Extranodal NK/T-Cell Lymphomas: The Role of Natural Killer Cells and EBV in Lymphomagenesis


Abstract
:1. Introduction to NK-Cell-Derived Hematolymphoid Malignancies
2. The Cell of Origin in ENKTL: NK Cells or T-Cells?
3. The Role of NK Cells in Lymphomagenesis
3.1. Presumed Pathogenetic Mechanisms Describing ENKTL Oncogenesis
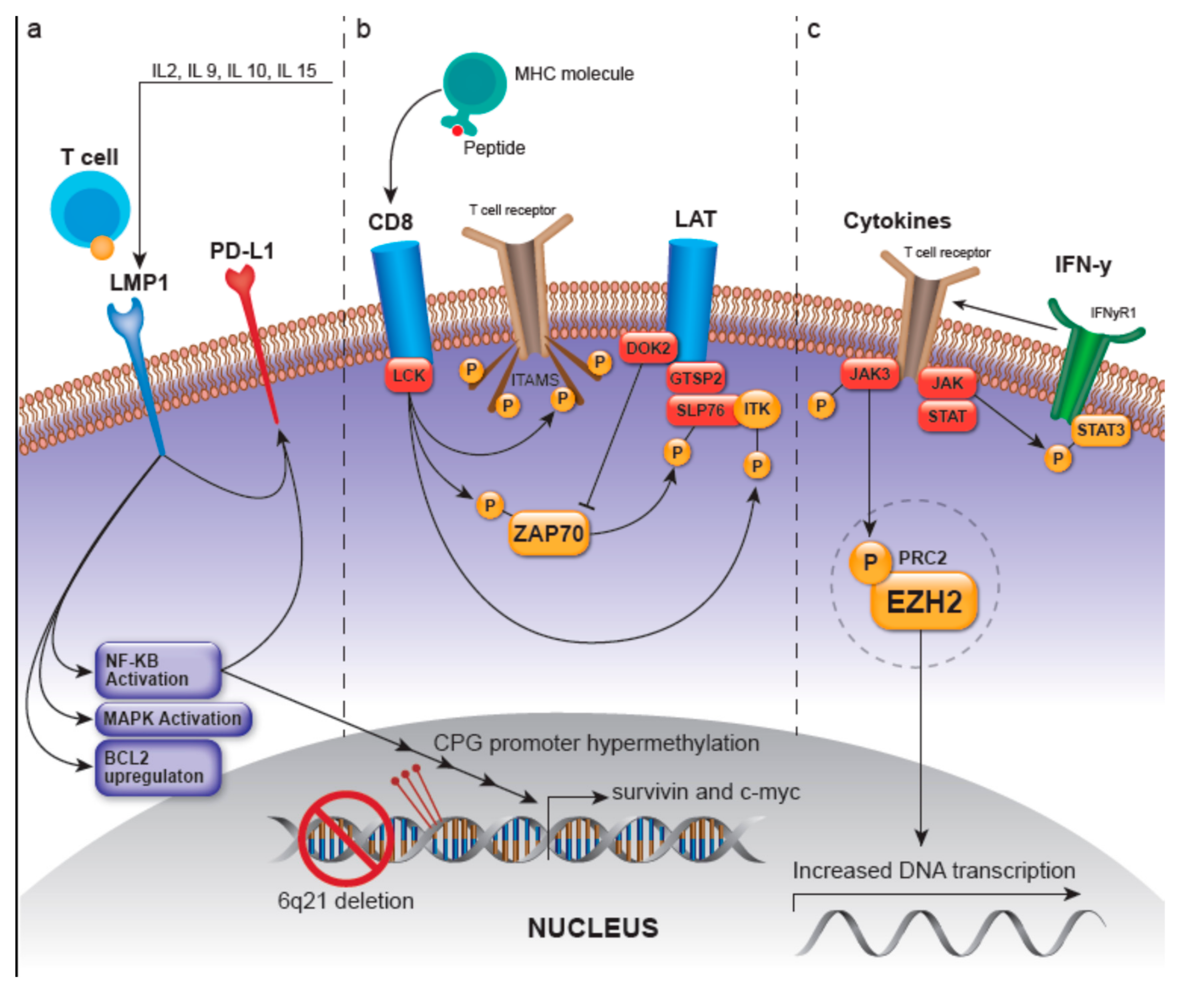
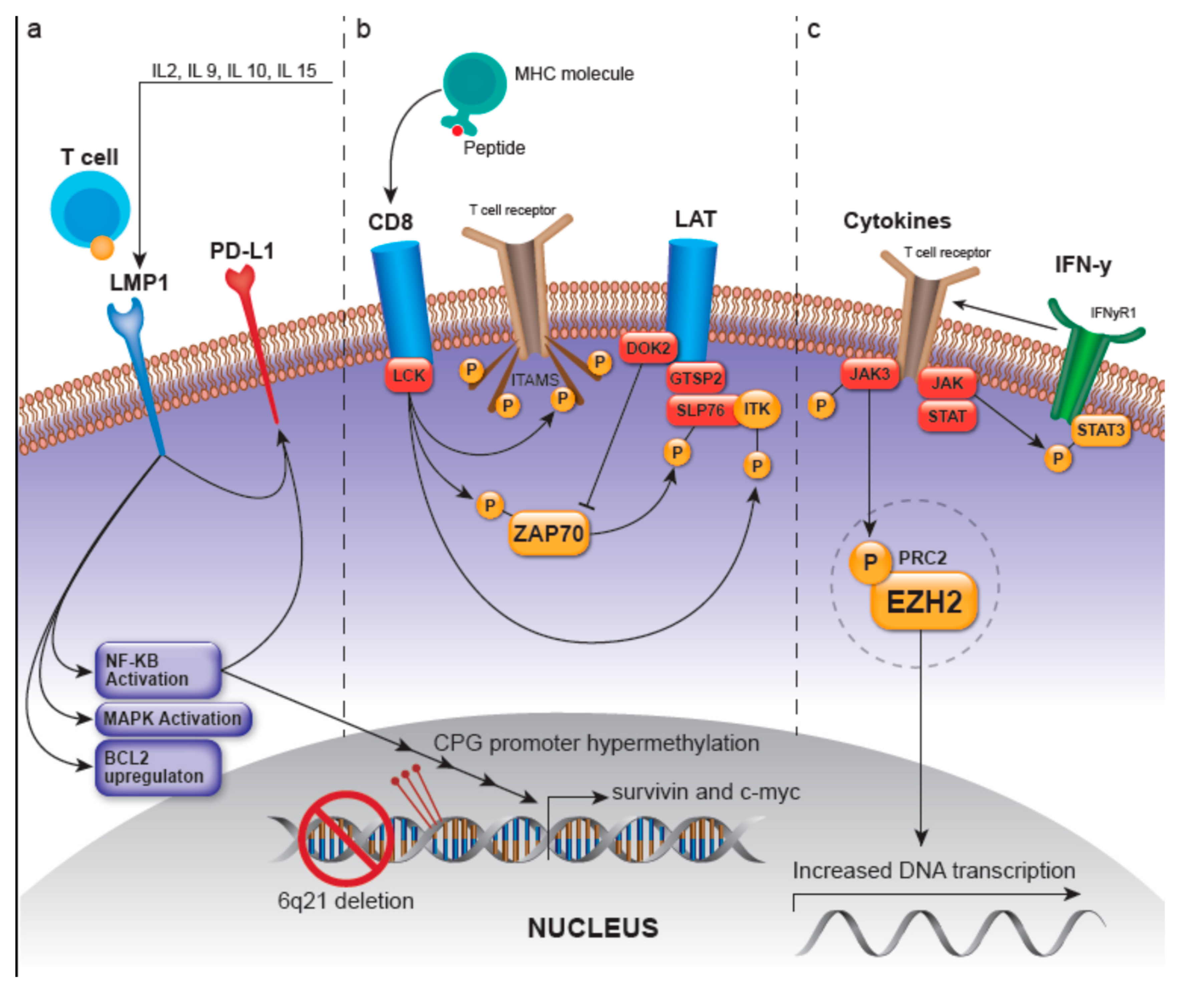
3.1.1. LMP-1-Related Pathways
3.1.2. LCK/ZAP70-Related Pathways
- (1)
- Autophosphorylation of ZAP70, deeming it active and able to phosphorylate the linker for activation of T-cells (LAT) and SH2 domain containing leukocyte protein of 76kDa (SLP-76), where SLP-76 can subsequently activate the PLC-gamma pathway and the MEK/extracellular signal-regulated kinase (ERK) pathway [2,21]. LAT is a phosphoprotein that localizes to glycosphingolipid-enriched microdomains (GEMs) and acts as a docking site for proteins that contain an Src homology-2 (SH2) domain [22]. The LAT signalosome, which ultimately promotes T-cell signaling, is composed of LAT, and GRAP2/GADS (GRAP2 protein is involved in leukocyte-specific protein-tyrosine kinase signaling and contains an SH2 domain) [22]. The protein tyrosine kinase ZAP70 along with GRAP2/GADS have been shown to be expressed in greater than 90% and 68% of ENKTLs, respectively, with the GRAP2/GADS expression rate being higher in those ENKTLs of T lineage [23]. Moreover, GRAP2/GADS-positive ENKTLs frequently co-express docking protein 2 (DOK2). DOK2 protein may be involved in modulating BCR-ABL signaling and cellular proliferation induced by IL-4, 2, and 3 [22]; it is involved in negatively regulating GRAP2/GADS and NK-cell activation via inhibition of ZAP70 [23,24].
- (2)
- Recruitment and activation of LAT which then recruits several molecules including GRAP2 and the tyrosine kinase IL-2 inducible T-cell kinase (ITK) to ultimately modulate T-cell transcriptional regulation and gene expression of survival, apoptosis, and proliferation-related genes as well as those related to the migration of T-cells [23].
- (3)
- Phosphorylation and activation of ITK [25]
3.1.3. JAK/STAT- and EZH2-Involved Pathways
3.1.4. Other Ungrouped Pathways
4. Ethnogenetic Predisposition of ENKTL
5. Therapeutic Strategies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sinkovics, J.G.; Horvath, J.C. Human Natural Killer Cells: A Comprehensive Review. Int. J. Oncol. 2005, 27, 5–47. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljunggren, H.G.; Kärre, K. In Search of the “Missing Self”: MHC Molecules and NK Cell Recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Brilot, F.; Münz, C. Non-Cytotoxic Functions of Natural Killer Cells: Direct Pathogen Restriction and Assistance to Adaptive Immunity. J. Immunol. Baltim. Md 1950 2008, 180, 7785–7791. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; World Health Organization: Lyon, France, 2017; pp. 351–371. [Google Scholar]
- International T-Cell Lymphoma Project. International Peripheral T-Cell and Natural Killer/T-Cell Lymphoma Study: Pathology Findings and Clinical Outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Pongpruttipan, T.; Sukpanichnant, S.; Assanasen, T.; Wannakrairot, P.; Boonsakan, P.; Kanoksil, W.; Kayasut, K.; Mitarnun, W.; Khuhapinant, A.; Bunworasate, U.; et al. Extranodal NK/T-Cell Lymphoma, Nasal Type, Includes Cases of Natural Killer Cell and Aβ, Γδ, and Aβ/Γδ T-Cell Origin: A Comprehensive Clinicopathologic and Phenotypic Study. Am. J. Surg. Pathol. 2012, 36, 481–499. [Google Scholar] [CrossRef]
- Lo Bello, G.; Akarca, A.U.; Ambrosio, M.R.; Agostinelli, C.; Molina-Kirsch, H.; Ramsay, A.; Rodriguez-Justo, M.; Pugh, M.; Zhao, S.; DeLisser, M.; et al. Granulysin, a Novel Marker for Extranodal NK/T Cell Lymphoma, Nasal Type. Virchows Arch. Int. J. Pathol. 2018, 473, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Howley, P. Fields Virology; Wolters Kluwer: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Fox, C.P.; Shannon-Lowe, C.; Rowe, M. Deciphering the Role of Epstein-Barr Virus in the Pathogenesis of T and NK Cell Lymphoproliferations. Herpesviridae 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Haverkos, B.M.; Coleman, C.; Gru, A.A.; Pan, Z.; Brammer, J.; Rochford, R.; Mishra, A.; Oakes, C.C.; Baiocchi, R.A.; Freud, A.G.; et al. Emerging Insights on the Pathogenesis and Treatment of Extranodal NK/T Cell Lymphomas (ENKTL). Discov. Med. 2017, 23, 189–199. [Google Scholar]
- Takayama, T.; Shin, S.; Kang, S.; Kim, S.J.; Kim, W.S.; Ko, Y.H. Identification of T-Cell Receptor Expression in EBV-Positive Neoplastic Cells in Extranodal NK/T-Cell Lymphoma, Nasal-Type, and Comparison with T-Cell Receptor Gene Rearrangement by BIOMED-2 Assay. Hum. Pathol. 2018, 73, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Kis, L.L.; Nagy, N.; Liu, A.; Harabuchi, Y.; Klein, G.; Klein, E. Concomitant Increase of LMP1 and CD25 (IL-2-Receptor Alpha) Expression Induced by IL-10 in the EBV-Positive NK Lines SNK6 and KAI3. Int. J. Cancer 2006, 119, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-B.; Selvarajan, V.; Huang, G.; Zhou, J.; Feldman, A.L.; Law, M.; Kwong, Y.-L.; Shimizu, N.; Kagami, Y.; Aozasa, K.; et al. Activated Oncogenic Pathways and Therapeutic Targets in Extranodal Nasal-Type NK/T Cell Lymphoma Revealed by Gene Expression Profiling. J. Pathol. 2011, 223, 496–510. [Google Scholar] [CrossRef]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial Survivin Inhibits Apoptosis and Promotes Tumorigenesis. J. Clin. Invest 2004, 114, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Floettmann, J.E.; Eliopoulos, A.G.; Jones, M.; Young, L.S.; Rowe, M. Epstein–Barr Virus Latent Membrane Protein-1 (LMP1) Signalling Is Distinct from CD40 and Involves Physical Cooperation of Its Two C-Terminus Functional Regions. Oncogene 1998, 17, 2383–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Young, L.S. LMP1 Structure and Signal Transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.-W.; Wang, H.; Zhang, W.-W.; Wang, J.-H.; Liu, W.-J.; Xia, Z.-J.; Huang, H.-Q.; Jiang, W.-Q.; Zhang, Y.-J.; Wang, L. PD-L1 Is Upregulated by EBV-Driven LMP1 through NF-ΚB Pathway and Correlates with Poor Prognosis in Natural Killer/T-Cell Lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Nong, L.; Zhang, S.; Zhao, J.; Ti, H.; Dong, Y.; Zhang, B.; Li, T. The Downregulation of PRDM1/Blimp-1 Is Associated with Aberrant Expression of MiR-223 in Extranodal NK/T-Cell Lymphoma, Nasal Type. J. Exp. Clin. Cancer Res. CR 2014, 33, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-Cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [Green Version]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems Database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [Green Version]
- Miyata-Takata, T.; Chuang, S.-S.; Takata, K.; Toji, T.; Maeda, Y.; Sato, Y.; Yoshino, T. Expression of T-Cell Receptor Signalling Pathway Components in Extranodal NK/T-Cell Lymphoma. Histopathology 2018, 73, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Bundo, K.; Hino, A.; Honda, K.; Inoue, A.; Shirakata, M.; Osawa, M.; Tamura, T.; Nariuchi, H.; Oda, H.; et al. Dok-1 and Dok-2 Are Negative Regulators of T Cell Receptor Signaling. Int. Immunol. 2007, 19, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Heyeck, S.D.; Wilcox, H.M.; Bunnell, S.C.; Berg, L.J. Lck Phosphorylates the Activation Loop Tyrosine of the Itk Kinase Domain and Activates Itk Kinase Activity. J. Biol. Chem. 1997, 272, 25401–25408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Park, H.Y.; Kang, S.Y.; Kim, S.J.; Hwang, J.; Lee, S.; Kwak, S.H.; Park, K.S.; Yoo, H.Y.; Kim, W.S.; et al. Genetic Alterations of JAK/STAT Cascade and Histone Modification in Extranodal NK/T-Cell Lymphoma Nasal Type. Oncotarget 2015, 6, 17764–17776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mel, S.; Hue, S.S.-S.; Jeyasekharan, A.D.; Chng, W.-J.; Ng, S.-B. Molecular Pathogenic Pathways in Extranodal NK/T Cell Lymphoma. J. Hematol. Oncol. 2019, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, L.; Huang, S.; Nong, L.; Li, D.; Zhang, B.; Li, T. Aberrant Differential Expression of EZH2 and H3K27me3 in Extranodal NK/T-Cell Lymphoma, Nasal Type, Is Associated with Disease Progression and Prognosis. Hum. Pathol. 2019, 83, 166–176. [Google Scholar] [CrossRef]
- Nakashima, Y.; Tagawa, H.; Suzuki, R.; Karnan, S.; Karube, K.; Ohshima, K.; Muta, K.; Nawata, H.; Morishima, Y.; Nakamura, S.; et al. Genome-Wide Array-Based Comparative Genomic Hybridization of Natural Killer Cell Lymphoma/Leukemia: Different Genomic Alteration Patterns of Aggressive NK-Cell Leukemia and Extranodal Nk/T-Cell Lymphoma, Nasal Type. Genes Chromosomes Cancer 2005, 44, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Nakagawa, M.; Tsuzuki, S.; Takeuchi, I.; Honma, K.; Nakashima, Y.; Shimizu, N.; Ko, Y.-H.; Morishima, Y.; Ohshima, K.; et al. Identification of FOXO3 and PRDM1 as Tumor-Suppressor Gene Candidates in NK-Cell Neoplasms by Genomic and Functional Analyses. Blood 2011, 118, 3195–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Current Topics in Microbiology and Immunology. In Natural Killer Cells; Vivier, E.; Santo, J.D.; Moretta, A. (Eds.) Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Godshalk, S.E.; Bhaduri-McIntosh, S.; Slack, F.J. Epstein-Barr Virus-Mediated Dysregulation of Human MicroRNA Expression. Cell Cycle Georget. Tex 2008, 7, 3595–3600. [Google Scholar] [CrossRef]
- Gatto, G.; Rossi, A.; Rossi, D.; Kroening, S.; Bonatti, S.; Mallardo, M. Epstein-Barr Virus Latent Membrane Protein 1 Trans-Activates MiR-155 Transcription through the NF-KappaB Pathway. Nucleic Acids Res. 2008, 36, 6608–6619. [Google Scholar] [CrossRef]
- Ishii, H.; Takahara, M.; Nagato, T.; Kis, L.L.; Nagy, N.; Kishibe, K.; Harabuchi, Y.; Klein, E. Monocytes Enhance Cell Proliferation and LMP1 Expression of Nasal Natural Killer/T-Cell Lymphoma Cells by Cell Contact-Dependent Interaction through Membrane-Bound IL-15. Int. J. Cancer 2012, 130, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xia, Y.; Feng, L.-N.; Chen, J.-R.; Li, H.-M.; Cui, J.; Cai, Q.-Q.; Sim, K.S.; Nairismägi, M.-L.; Laurensia, Y.; et al. Genetic Risk of Extranodal Natural Killer T-Cell Lymphoma: A Genome-Wide Association Study. Lancet Oncol. 2016, 17, 1240–1247. [Google Scholar] [CrossRef]
- Midgley, R.S.; Bell, A.I.; Yao, Q.Y.; Croom-Carter, D.; Hislop, A.D.; Whitney, B.M.; Chan, A.T.C.; Johnson, P.J.; Rickinson, A.B. HLA-A11-Restricted Epitope Polymorphism among Epstein-Barr Virus Strains in the Highly HLA-A11-Positive Chinese Population: Incidence and Immunogenicity of Variant Epitope Sequences. J. Virol. 2003, 77, 11507–11516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Rijn, M.; Bhargava, V.; Molina-Kirsch, H.; Carlos-Bregni, R.; Warnke, R.A.; Cleary, M.L.; Kamel, O.W. Extranodal Head and Neck Lymphomas in Guatemala: High Frequency of Epstein-Barr Virus-Associated Sinonasal Lymphomas. Hum. Pathol. 1997, 28, 834–839. [Google Scholar] [CrossRef]
- Kimura, H. EBV in T-/NK-Cell Tumorigenesis. In Human Herpesviruses; Kawaguchi, Y., Mori, Y., Kimura, H., Eds.; Springer: Singapore, 2018; pp. 459–475. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Ansell, S.M.; Ai, W.Z.; Barnes, J.; Barta, S.K.; Choi, M.; Clemens, M.W.; Dogan, A.; Greer, J.P.; Halwani, A.; et al. NCCN Guidelines Insights: T-Cell Lymphomas, Version 2. J. Natl. Compr. Cancer Netw. 2018, 16, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Oki, Y. Novel Immunotherapy Options for Extranodal NK/T-Cell Lymphoma. Front. Oncol. 2018, 8, 139. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Suzuki, R.; Oguchi, M. Advances in the Treatment of Extranodal NK/T-Cell Lymphoma, Nasal Type. Blood 2018, 131, 2528–2540. [Google Scholar] [CrossRef]
- Weigel, C.; Mundy-Bosse, B.L.; Wu, Y.-Z.; McConnell, K.; Mishra, A.; Caligiuri, M.A.; Baiocchi, R.A.; Natkunam, Y.; Porcu, P.; Brammer, J.; et al. Abstract LB-102: Extranodal Natural Killer/T Cell Lymphoma (ENKTL) Exhibits an Unprecedented Degree of Global DNA Hypermethylation, Providing a Potent Targeted Therapy in Vivo. Cancer Res. 2019, 79 (Suppl. S13), LB-102. [Google Scholar] [CrossRef]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.-H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.-B.; et al. EZH2 Phosphorylation by JAK3 Mediates a Switch to Noncanonical Function in Natural Killer/T-Cell Lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Nairismägi, M.-L.; Gerritsen, M.E.; Li, Z.M.; Wijaya, G.C.; Chia, B.K.H.; Laurensia, Y.; Lim, J.Q.; Yeoh, K.W.; Yao, X.S.; Pang, W.L.; et al. Oncogenic Activation of JAK3-STAT Signaling Confers Clinical Sensitivity to PRN371, a Novel Selective and Potent JAK3 Inhibitor, in Natural Killer/T-Cell Lymphoma. Leukemia 2018, 32, 1147–1156. [Google Scholar] [CrossRef]
- Liu, J.; Liang, L.; Li, D.; Nong, L.; Zheng, Y.; Huang, S.; Zhang, B.; Li, T. JAK3/STAT3 Oncogenic Pathway and PRDM1 Expression Stratify Clinicopathologic Features of Extranodal NK/T-cell Lymphoma, Nasal Type. Oncol. Rep. 2019, 41, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Zhang, L.; Feng, Y.; Zou, L.-Q. Bortezomib Inhibits Extranodal Natural Killer/T Cell Lymphoma, Nasal Type by Targeting NF-κB Signaling Pathway. Sichuan Da Xue Xue Bao Yi Xue Ban 2019, 50, 311–316. [Google Scholar] [PubMed]
- Li, X.; Cheng, Y.; Zhang, M.; Yan, J.; Li, L.; Fu, X.; Zhang, X.; Chang, Y.; Sun, Z.; Yu, H.; et al. Activity of Pembrolizumab in Relapsed/Refractory NK/T-Cell Lymphoma. J. Hematol. Oncol. 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Guan, P.; Shan, T.; Ye, Y.; Gao, L.; Wang, Z.; Zhao, S.; Zhang, W.; Zhang, L.; Pan, L.; et al. CD30 Expression and Survival in Extranodal NK/T-Cell Lymphoma: A Systematic Review and Meta-Analysis. Oncotarget 2018, 9, 16547–16556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, M.; Ando, J.; Yamazaki, S.; Ishii, M.; Sakiyama, Y.; Harada, S.; Honda, T.; Yamaguchi, T.; Nojima, M.; Ohshima, K.; et al. Long-Term Eradication of Extranodal NK/T Cell Lymphoma, Nasal Type, by Induced Pluripotent Stem Cell-Derived Epstein-Barr Virus-Specific Rejuvenated T Cells in Vivo. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-G.; Kim, N.; Sohn, H.-J.; Lee, S.K.; Oh, S.T.; Lee, H.-J.; Cho, H.-I.; Yim, H.W.; Jung, S.E.; Park, G.; et al. Long-Term Outcome of Extranodal NK/T Cell Lymphoma Patients Treated With Postremission Therapy Using EBV LMP1 and LMP2a-Specific CTLs. Mol. Ther. 2015, 23, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Nagato, T.; Ueda, S.; Takahara, M.; Kishibe, K.; Komabayashi, Y.; Kumai, T.; Ohara, K.; Hirata-Nozaki, Y.; Harabuchi, S.; Hayashi, R.; et al. Cyclin-Dependent Kinase 1 and Survivin as Potential Therapeutic Targets against Nasal Natural Killer/T-Cell Lymphoma. Lab. Invest. 2019, 99, 612–624. [Google Scholar] [CrossRef]
- Go, H.; Jang, J.-Y.; Kim, C.-W.; Huh, J.; Kim, P.-J.; Jeon, Y.K. Identification of MicroRNAs Modulated by DNA Hypomethylating Drugs in Extranodal NK/T-Cell Lymphoma. Leuk. Lymphoma 2020, 61, 66–74. [Google Scholar] [CrossRef]

{kind=link}
| Pathway | Therapeutic | References |
|---|---|---|
| JAK/STAT | Tofacitinib (JAK inhibitor)/PRN371 (JAK3 selective inhibitor)/Stattic (STAT3 inhibitor) | [44,45,46] |
| EZH2 | 3-deazaneplanocin A (EZH2 inhibitor) | [29,44] |
| NF-κB | Bortezomib | [47] |
| PD-1 | Pembrolizumab | [28,48] |
| CD30 | Brentuximab | [49] |
| LMP-1 | LMP1/2a-specific CTL | [51] |
| Survivin | Terameprocol | [15,52] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleem, A.; Natkunam, Y. Extranodal NK/T-Cell Lymphomas: The Role of Natural Killer Cells and EBV in Lymphomagenesis. Int. J. Mol. Sci. 2020, 21, 1501. https://doi.org/10.3390/ijms21041501
Saleem A, Natkunam Y. Extranodal NK/T-Cell Lymphomas: The Role of Natural Killer Cells and EBV in Lymphomagenesis. International Journal of Molecular Sciences. 2020; 21(4):1501. https://doi.org/10.3390/ijms21041501
Chicago/Turabian StyleSaleem, Atif, and Yasodha Natkunam. 2020. "Extranodal NK/T-Cell Lymphomas: The Role of Natural Killer Cells and EBV in Lymphomagenesis" International Journal of Molecular Sciences 21, no. 4: 1501. https://doi.org/10.3390/ijms21041501
APA StyleSaleem, A., & Natkunam, Y. (2020). Extranodal NK/T-Cell Lymphomas: The Role of Natural Killer Cells and EBV in Lymphomagenesis. International Journal of Molecular Sciences, 21(4), 1501. https://doi.org/10.3390/ijms21041501