chronic Migraines: A Comprehensive Review. Curr. Pain Headache Rep. 2019, 23, 29. [Google Scholar] [CrossRef]
- Miranda, L.P.; Shi, L.; Holder, J.R.; Wright, M.; Gegg, C.V.; Johnson, E.; Wild, K.; Stenkilsson, M.; Doellgast, G.; Manning, B.H.; et al. Peptide antagonists of the calcitonin gene-related peptide (CGRP) receptor with improved pharmacokinetics and pharmacodynamics. Pept. Sci. 2013, 100, 422–430. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies—Successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Kyani, A.; Mehrabian, M.; Jenssen, H. Quantitative Structure-Activity Relationships and Docking Studies of Calcitonin Gene-Related Peptide Antagonists. Chem. Biol. Drug Des. 2012, 79, 166–176. [Google Scholar] [CrossRef]
- Miranda, L.P.; Holder, J.R.; Shi, L.; Bennett, B.; Aral, J.; Gegg, C.V.; Wright, M.; Walker, K.; Doellgast, G.; Rogers, R.; et al. Identification of Potent, Selective, and Metabolically Stable Peptide Antagonists to the Calcitonin Gene-Related Peptide (CGRP) Receptor. J. Med. Chem. 2008, 51, 7889–7897. [Google Scholar] [CrossRef]
- Nilsson, C.; Hansen, T.K.; Rosenquist, C.; Hartmann, B.; Kodra, J.T.; Lau, J.F.; Clausen, T.R.; Raun, K.; Sams, A. Long acting analogue of the calcitonin gene-related peptide induces positive metabolic effects and secretion of the glucagon-like peptide-1. Eur. J. Pharmacol. 2016, 773, 24–31. [Google Scholar] [CrossRef]
- Sams-Nielsen, A.; Orskov, C.; Jansen-Olesen, I. Pharmacological evidence for CGRP uptake into perivascular capsaicin sensitive nerve terminals. Br. J. Pharmacol. 2001, 132, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Amrutkar, D.V.; Mataji, A.; Salmasi, H.; Hay-Schmidt, A.; Sheykhzade, M.; Messlinger, K.; Olesen, J.; Jansen-Olesen, I. Evidence for CGRP re-uptake in rat dura mater encephali. Br. J. Pharmacol. 2010, 161, 1885–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheykhzade, M.; Gupta, S.; Sorensen, T.; Sorensen, O.A.; Koch, H.; Boonen, H.C.M.; Back, O.; Fjalland, B. Characterization of capsaicin induced responses in mice vas deferens: Evidence of CGRP uptake. Eur. J. Pharmacol. 2011, 667, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; Kneib-Cordonier, N.; Biancalana, S.; Gera, L.; Masada, R.I.; Hudson, D.; Barany, G. Preparation and Application of the PAL Handle for the Solid-Phase Peptide Synthesis of C-terminal Peptide Amides under Mild Conditions. J. Org. Chem. 1990, 55, 3730–3743. [Google Scholar] [CrossRef]
- Mutter, M.; Nefzi, A.; Sato, T.; Sun, X.; Wahl, F.; Wöhr, T. Pseudo-Prolines (YPro) for Acessing "Inaccessible" Peptides. Pept. Res. 1995, 8, 145–153. [Google Scholar] [PubMed]
- Garcia-Martin, F.; White, P.; Steinauer, R.; Cote, S.; Tulla-Puche, J.; Albericio, F. The synergy of ChemMatrix resin (R) and pseudoproline building blocks renders RANTES, a complex aggregated chemokine. Biopolymers 2006, 84, 566–575. [Google Scholar]
- Gonçalves, M.S.T. Fluorescent Labeling of Biomolecules with Organic Probes. Chem. Rev. 2009, 109, 190–212. [Google Scholar] [CrossRef]
- Fernandez-Carneado, J.; Giralt, E. An efficient method for the solid-phase synthesis of fluorescently labelled peptides. Tetrahedron Lett. 2004, 45, 6079–6081. [Google Scholar] [CrossRef]
- Stahl, P.J.; Cruz, J.C.; Li, Y.; Yu, S.M.; Hristova, K. On-the-resin N-terminal modification of long synthetic peptides. Anal. Biochem. 2012, 424, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Arano, Y.; Akizawa, H.; Uezono, T.; Akaji, K.; Ono, M.; Funakoshi, S.; Koizumi, M.; Yokoyama, A.; Kiso, Y.; Saji, H. Conventional and High-Yield Synthesis of DTPA-Conjugated Peptides:Application of a Monoreactive DTPA to DTPA-d-Phe1-octreotide Synthesisâ€. Bioconjug. Chem. 1997, 8, 442–446. [Google Scholar] [CrossRef]
- Jullian, M.; Hernandez, A.; Maurras, A.; Puget, K.; Amblard, M.; Martinez, J.; Subra, G. N-terminus FITC labeling of peptides on solid support: The truth behind the spacer. Tetrahedron Lett. 2009, 50, 260–263. [Google Scholar] [CrossRef]
- Weber, P.J.A.; Bader, J.E.; Folkers, G.; Beck-Sickinger, A.G. A fast and inexpensive method for N-terminal fluorescein-labeling of peptides. Bioorg. Med. Chem. Lett. 1998, 8, 597–600. [Google Scholar] [CrossRef]
- Fischer, R.; Mader, O.; Jung, G.; Brock, R. Extending the applicability of carboxyfluorescein in solid-phase synthesis. Bioconjug. Chem. 2003, 14, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Anthoni, U.; Christophersen, C.; Nielsen, P.H.; Püschl, A.; Schaumburg, K. Structure of red and orange fluorescein. Struct. Chem. 1995, 6, 161–165. [Google Scholar] [CrossRef]
- Chhabra, S.R.; Hothi, B.; Evans, D.J.; White, P.D.; Bycroft, B.W.; Chan, W.C. An appraisal of new variants of Dde amine protecting group for solid phase peptide synthesis. Tetrahedron Lett. 1998, 39, 1603–1606. [Google Scholar] [CrossRef]
- Lau, J.; Kwon, D.; Rousseau, E.; Zhang, Z.; Zeisler, J.; Uribe, C.F.; Kuo, H.-T.; Zhang, C.; Lin, K.-S.; Bénard, F. [68Ga]Ga/[177Lu]Lu-BL01, a Novel Theranostic Pair for Targeting C-X-C Chemokine Receptor 4. Mol. Pharm. 2019, 16, 4688–4695. [Google Scholar] [CrossRef]
- Denton, E. Optimizing the Removal of an IvDde Protecting Group. Available online: http://www.peptidesynthesisblog.com/optimizing-the-removal-of-an-ivdde-protecting-group/ (accessed on 11 February 2020).
- Domenyuk, V.; Loskutov, A.; Johnston, S.A.; Diehnelt, C.W. A Technology for Developing Synbodies with Antibacterial Activity. PLoS ONE 2013, 8, e54162. [Google Scholar] [CrossRef]
- Góngora-Benítez, M.; Mendive-Tapia, L.; Ramos-Tomillero, I.; Breman, A.C.; Tulla-Puche, J.; Albericio, F. Acid-Labile Cys-Protecting Groups for the Fmoc/tBu Strategy: Filling the Gap. Org. Lett. 2012, 14, 5472–5475. [Google Scholar] [CrossRef]
- Bollhagen, R.; Schmiedberger, M.; Barlos, K.; Grell, E. A new reagent for the cleavage of fully protected peptides synthesized on 2-chlorotrityl chloride resin. J. Chem. Soc. Chem. Commun. 1994, 2559–2560. [Google Scholar] [CrossRef]
- Manning, M.C.; Illangasekare, M.; Woody, R. Circular dichroism studies of distorted α-helices, twisted β-sheets, and β-turns. Biophys. Chem. 1988, 31, 77–86. [Google Scholar] [CrossRef]
- Hubbard, J.A.M.; Martin, S.R.; Chaplin, L.C.; Bose, C.; Kelly, S.M.; Price, N.C. Solution structures of calcitonin-gene-related-peptide analogues of calcitonin-gene-related peptide and amylin. Biochem. J. 1991, 275, 785–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimeault, M.; St-Pierre, S.; Fournier, A. Conformational characterization by circular-dichroism spectroscopy of various fragments and analogs of calcitonin-gene-related peptide. Eur. J. Biochem. 1993, 213, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Simms, J.; Uddin, R.; Sakmar, T.P.; Gingell, J.J.; Garelja, M.L.; Hay, D.L.; Brimble, M.A.; Harris, P.W.; Reynolds, C.A.; Poyner, D.R. Photoaffinity Cross-Linking and Unnatural Amino Acid Mutagenesis Reveal Insights into Calcitonin Gene-Related Peptide Binding to the Calcitonin Receptor-like Receptor/Receptor Activity-Modifying Protein 1 (CLR/RAMP1) Complex. Biochemistry 2018, 57, 4915–4922. [Google Scholar] [CrossRef] [Green Version]
- Yule, L.R.; Garelja, M.L.; Hendrikse, E.R.; Gingell, J.J.; Poyner, D.R.; Harris, P.W.H.; Brimble, M.A.; Hay, D.L. A potent fluorescent calcitonin gene-related peptide analogue enables visualization of receptor internalization. Pept. Sci. 2019, 111, e24126. [Google Scholar] [CrossRef]
- Kenakin, T. Competitive Antagonism Pharmacologic Analysis of Drug-Receptor Interaction, 3rd ed.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1997; pp. 331–373. [Google Scholar]
- Poyner, D.R.; Andrew, D.P.; Brown, D.; Bose, C.; Hanley, M.R. Pharmacological characterization of a receptor for calcitonin gene-related peptide on rat, L6 myocytes. Br. J. Pharmacol. 1992, 105, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheykhzade, M.; Amandi, N.; Pla, M.V.; Abdolalizadeh, B.; Sams, A.; Warfvinge, K.; Edvinsson, L.; Pickering, D.S. Binding and functional pharmacological characteristics of gepant-type antagonists in rat brain and mesenteric arteries. Vasc. Pharmacol. 2017, 90, 36–43. [Google Scholar] [CrossRef] [PubMed]
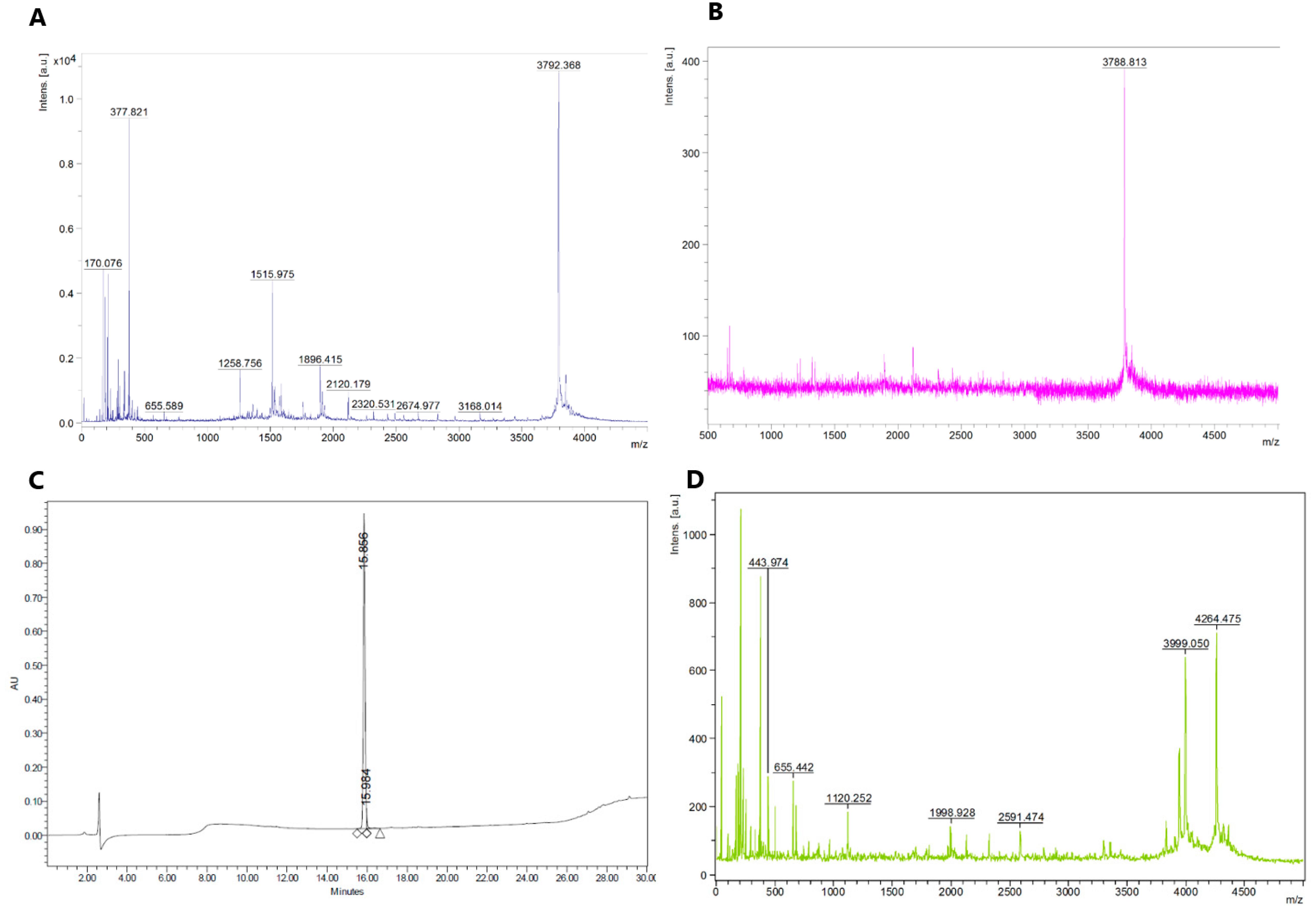
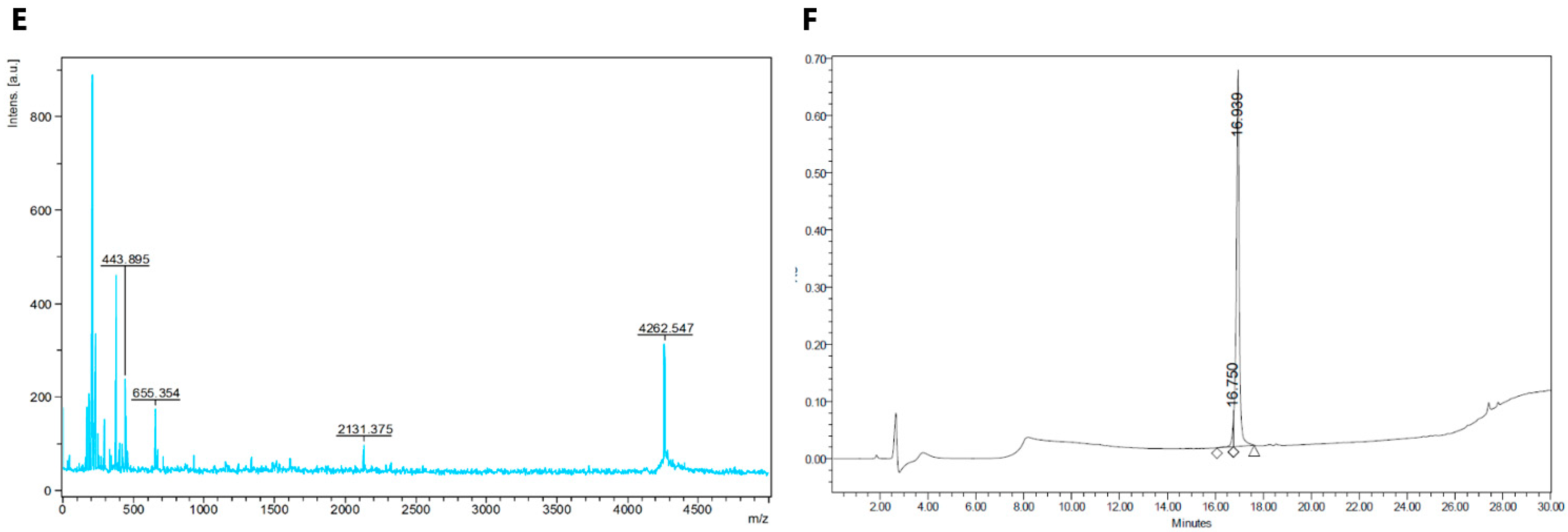
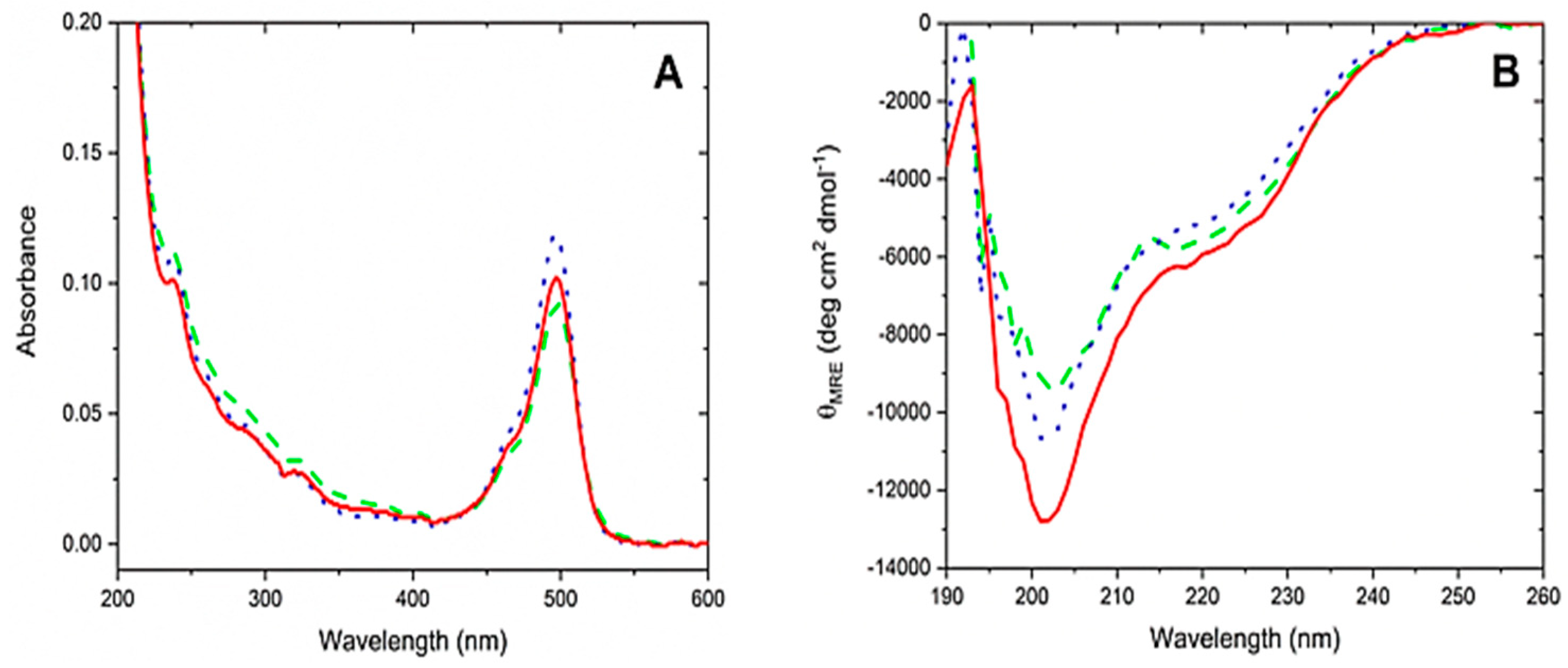

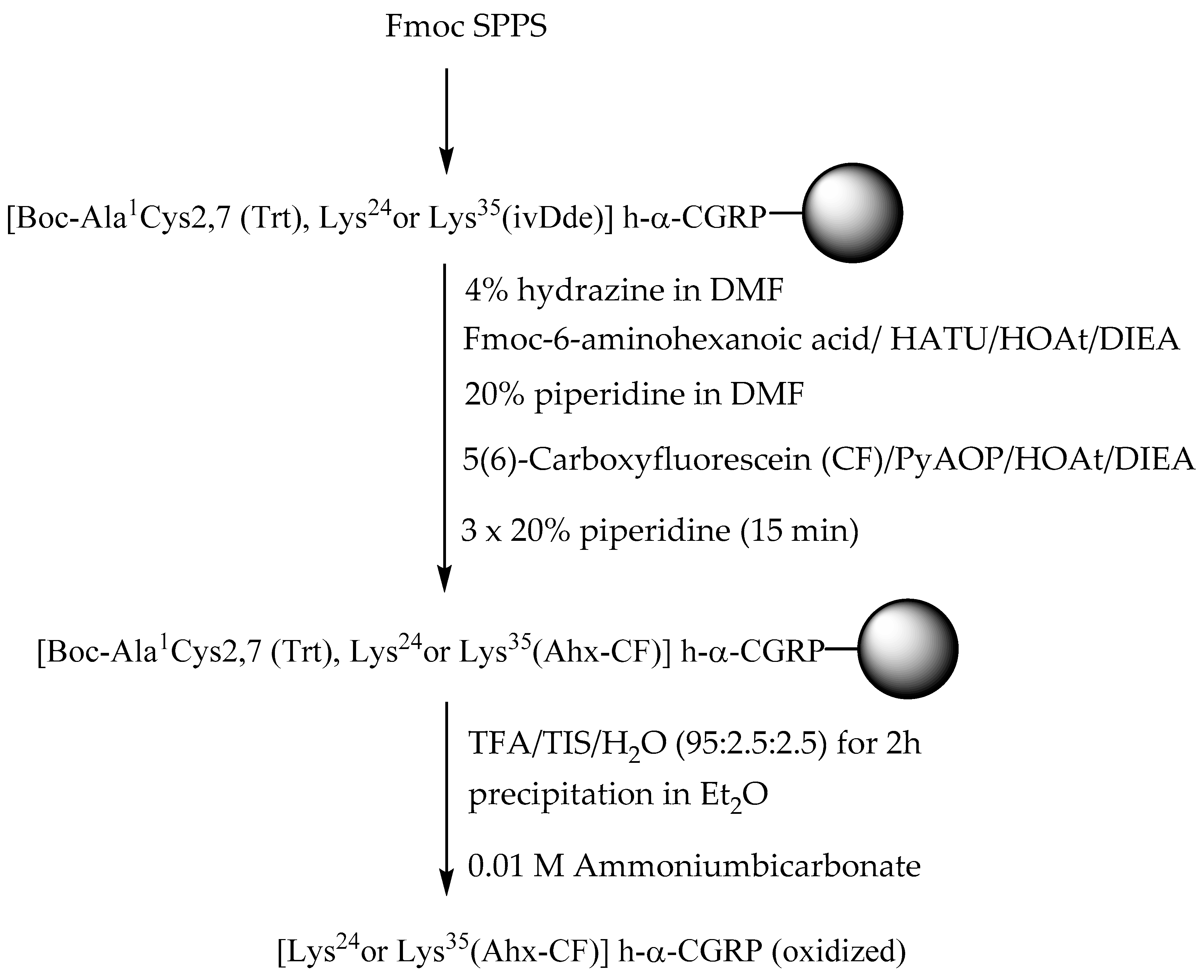

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Activity | Wild-Type CGRP | [N-terminal(Ahx-CF)] CGRP | [Lys24(Ahx-CF)] CGRP | [Lys35(Ahx-CF)] CGRP |
|---|---|---|---|---|
| Mouse mesenteric artery | ||||
| Emax, % | 99 ± 7 (n = 5) | 87 ± 13 (n = 5) | 82 ± 10 (n = 6) | 94 ± 8 (n = 6) |
| pIC50 | 9.14 ± 0.18 (n = 5) | 7.30 ± 0.23 (n = 5) *** | 7.96 ± 0.38 (n = 6) * | 8.57 ± 0.21 (n = 6) |
| Potency ratio | 1 | 70 | 15 | 4 |
| Hill slope | 1.0 ± 0.2 (n = 5) | 0.9 ± 0.3 (n = 5) | 1.3 ± 0.5 (n = 6) | 1.0 ± 0.3 (n = 6) |
| Rat mesenteric artery | ||||
| Emax, % | 67 ± 5 (n = 5) | 66 ± 5 (n = 5) | 74 ± 3 (n = 5) | 70 ± 6 (n = 5) |
| pIC50 | 8.61 ± 0.06 (n = 5) | 6.50 ± 0.50 (n = 5) *** | 7.35 ± 0.05 (n = 5) * | 8.13 ± 0.11 (n = 5) |
| Potency ratio | 1 | 128 | 19 | 3 |
| Hill slope | 1.3 ± 0.3 (n = 5) | 0.9 ± 0.5 (n = 5) | 1.1 ± 0.1 (n = 5) | 1.2 ± 0.4 (n = 5) |
| Human subcutaneous artery | ||||
| Emax, % | 100 ± 3 (n = 7) | 99 ± 8 (n = 8) | 96 ± 5 (n = 7) | 99 ± 4 (n = 5) |
| pIC50 | 8.62 ± 0.06 (n = 7) | 7.25 ± 0.16 (n = 8) *** | 7.85 ± 0.06 (n = 7) *** | 8.12 ± 0.12 (n = 5) |
| Potency ratio | 1 | 24 | 5 | 3 |
| Hill slope | 1.7 ± 0.2 (n = 7) | 2.0 ± 0.6 (n = 8) | 1.5 ± 0.3 (n = 7) | 1.6 ± 0.3 (n = 5) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Pedersen, M.D.; Ahmed, L.S.; Abdolalizadeh, B.; Grell, A.-S.; Berg, J.O.; Thulstrup, P.W.; Franzyk, H.; Edvinsson, L.; Sams, A.; et al. Fluorescent Analogues of Human α-Calcitonin Gene-Related Peptide with Potent Vasodilator Activity. Int. J. Mol. Sci. 2020, 21, 1343. https://doi.org/10.3390/ijms21041343
Zhu J, Pedersen MD, Ahmed LS, Abdolalizadeh B, Grell A-S, Berg JO, Thulstrup PW, Franzyk H, Edvinsson L, Sams A, et al. Fluorescent Analogues of Human α-Calcitonin Gene-Related Peptide with Potent Vasodilator Activity. International Journal of Molecular Sciences. 2020; 21(4):1343. https://doi.org/10.3390/ijms21041343
Chicago/Turabian StyleZhu, Jing, Mahdieh Dagina Pedersen, Laraib Sabbah Ahmed, Bahareh Abdolalizadeh, Anne-Sofie Grell, Jais Oliver Berg, Peter Waaben Thulstrup, Henrik Franzyk, Lars Edvinsson, Anette Sams, and et al. 2020. "Fluorescent Analogues of Human α-Calcitonin Gene-Related Peptide with Potent Vasodilator Activity" International Journal of Molecular Sciences 21, no. 4: 1343. https://doi.org/10.3390/ijms21041343