From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. R-Loops
2.1. R-Loops and Replication
2.2. R-Loops and Cancer
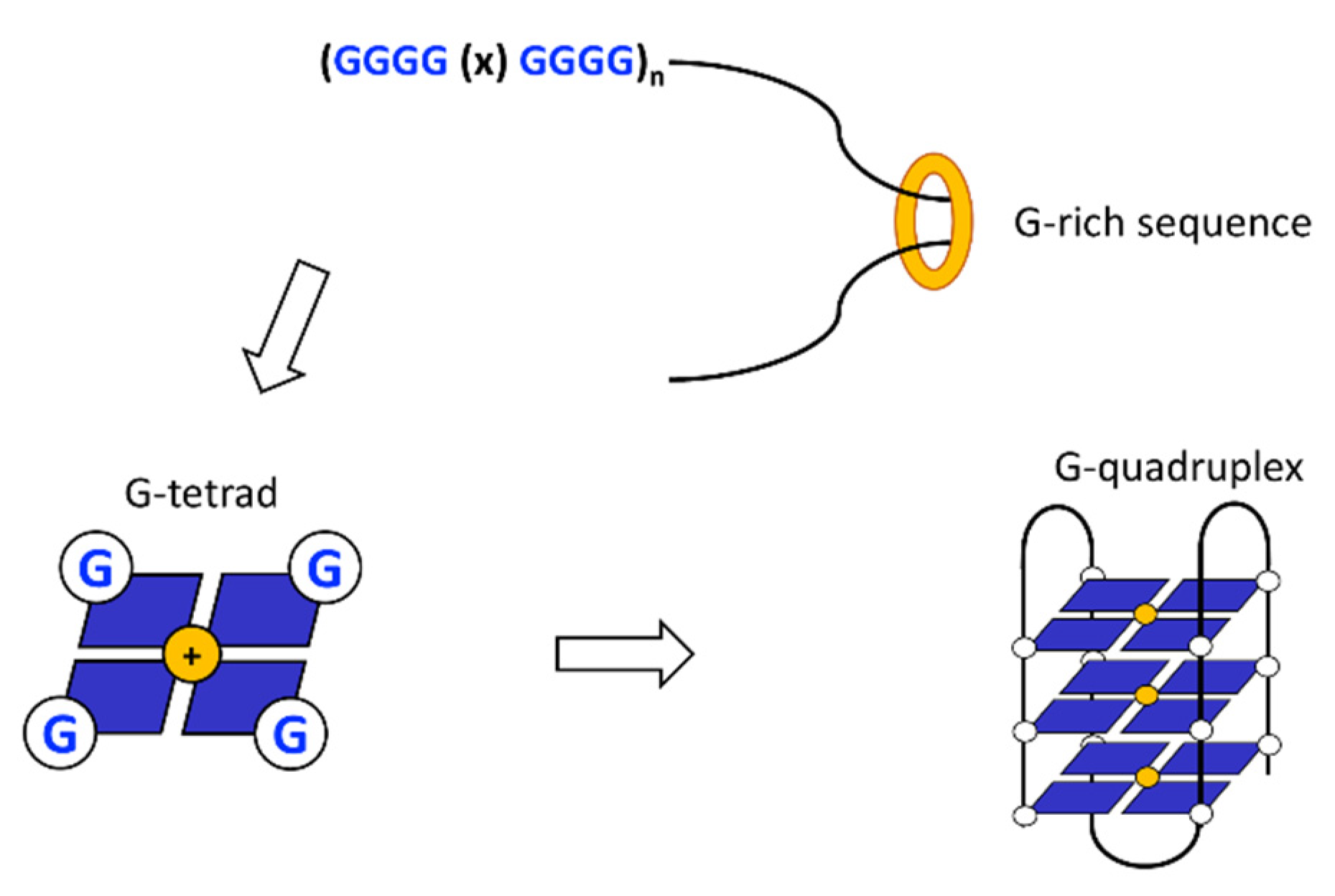
3. G-Quadruplex
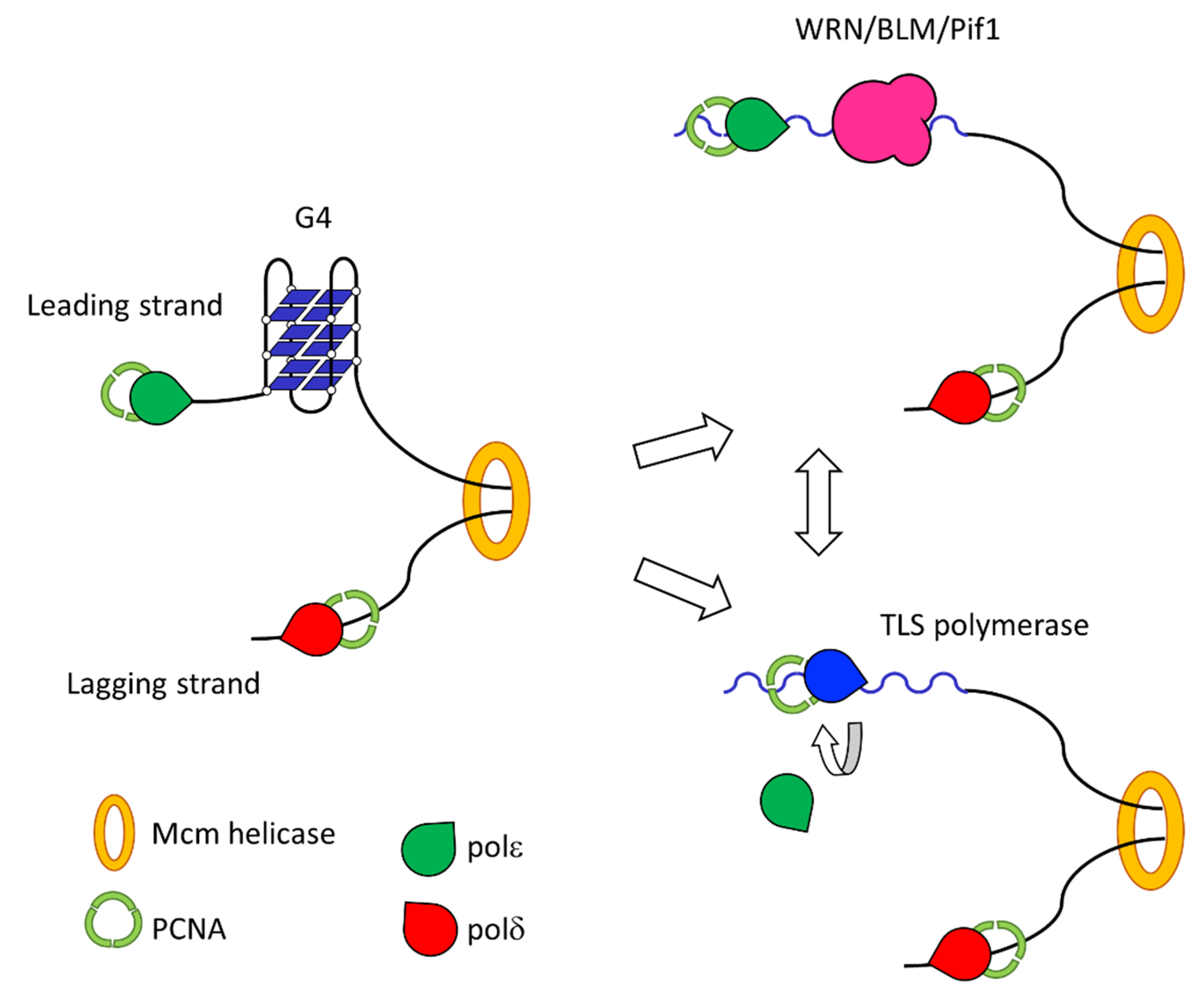
3.1. Replication of G-Quadruplex DNA
3.2. G-Quadruplex and Cancer
4. Common Fragile Sites
4.1. Replication of CFS
4.2. CFS and Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATR | Ataxia Telangectasia and Rad3 Related |
| FANCM | Fanconi Anemia Complementation Group M |
| FANCD2 | Fanconi Anemia Complementation Group D2 |
| FANCI | Fanconi Anemia Complementation Group I |
| SLX4 | SLX4 Structure-Specific Endonuclease Subunit |
| MUS81 | gene name |
| XPF | Xeroderma Pigmentosum Group F |
| Rad6 | Radiation gene 6 homolog |
| SMARCAL1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1 |
| ZRANB3 | zinc finger RANBP2-type containing 3 |
| HLTF | helicase like transcription factor |
| SHPRH | SNF2 histone linker PHD RING helicase |
| BRCA1 | BReast CAncer 1 |
| BRCA2 | BReast CAncer 2 |
| WRN | Werner syndrome RecQ like helicase |
| RECQ1 | RecQ Protein-Like 1 |
| ChIP | Chromatin ImmunoPrecipitation |
| AQR | aquarius intron-binding spliceosomal factor |
| DDX19 | DEAD-box helicase 19 |
| DDX23 | DEAD-box helicase 23 |
| DDX1 | DEAD-box helicase 1 |
| DDX5 | DEAD-box helicase 5 |
| DHX9 | DExH-box helicase 9 |
| BLM | Bloom syndrome RecQ like helicase |
| XPG | Xeroderma Pigmentosum Group F |
| PCID2 | PCI domain containing 2 |
| TREX-2 | Transcription and export complex 2 |
| Mre11 | meiotic recombination 11 homolog |
| EME1 | essential meiotic structure-specific endonuclease 1 |
| Chk1 | checkpoint kinase 1 |
| HRAS | Harvey rat sarcoma viral oncogene homolog |
| EWS-FLI1 | EWS RNA binding protein 1- Friend leukemia virus integration 1 fusion gene |
| PARP | poly(ADP-ribose) polymerases |
| Pif1 | PIF1 5′-to-3′ DNA helicase |
| Rev1 | REV1 DNA directed polymerase |
| DT40 | chicken B cell line derived from an avian leukosis virus (ALV)-induced bursal lymphoma |
| BU-1 | Chicken B-cell marker chB6 |
| DNMT1 | DNA methyltransferase 1 |
| cMYC | MYC proto-oncogene, bHLH transcription factor |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| c-KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog |
| BCL2 | BCL2 apoptosis regulator |
| VEGF | vascular endothelial growth factor A |
| RHSP4 | Telomerase inhibitor |
| EMICORON | G4-interactive molecule |
| HCT116 Human | COLORECTAL CARCINOMA cell line |
| HEK293T | human embryonic kidney 293 T |
| FHIT | ragile histidine triad diadenosine triphosphatase |
| WWOX | WW domain containing oxidoreductase |
| POLD3 | DNA polymerase delta 3 |
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: what makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. Dna Repair 2016, 44, 68–75. [Google Scholar] [CrossRef]
- Quinet, A.; Lemacon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Rondón, A.G.; Aguilera, A. What causes an RNA-DNA hybrid to compromise genome integrity? DNA Repair(Amst) 2019, 81, 102660. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Vanoosthuyse, V. Strengths and weaknesses of the current strategies to map and characterize R-loops. Non-coding RNA 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA: DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. Tig. 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Gish, G.; Braunschweig, U.; Li, Y.; Ni, Z.; Schmitges, F.W.; Zhong, G.; Liu, K.; Li, W.; Moffat, J.; et al. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 2016, 529, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martinez, S.; Tsanov, N.; Abou Merhi, R.; Maiorano, D. An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. Embo J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef]
- Bonnet, A.; Grosso, A.R.; Elkaoutari, A.; Coleno, E.; Presle, A.; Sridhara, S.C.; Janbon, G.; Geli, V.; de Almeida, S.F.; Palancade, B. Introns Protect Eukaryotic Genomes from Transcription-Associated Genetic Instability. Mol. Cell 2017, 67, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro de Almeida, C.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol. Cell 2018, 70, 650–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, F.A.; Cloutier, S.C.; Tran, E.J.; Jankowsky, E. The DEAD-box protein Dbp2p is linked to noncoding RNAs, the helicase Sen1p, and R-loops. RNA 2018, 24, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.Y.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef] [PubMed]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [Green Version]
- Zatreanu, D.; Han, Z.; Mitter, R.; Tumini, E.; Williams, H.; Gregersen, L.; Dirac-Svejstrup, A.B.; Roma, S.; Stewart, A.; Aguilera, A.; et al. Elongation Factor TFIIS Prevents Transcription Stress and R-Loop Accumulation to Maintain Genome Stability. Mol. Cell 2019, 76, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rubio, M.; Aguilera, P.; Lafuente-Barquero, J.; Ruiz, J.F.; Simon, M.N.; Geli, V.; Rondon, A.G.; Aguilera, A. Yra1-bound RNA-DNA hybrids cause orientation-independent transcription-replication collisions and telomere instability. Genes Dev. 2018, 32, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Soo Shin, J.H.; Hanawalt, P.C. Strong transcription blockage mediated by R-loop formation within a G-rich homopurine-homopyrimidine sequence localized in the vicinity of the promoter. Nucleic Acids Res. 2017, 45, 6589–6599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 2017, 170, 787–799.e718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PloS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: the interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 2020, 77, 528–541.e528. [Google Scholar] [CrossRef] [PubMed]
- Matos, D.A.; Zhang, J.M.; Ouyang, J.; Nguyen, H.D.; Genois, M.M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2020, 77, 514–527.e514. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PloS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Andreassen, P.R.; D’Andrea, A.D. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell. Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef] [Green Version]
- Costantino, L.; Koshland, D. Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol. Cell 2018, 71, 487–497.e483. [Google Scholar] [CrossRef] [Green Version]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Grabczyk, E.; Mancuso, M.; Sammarco, M.C. A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007, 35, 5351–5359. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Dent, S.Y.; Wilson, J.H.; Wells, R.D.; Napierala, M. R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. USA 2010, 107, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquette, M.L.; Handa, P.; Vincent, J.A.; Taylor, A.F.; Maizels, N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, V.A.; Bondarenko, V.; Wongsurawat, T.; Yenamandra, S.P.; Jenjaroenpun, P. Toward predictive R-loop computational biology: genome-scale prediction of R-loops reveals their association with complex promoter structures, G-quadruplexes and transcriptionally active enhancers. Nucleic Acids Res. 2018, 46, 7566–7585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118.e1115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chiang, H.C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Jones, S.E.; Fleuren, E.D.G.; Frankum, J.; Konde, A.; Williamson, C.T.; Krastev, D.B.; Pemberton, H.N.; Campbell, J.; Gulati, A.; Elliott, R.; et al. ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res. 2017, 77, 7014–7026. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, S.M. Discovery of alternative DNA structures: a heroic decade (1979-1989). Front. Biosci. A J. Virtual Libr. 2008, 13, 1064–1071. [Google Scholar] [CrossRef]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinog. 2009, 48, 273–285. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M., Jr.; Sen, D.; Lansdorp, P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Capra, J.A.; Paeschke, K.; Singh, M.; Zakian, V.A. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010, 6, e1000861. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guedin, A.; Mergny, J.L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, S.Q.; Ghanbarian, A.T.; Spiegel, J.; Martinez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Mulhearn, D.S.; Adhikari, S.; Martinez Cuesta, S.; Di Antonio, M.; Erard, N.; Hannon, G.J.; Tannahill, D.; Balasubramanian, S. Genetic interactions of G-quadruplexes in humans. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Woodford, K.J.; Howell, R.M.; Usdin, K. A novel K (+)-dependent DNA synthesis arrest site in a commonly occurring sequence motif in eukaryotes. J. Biol. Chem. 1994, 269, 27029–27035. [Google Scholar]
- Kamath-Loeb, A.S.; Loeb, L.A.; Johansson, E.; Burgers, P.M.; Fry, M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001, 276, 16439–16446. [Google Scholar] [CrossRef] [Green Version]
- Kruisselbrink, E.; Guryev, V.; Brouwer, K.; Pontier, D.B.; Cuppen, E.; Tijsterman, M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr. Biol. Cb 2008, 18, 900–905. [Google Scholar] [CrossRef] [Green Version]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom’s syndrome helicase unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592. [Google Scholar] [CrossRef] [Green Version]
- Fry, M.; Loeb, L.A. Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem. 1999, 274, 12797–12802. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Deoxycytidyl transferase activity of yeast REV1 protein. Nature 1996, 382, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Svikovic, S.; Sale, J.E. The Effects of Replication Stress on S Phase Histone Management and Epigenetic Memory. J. Mol. Biol. 2017, 429, 2011–2029. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.D.; Simpson, M.; Gaszner, M.; Allis, C.D.; Felsenfeld, G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 2001, 293, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Reams, C.; Simpson, L.J.; Sale, J.E. Epigenetic instability due to defective replication of structured DNA. Mol. Cell 2010, 40, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Guilbaud, G.; Murat, P.; Papadopoulou, C.; Sarkies, P.; Prioleau, M.N.; Balasubramanian, S.; Sale, J.E. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. Embo J. 2014, 33, 2507–2520. [Google Scholar] [CrossRef]
- Eddy, S.; Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Choi, J.Y.; Eoff, R.L. Human Rev1 polymerase disrupts G-quadruplex DNA. Nucleic Acids Res. 2014, 42, 3272–3285. [Google Scholar] [CrossRef] [Green Version]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.; Maddukuri, L.; Ketkar, A.; Zafar, M.K.; Henninger, E.E.; Pursell, Z.F.; Eoff, R.L. Evidence for the kinetic partitioning of polymerase activity on G-quadruplex DNA. Biochemistry 2015, 54, 3218–3230. [Google Scholar] [CrossRef] [Green Version]
- Cea, V.; Cipolla, L.; Sabbioneda, S. Replication of Structured DNA and its implication in epigenetic stability. Front. Genet. 2015, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Kuryavyi, V.; Phan, A.T.; Patel, D.J. Solution structures of all parallel-stranded monomeric and dimeric G-quadruplex scaffolds of the human c-kit2 promoter. Nucleic Acids Res. 2010, 38, 6757–6773. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.; Hatzakis, E.; Guo, K.; Carver, M.; Yang, D. Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013, 41, 10584–10592. [Google Scholar] [CrossRef]
- Agrawal, P.; Lin, C.; Mathad, R.I.; Carver, M.; Yang, D. The major G-quadruplex formed in the human BCL-2 proximal promoter adopts a parallel structure with a 13-nt loop in K+ solution. J. Am. Chem. Soc. 2014, 136, 1750–1753. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Hermetz, K.E.; Conneely, K.N.; Rudd, M.K. Tandem repeats and G-rich sequences are enriched at human CNV breakpoints. PLoS ONE 2014, 9, e101607. [Google Scholar] [CrossRef]
- De, S.; Michor, F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011, 29, 1103–1108. [Google Scholar] [CrossRef]
- Katapadi, V.K.; Nambiar, M.; Raghavan, S.C. Potential G-quadruplex formation at breakpoint regions of chromosomal translocations in cancer may explain their fragility. Genomics 2012, 100, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Tannahill, D.; Miller, J.; Howat, W.J.; Balasubramanian, S. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS ONE 2014, 9, e102711. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Salvati, E.; Scarsella, M.; Porru, M.; Rizzo, A.; Iachettini, S.; Tentori, L.; Graziani, G.; D’Incalci, M.; Stevens, M.F.; Orlandi, A.; et al. PARP1 is activated at telomeres upon G4 stabilization: possible target for telomere-based therapy. Oncogene 2010, 29, 6280–6293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheli, E.; Altieri, A.; Cianni, L.; Cingolani, C.; Iachettini, S.; Bianco, A.; Leonetti, C.; Cacchione, S.; Biroccio, A.; Franceschin, M.; et al. Perylene and coronene derivatives binding to G-rich promoter oncogene sequences efficiently reduce their expression in cancer cells. Biochimie 2016, 125, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Franceschin, M.; Rizzo, A.; Casagrande, V.; Salvati, E.; Alvino, A.; Altieri, A.; Ciammaichella, A.; Iachettini, S.; Leonetti, C.; Ortaggi, G.; et al. Aromatic core extension in the series of N-cyclic bay-substituted perylene G-quadruplex ligands: increased telomere damage, antitumor activity, and strong selectivity for neoplastic over healthy cells. ChemMedChem 2012, 7, 2144–2154. [Google Scholar] [CrossRef]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef] [Green Version]
- McLuckie, K.I.; Di Antonio, M.; Zecchini, H.; Xian, J.; Caldas, C.; Krippendorff, B.F.; Tannahill, D.; Lowe, C.; Balasubramanian, S. G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J. Am. Chem. Soc. 2013, 135, 9640–9643. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Boldog, F.; Gemmill, R.M.; West, J.; Robinson, M.; Robinson, L.; Li, E.; Roche, J.; Todd, S.; Waggoner, B.; Lundstrom, R.; et al. Chromosome 3p14 homozygous deletions and sequence analysis of FRA3B. Hum. Mol. Genet. 1997, 6, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Mishmar, D.; Rahat, A.; Scherer, S.W.; Nyakatura, G.; Hinzmann, B.; Kohwi, Y.; Mandel-Gutfroind, Y.; Lee, J.R.; Drescher, B.; Sas, D.E.; et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. USA 1998, 95, 8141–8146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. Mmbr 2007, 71, 13–35. [Google Scholar] [CrossRef] [Green Version]
- Shima, N.; Pederson, K.D. Dormant origins as a built-in safeguard in eukaryotic DNA replication against genome instability and disease development. DNA Repair 2017, 56, 166–173. [Google Scholar] [CrossRef]
- Palakodeti, A.; Lucas, I.; Jiang, Y.; Young, D.J.; Fernald, A.A.; Karrison, T.; Le Beau, M.M. Impaired replication dynamics at the FRA3B common fragile site. Hum. Mol. Genet. 2010, 19, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachages, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Miotto, B.; Ji, Z.; Struhl, K. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, E4810–E4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, C.M.; Guo, S.W.; Hall, B.K.; Boldog, F.; Gemmill, R.M.; Chandrasekharappa, S.C.; Barcroft, C.L.; Drabkin, H.A.; Glover, T.W. Multicolor FISH mapping of YAC clones in 3p14 and identification of a YAC spanning both FRA3B and the t(3;8) associated with hereditary renal cell carcinoma. Genomics 1994, 22, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradee, W.; Mullins, C.; He, Z.; Glover, T.; Wilke, C.; Opalka, B.; Schutte, J.; Smith, D.I. Precise localization of aphidicolin-induced breakpoints on the short arm of human chromosome 3. Genomics 1995, 27, 358–361. [Google Scholar] [CrossRef]
- Mangelsdorf, M.; Ried, K.; Woollatt, E.; Dayan, S.; Eyre, H.; Finnis, M.; Hobson, L.; Nancarrow, J.; Venter, D.; Baker, E.; et al. Chromosomal fragile site FRA16D and DNA instability in cancer. Cancer Res. 2000, 60, 1683–1689. [Google Scholar]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Le Beau, M.M.; Drabkin, H.; Glover, T.W.; Gemmill, R.; Rassool, F.V.; McKeithan, T.W.; Smith, D.I. AnFHIT tumor suppressor gene? Genes Chromosomes Cancer 1998, 21, 281–289. [Google Scholar] [CrossRef]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 2010, 119, 575–587. [Google Scholar] [CrossRef]
- Pelliccia, F.; Bosco, N.; Curatolo, A.; Rocchi, A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 2008, 121, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Hickson, I.D. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol. 2011, 22, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Rajendran, S.; Birkeland, S.R.; Wilson, T.E.; Glover, T.W. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLoS Genet. 2012, 8, e1002981. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Carvalho, C.M.; Hastings, P.J.; Lupski, J.R. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 2012, 22, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Schwarzbraun, T.; Speicher, M.R.; Nigg, E.A. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma 2008, 117, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Pladevall-Morera, D.; Munk, S.; Ingham, A.; Garribba, L.; Albers, E.; Liu, Y.; Olsen, J.V.; Lopez-Contreras, A.J. Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res. 2019, 47, 8004–8018. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Barnes, R.P.; Hile, S.E.; Lee, M.Y.; Eckert, K.A. DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair 2017, 57, 1–11. [Google Scholar] [CrossRef]
- Karras, J.R.; Schrock, M.S.; Batar, B.; Huebner, K. Fragile Genes That Are Frequently Altered in Cancer: Players Not Passengers. Cytogenet. Genome Res. 2016, 150, 208–216. [Google Scholar] [CrossRef]
- Schrock, M.S.; Huebner, K. WWOX: a fragile tumor suppressor. Exp. Biol. Med. 2015, 240, 296–304. [Google Scholar] [CrossRef]
- Hussain, T.; Liu, B.; Shrock, M.S.; Williams, T.; Aldaz, C.M. WWOX, the FRA16D gene: A target of and a contributor to genomic instability. Genes Chromosomes Cancer 2019, 58, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Tanna, M.; Aqeilan, R.I. Modeling WWOX Loss of Function in vivo: What Have We Learned? Front. Oncol. 2018, 8, 420. [Google Scholar] [CrossRef] [Green Version]
- Aqeilan, R.I.; Hagan, J.P.; Aqeilan, H.A.; Pichiorri, F.; Fong, L.Y.; Croce, C.M. Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. 2007, 67, 5606–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanesi, N.; Fidanza, V.; Fong, L.Y.; Mancini, R.; Druck, T.; Valtieri, M.; Rudiger, T.; McCue, P.A.; Croce, C.M.; Huebner, K. The tumor spectrum in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10250–10255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 2011, 18, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Wu, X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers 2019, 11, 1467. [Google Scholar] [CrossRef] [Green Version]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 2018, 71, 158–163. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffia, A.; Ranise, C.; Sabbioneda, S. From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. Int. J. Mol. Sci. 2020, 21, 1506. https://doi.org/10.3390/ijms21041506
Maffia A, Ranise C, Sabbioneda S. From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. International Journal of Molecular Sciences. 2020; 21(4):1506. https://doi.org/10.3390/ijms21041506
Chicago/Turabian StyleMaffia, Antonio, Cecilia Ranise, and Simone Sabbioneda. 2020. "From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork" International Journal of Molecular Sciences 21, no. 4: 1506. https://doi.org/10.3390/ijms21041506
APA StyleMaffia, A., Ranise, C., & Sabbioneda, S. (2020). From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. International Journal of Molecular Sciences, 21(4), 1506. https://doi.org/10.3390/ijms21041506