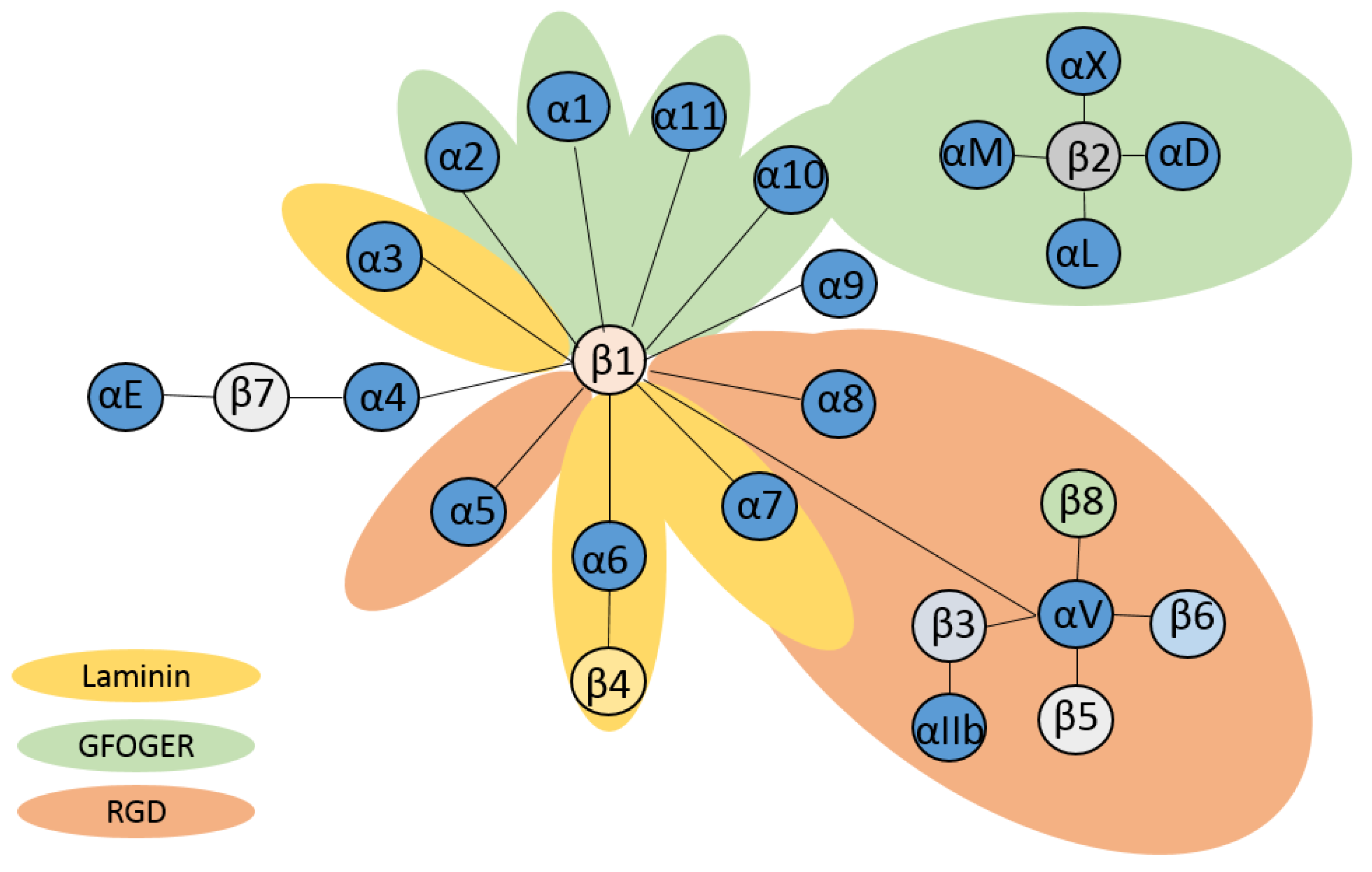
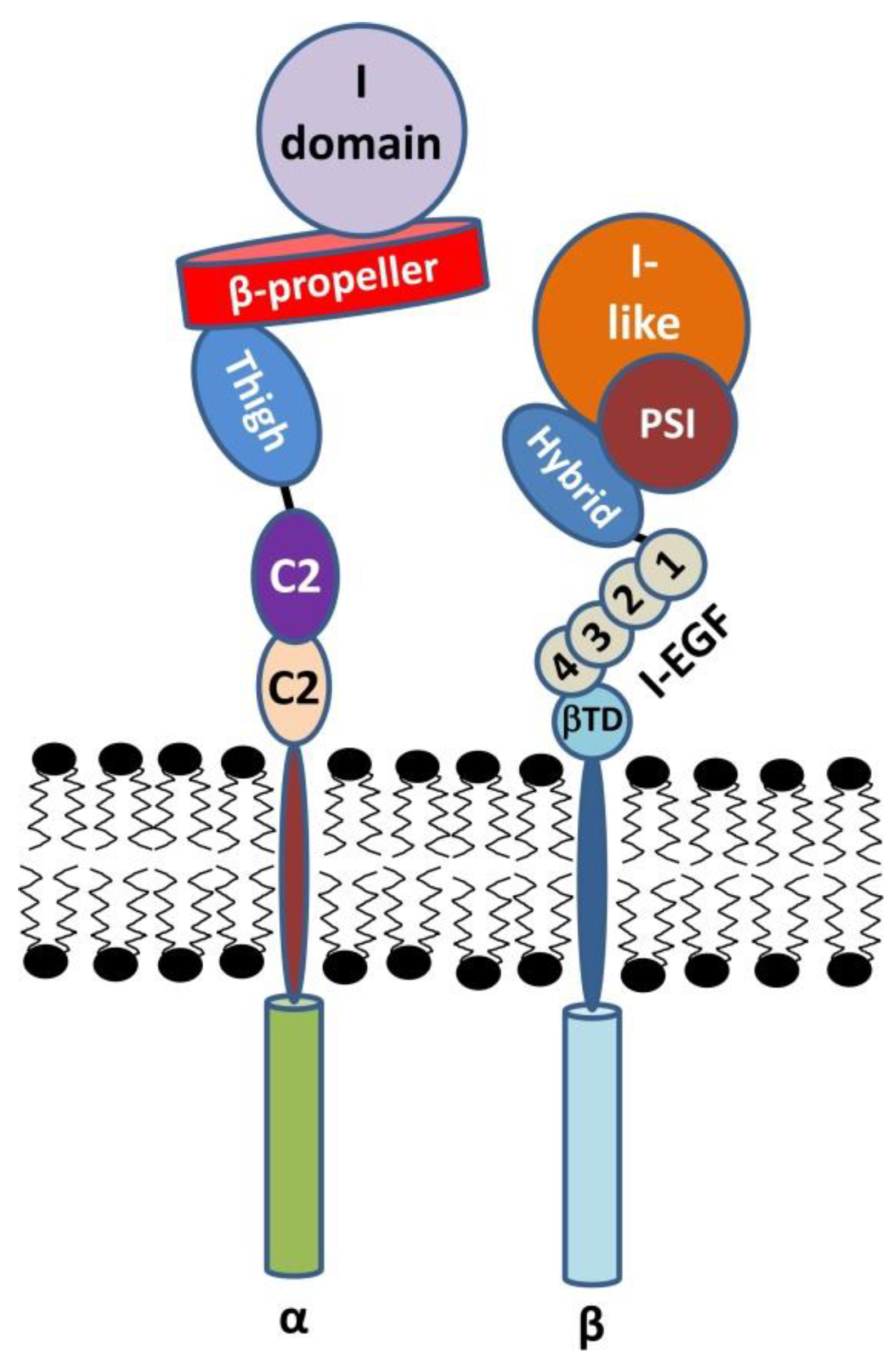
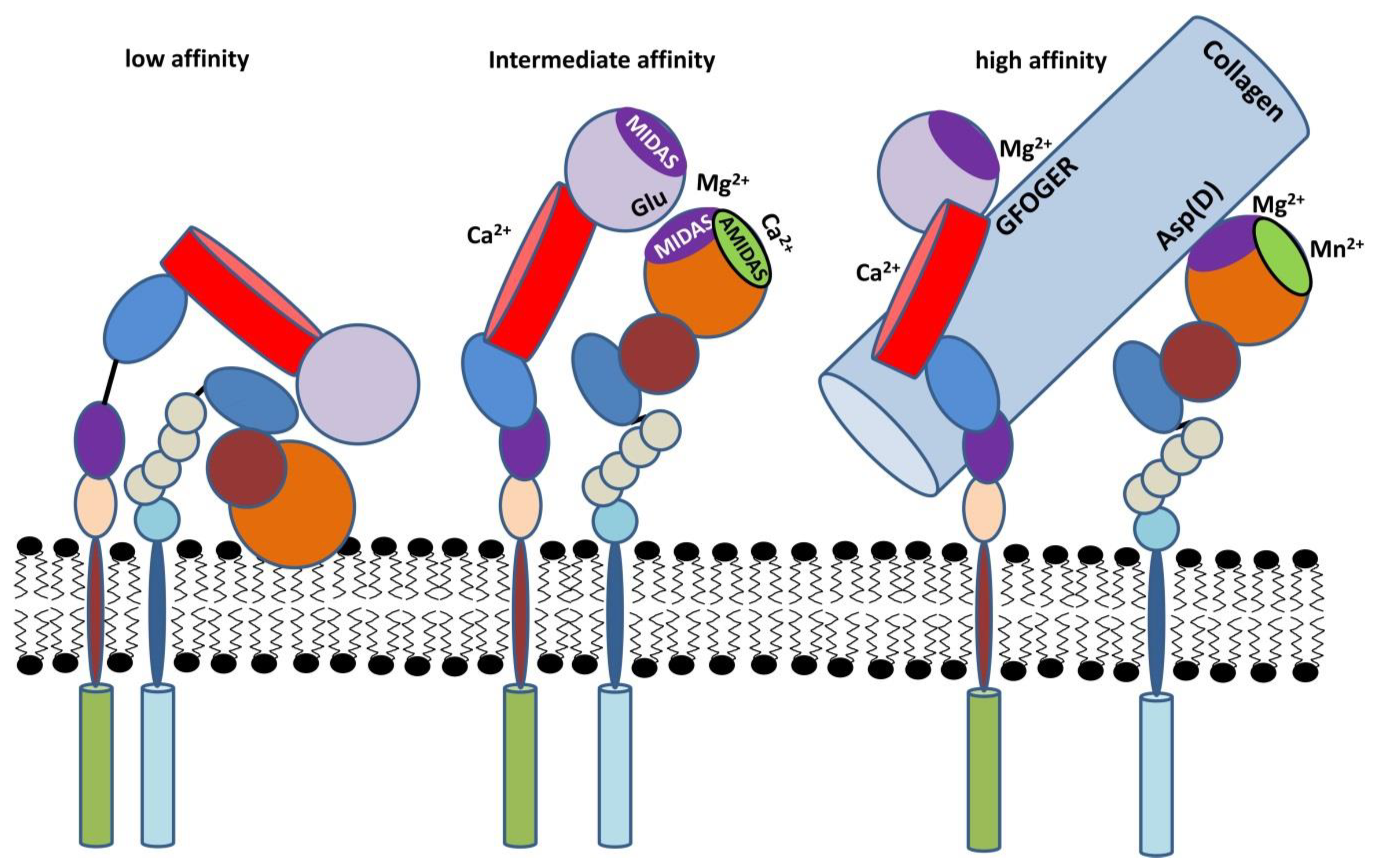
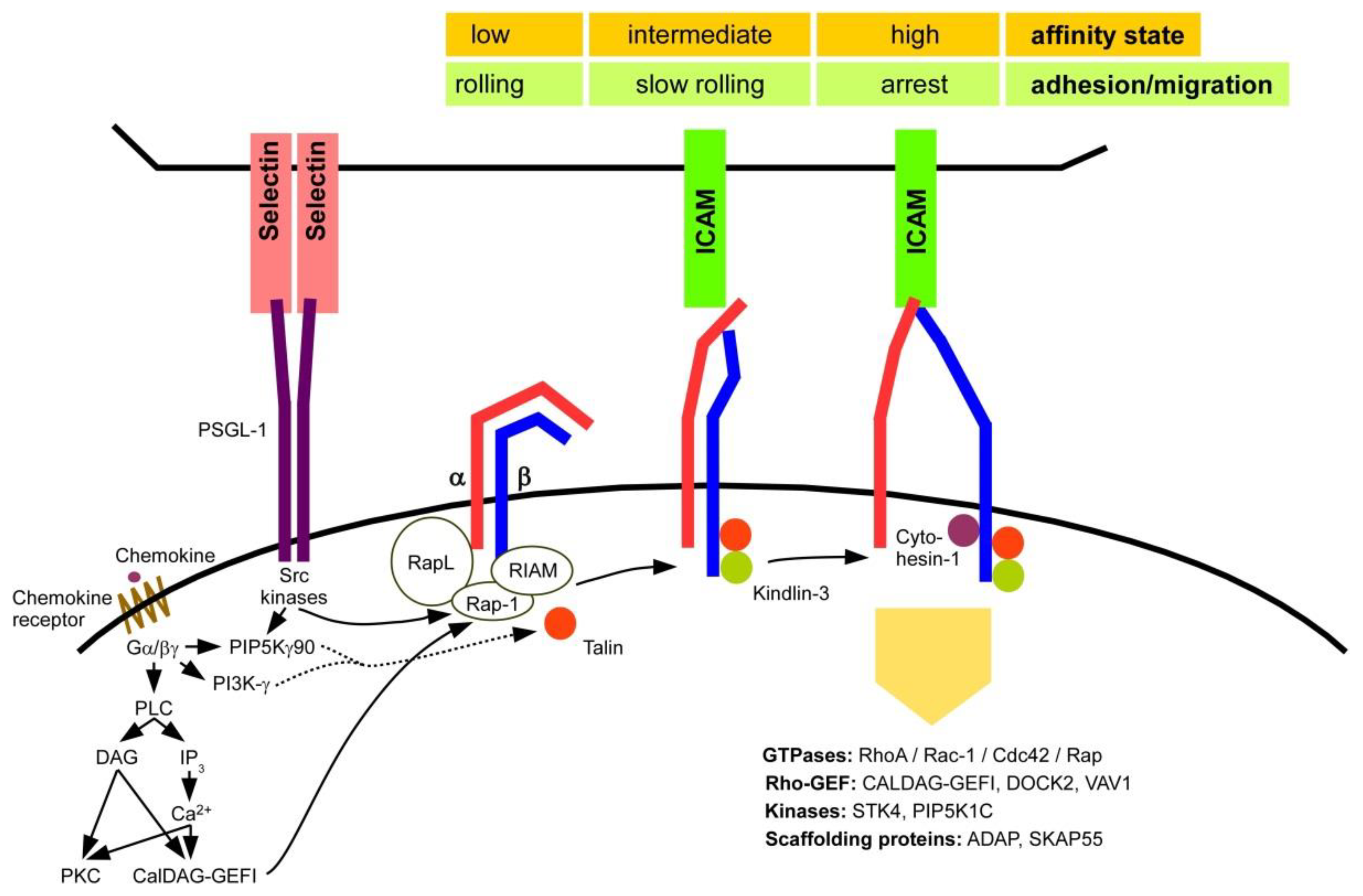
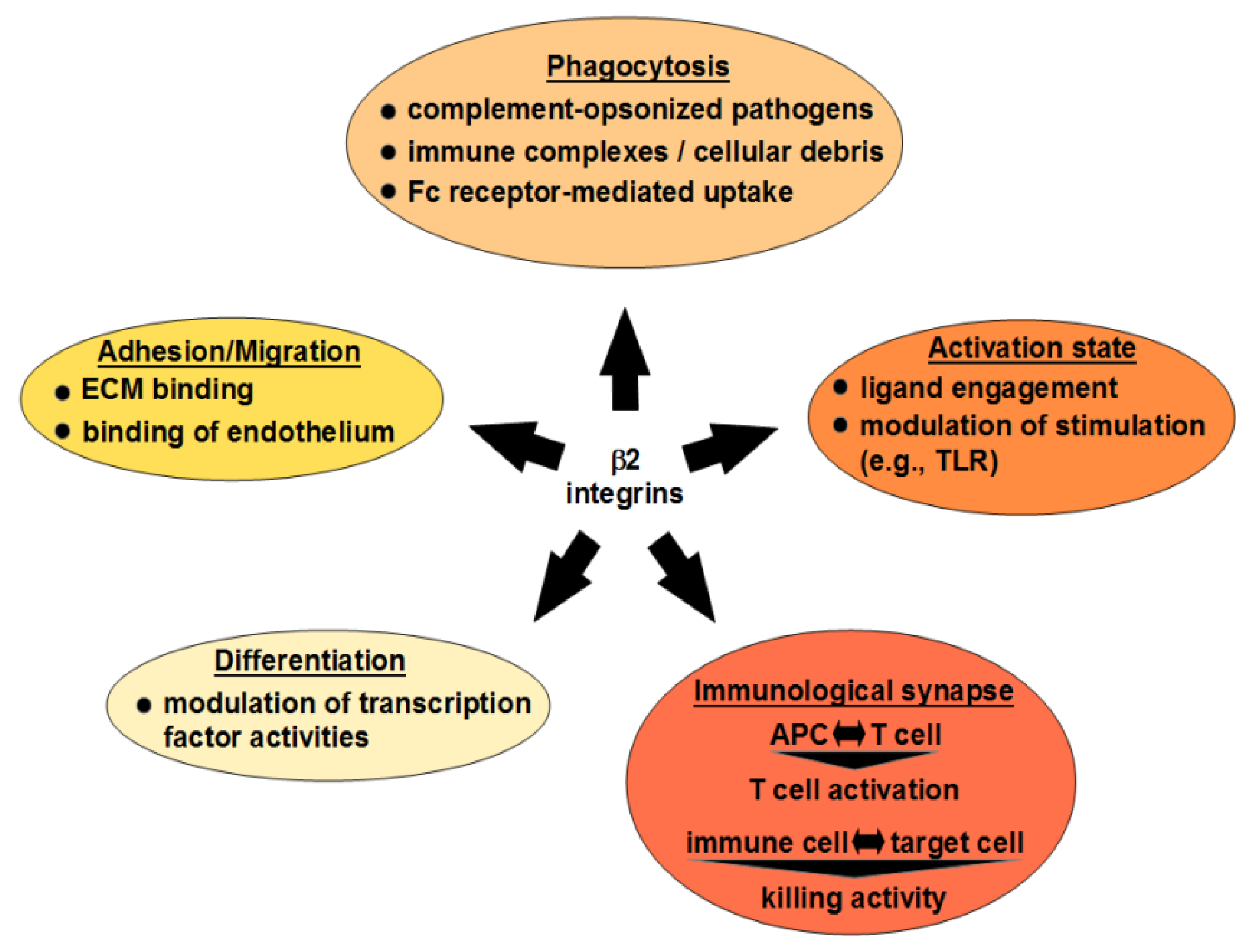
β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. https://doi.org/10.3390/ijms21041402
Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. International Journal of Molecular Sciences. 2020; 21(4):1402. https://doi.org/10.3390/ijms21041402
Chicago/Turabian StyleBednarczyk, Monika, Henner Stege, Stephan Grabbe, and Matthias Bros. 2020. "β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease" International Journal of Molecular Sciences 21, no. 4: 1402. https://doi.org/10.3390/ijms21041402
APA StyleBednarczyk, M., Stege, H., Grabbe, S., & Bros, M. (2020). β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. International Journal of Molecular Sciences, 21(4), 1402. https://doi.org/10.3390/ijms21041402