Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML


Abstract
:1. Introduction
2. Fms-Like Tyrosine Kinase 3 (FLT3)
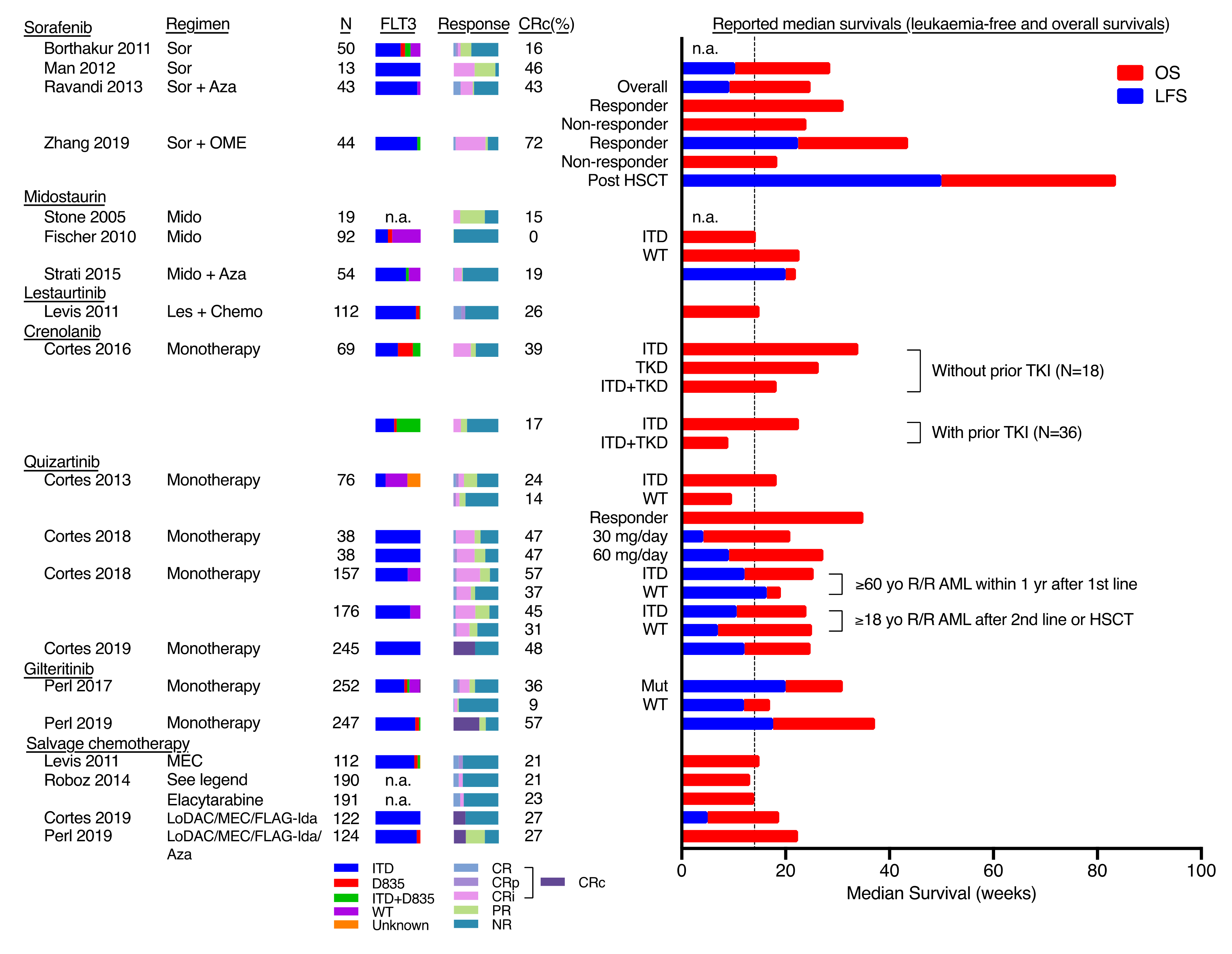
3. FLT3 Inhibitors
4. Clonal Evolution
4.1. Emergence of FL3-TKD Mutations
4.2. Emergence of Non-FLT3 Mutations
5. Adaptive Cellular Mechanisms
5.1. Upregulation of FLT3 Ligand
5.2. Increase in Intracellular pH
5.3. Upregulation of other Cooperative Kinases
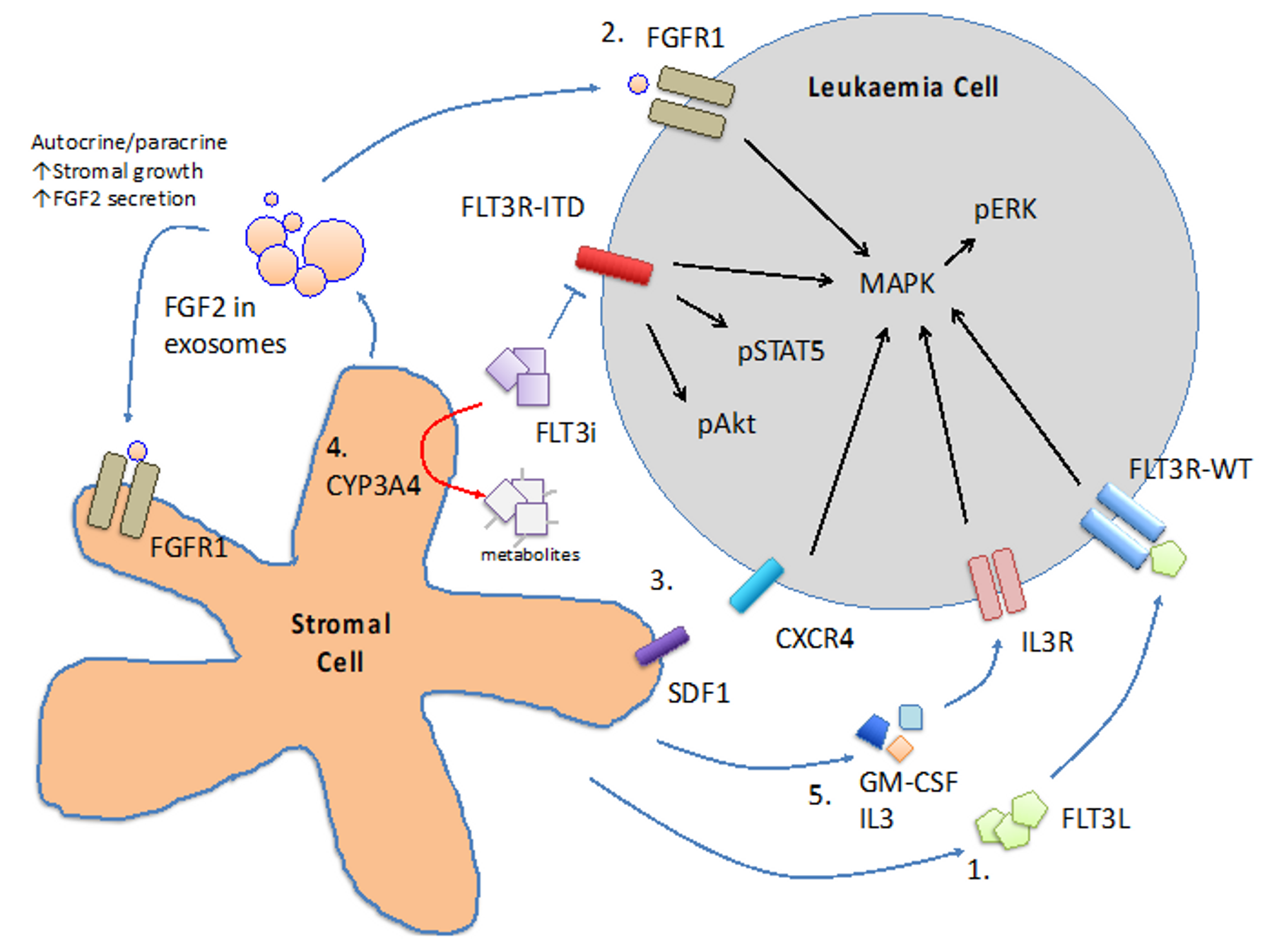
6. Microenvironment Protection
6.1. Cytokines
6.2. CYP3A4 in BM Stromal Cells
7. Clinical Strategies to Overcome Drug Resistance in FLT3-ITD AML
7.1. Upfront FLT3 Inhibitors in Combination with Chemotherapy
7.2. Combination of FLT3 Inhibitors with Low Intensity Regimen
7.3. HSCT with FLT3 Inhibitor as Maintenance Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukaemia |
| CR | Complete remission |
| CRc | Composite complete remission |
| CRi | Complete remission with incomplete haematological response |
| CRp | Complete remission with incomplete platelet count |
| FLT3L | FLT3 ligand |
| HSCT | Haematopoietic stem cell transplantation |
| HSPC | Haematopoietic stem and progenitor cell |
| ITD | Internal tandem duplication |
| OME | Omacetaxine mepesuccinate |
| PR | Partial response |
| R/R | Relapse or refractory |
| TKD | Tyrosine kinase domain |
| TKI | Tyrosine kinase inhibitor |
References
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Kramer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Santos, F.P.; Jones, D.; Qiao, W.; Cortes, J.E.; Ravandi, F.; Estey, E.E.; Verma, D.; Kantarjian, H.; Borthakur, G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer 2011, 117, 2145–2155. [Google Scholar] [CrossRef] [Green Version]
- Meshinchi, S.; Stirewalt, D.L.; Alonzo, T.A.; Boggon, T.J.; Gerbing, R.B.; Rocnik, J.L.; Lange, B.J.; Gilliland, D.G.; Radich, J.P. Structural and numerical variation of FLT3/ITD in pediatric AML. Blood 2008, 111, 4930–4933. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. Medical Research Council Adult Leukaemia Working Party. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters—An analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.; Chow, H.C.; Ma, A.C.; Choi, W.W.; Lok, S.; Cheung, A.M.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Schwable, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. AML-Associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef]
- Leischner, H.; Albers, C.; Grundler, R.; Razumovskaya, E.; Spiekermann, K.; Bohlander, S.; Ronnstrand, L.; Gotze, K.; Peschel, C.; Duyster, J. SRC is a signaling mediator in FLT3-ITD-but not in FLT3-TKD-positive AML. Blood 2012, 119, 4026–4033. [Google Scholar] [CrossRef]
- Battipaglia, G.; Ruggeri, A.; Massoud, R.; El Cheikh, J.; Jestin, M.; Antar, A.; Ahmed, S.O.; Rasheed, W.; Shaheen, M.; Belhocine, R.; et al. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer 2017, 123, 2867–2874. [Google Scholar] [CrossRef]
- Brunner, A.M.; Li, S.; Fathi, A.T.; Wadleigh, M.; Ho, V.T.; Collier, K.; Connolly, C.; Ballen, K.K.; Cutler, C.S.; Dey, B.R.; et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br. J. Haematol. 2016, 175, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Burchert, A.; Bug, G.; Finke, J.; Stelljes, M.; Rollig, C.; Wäsch, R.; Bornhäuser, M.; Berg, T.; Lang, F.; Ehninger, G.; et al. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3-ITD positive AML: Results from the randomized, double-blind, placebo-controlled multicentre sormain trial. Blood 2018, 132, 661. [Google Scholar] [CrossRef]
- Fischer, T.; Stone, R.M.; Deangelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Zhang, C.; Lam, S.S.Y.; Leung, G.M.K.; Tsui, S.P.; Yang, N.; Ng, N.K.L.; Ip, H.W.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; et al. Sorafenib and omacetaxine mepesuccinate as a safe and effective treatment for acute myeloid leukemia carrying internal tandem duplication of Fms-like tyrosine kinase 3. Cancer 2019, 126, 344–353. [Google Scholar] [CrossRef]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef]
- Strati, P.; Kantarjian, H.; Ravandi, F.; Nazha, A.; Borthakur, G.; Daver, N.; Kadia, T.; Estrov, Z.; Garcia-Manero, G.; Konopleva, M.; et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am. J. Hematol. 2015, 90, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.M.; Kadia, T.M.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Daver, N.G.; Pemmaraju, N.; Jabbour, E.; Estrov, Z. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. Am. Soc. Clin. Oncol. J. 2016, 34, 7008. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Dohner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Roboz, G.J.; Rosenblat, T.; Arellano, M.; Gobbi, M.; Altman, J.K.; Montesinos, P.; O’Connell, C.; Solomon, S.R.; Pigneux, A.; Vey, N.; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2014, 32, 1919–1926. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal selection with Ras pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Lasater, E.A.; Zhu, X.; Lin, K.C.; Stewart, W.K.; Damon, L.E.; Salerno, S.; Shah, N.P. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood 2013, 121, 3165–3171. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “Gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and In Vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ishikawa, Y.; Akashi, A.; Naoe, T.; Kiyoi, H. Co-Expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget 2016, 7, 47018–47032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, C.H.; Lam, S.S.; Sun, M.K.; Chow, H.C.; Gill, H.; Kwong, Y.L.; Leung, A.Y. A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood 2014, 123, 2530–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.S.; Maciel, T.T.; Hospital, M.A.; Yin, C.; Mazed, F.; Townsend, E.C.; Pilorge, S.; Lambert, M.; Paubelle, E.; Jacquel, A.; et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci. Adv. 2015, 1, e1500221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Dumas, P.Y.; Naudin, C.; Martin-Lanneree, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossee, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [Green Version]
- Ueno, Y.; Mori, M.; Kamiyama, Y.; Saito, R.; Kaneko, N.; Isshiki, E.; Kuromitsu, S.; Takeuchi, M. Evaluation of gilteritinib in combination with chemotherapy in preclinical models of FLT3-ITD+ acute myeloid leukemia. Oncotarget 2019, 10, 2530–2545. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Scadden, D.T. Nice neighborhood: Emerging concepts of the stem cell niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 1915, 177, 1915–1932.e1916. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.J.; Sugita, M.; Koblish, H.; Perl, A.E.; Carroll, M. Hematopoietic cytokines mediate resistance to targeted therapy in FLT3-ITD acute myeloid leukemia. Blood Adv. 2019, 3, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traer, E.; Martinez, J.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Kovacsovics, T.; Tyner, J.W.; Wong, M.; Druker, B.J. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016, 76, 6471–6482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribeiro, R.; Viola, S.K.; Edwards, D.K.T.; Agarwal, A.; Lopez, C.; Jorgens, D.; et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. eLife 2019, 8, e40033. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Broxmeyer, H.E.; Pelus, L.M. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood 2005, 105, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; McQueen, T.; Chen, Y.; Jacamo, R.; Konopleva, M.; Shinojima, N.; Shpall, E.; Huang, X.; Andreeff, M. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood 2011, 118, 4431–4439. [Google Scholar] [CrossRef] [Green Version]
- Teo, Y.L.; Ho, H.K.; Chan, A. Metabolism-Related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br. J. Clin. Pharm. 2015, 79, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Kadia, T.; Patel, K.; Loghavi, S.; Garcia-Manero, G.; Jabbour, E.J.; DiNardo, C.; Pemmaraju, N.; Daver, N.; et al. Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed, FLT3-internal tandem duplication mutation-positive acute myeloid leukemia. Cancer 2019, 125, 3755–3766. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Coombs, C.C.; Wang, E.S.; Walter, R.B.; Karanes, C.; Vigil, C.E.; Messahel, B.; Stone, R.M.; Collins, R.H. Younger patients with newly diagnosed FLT3-mutant AML treated with crenolanib plus chemotherapy achieve adequate free crenolanib levels and durable remissions. Blood 2019, 134, 1326. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.J.; Tardi, P.; Loriaux, M.M.; Spurgeon, S.E.; Traer, E.; Kovacsovics, T.; Mayer, L.D.; Tyner, J.W. CPX-351 exhibits potent and direct ex vivo cytotoxicity against AML blasts with enhanced efficacy for cells harboring the FLT3-ITD mutation. Leuk. Res. 2017, 53, 39–49. [Google Scholar] [CrossRef]
- Lam, S.S.; Ho, E.S.; He, B.L.; Wong, W.W.; Cher, C.Y.; Ng, N.K.; Man, C.H.; Gill, H.; Cheung, A.M.; Ip, H.W.; et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 359ra129. [Google Scholar] [CrossRef] [PubMed]
- Gurel, G.; Blaha, G.; Moore, P.B.; Steitz, T.A. U2504 determines the species specificity of the A-site cleft antibiotics: The structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009, 389, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Leung, G.M.; Tsui, S.P.; Ip, H.-W.; Ma, E.S.K.; Au, C.H.; Chan, C.T.L.; Ng, K.L.N.; Yip, S.F.; Kho, B.; et al. A phase II single-arm open-labeled study evaluating combination of Quizartinib and Omacetaxine Mepesuccinate (QUIZOM) in newly diagnosed or relapsed/refractory AML carrying FlT3-ITD. Blood 2019, 134, 3825. [Google Scholar] [CrossRef]
- Gill, H.; Man, C.H.; Ip, A.H.; Choi, W.W.; Chow, H.C.; Kwong, Y.L.; Leung, A.Y. Azacitidine as post-remission consolidation for sorafenib-induced remission of Fms-like tyrosine kinase-3 internal tandem duplication positive acute myeloid leukemia. Haematologica 2015, 100, e250–e253. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, M.R.; Portwood, S.; Griffiths, E.A.; Thompson, J.E.; Ford, L.A.; Freyer, C.W.; Wetzler, M.; Wang, E.S. Decitabine and Sorafenib therapy in FLT-3 ITD-mutant acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15 (Suppl. S73–S79). [Google Scholar] [CrossRef]
- Chang, E.; Ganguly, S.; Rajkhowa, T.; Gocke, C.D.; Levis, M.; Konig, H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia 2016, 30, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, M.; Kantarjian, H.M.; Daver, N.; Borthakur, G.; Ohanian, M.; Kadia, T.; DiNardo, C.D.; Jain, N.; Estrov, Z.; Ferrajoli, A.; et al. The combination of Quizartinib with Azacitidine or low dose Cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: Interim report of a phase I/II trial. Blood 2017, 130, 723. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib combined with 5-azacytidine in older patients with untreated FLT3-ITD mutated acute myeloid leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornhauser, M.; Illmer, T.; Schaich, M.; Soucek, S.; Ehninger, G.; Thiede, C. AML SHG 96 Study Group. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood 2007, 109, 2264–2265, author reply 2265. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, S.; Ferrara, F.; Leoni, F.; Ciolli, S.; Pollio, F.; D’Amico, M.R.; Celentano, M.; Viola, A.; Vicari, L.; Izzo, B.; et al. Myeloablative chemotherapy followed by autologous stem cell infusion may overcome the adverse prognostic impact of FLT3 (foetal liver tyrosine kinase 3) mutations in patients with acute myeloid leukaemia and normal karyotype. Hematol. Oncol. 2007, 25, 1–5. [Google Scholar] [CrossRef]
- DeZern, A.E.; Sung, A.; Kim, S.; Smith, B.D.; Karp, J.E.; Gore, S.D.; Jones, R.J.; Fuchs, E.; Luznik, L.; McDevitt, M.; et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: Outcomes from 133 consecutive newly diagnosed patients from a single institution. Biol. Blood Marrow Transplant. 2011, 17, 1404–1409. [Google Scholar] [CrossRef] [Green Version]
- Canaani, J.; Labopin, M.; Huang, X.J.; Arcese, W.; Ciceri, F.; Blaise, D.; Irrera, G.; Corral, L.L.; Bruno, B.; Santarone, S.; et al. T-Cell replete haploidentical stem cell transplantation attenuates the prognostic impact of FLT3-ITD in acute myeloid leukemia: A report from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Am. J. Hematol. 2018, 93, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Yamaguchi, H.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; Kobayashi, Y.; et al. Prognostic impact of low allelic ratio FLT3-ITD and NPM1 mutation in acute myeloid leukemia. Blood Adv. 2018, 2, 2744–2754. [Google Scholar] [CrossRef] [Green Version]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Muller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef]
- Maziarz, R.T.; Fernandez, H.; Patnaik, M.M.; Scott, B.L.; Mohan, S.; Deol, A.; Rowley, S.D.; Kim, D.D.; Rajkhowa, T.; Haines, K.; et al. Radius: Midostaurin (mido) plus standard of care (SOC) after allogeneic stem cell transplant (alloSCT) in patients (pts) with FLT3-internal tandem duplication (ITD)-mutated acute myeloid leukemia (AML). Biol. Blood Marrow Transplant. 2019, 25, S11–S12. [Google Scholar] [CrossRef] [Green Version]
- Sandmaier, B.M.; Khaled, S.; Oran, B.; Gammon, G.; Trone, D.; Frankfurt, O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am. J. Hematol. 2018, 93, 222–231. [Google Scholar] [CrossRef]
- Lange, A.; Jaskula, E.; Lange, J.; Dworacki, G.; Nowak, D.; Simiczyjew, A.; Mordak-Domagala, M.; Sedzimirska, M. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+PD-1+ (CD279+) lymphocytes in marrow. PLoS ONE 2018, 13, e0190525. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Therapeutic Agents | Indications |
|---|---|
| FLT3 inhibitors | |
| 1. Midostaurin | Newly diagnosed FLT3-mutated AML |
| 2. Gilteritinib | Relapsed/refractory FLT3-mutated AML |
| IDH inhibitors | |
| 3. Ivosidenib | Relapsed/refractory IDH1-mutated AML |
| 4. Enasidenib | Newly diagnosed or relapsed/refractory IDH2-mutated AML |
| BCL2 inhibitor | |
| 5. Venetoclax + hypomethylating agents or LoDAC | Newly diagnosed AML aged ≥ 75 |
| Hedgehog pathway inhibitor | |
| 6. Glasdegib + LoDAC | Newly diagnosed AML aged ≥ 75 |
| Liposomal combination of daunorubicin and cytarabine | |
| 7. CPX-351 | Newly diagnosed AML-MRC and t-AML |
| Antibody-chemotherapy adjunct | |
| 8. Gemtuzumab ozogamicin | Newly diagnosed and relapsed/refractory CD33-positive AML |
| TKD Mutations | FLT3 Inhibitor Resistance |
|---|---|
| N676K | Resistance to midostaurin [35] |
| D835 I836 Y842 | Resistance to type II FLT3 inhibitors [36,37] (sorafenib, quizartinib, ponatinib) |
| F691L | Resistance to crenolanib, but not to ponatinib [38] and pexidartinib [39] |
| K429E | Resistance to crenolanib [33] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. https://doi.org/10.3390/ijms21041537
Lam SSY, Leung AYH. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. International Journal of Molecular Sciences. 2020; 21(4):1537. https://doi.org/10.3390/ijms21041537
Chicago/Turabian StyleLam, Stephen S.Y., and Anskar Y.H. Leung. 2020. "Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML" International Journal of Molecular Sciences 21, no. 4: 1537. https://doi.org/10.3390/ijms21041537
APA StyleLam, S. S. Y., & Leung, A. Y. H. (2020). Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. International Journal of Molecular Sciences, 21(4), 1537. https://doi.org/10.3390/ijms21041537