2. Intracellular Ca2+ Dyshomeostasis Is Involved in Neuronal Damage
Intracellular free Ca
2+ concentration is involved in the control of many physiological functions in all cell types, and neurons are not an exception in this regard. Rises in cytosolic Ca
2+ levels are involved primarily in neurotransmitter release, which takes place after activation of voltage-dependent Ca
2+ channels following depolarization; and/or after activation of inositol triphosphate receptors (IP
3Rs) and/or Ca
2+-sensitive ryanodine receptors, which releases Ca
2+ from intracellular stores. In addition, Ca
2+ is also involved in synaptic plasticity that depends on NMDARs activation [
8]. Synaptic plasticity also relies on spine stability that is modulated by store-operated Ca
2+ entry (SOCE), mediated by Orai1 channels [
9]. In the absence of such entry, mushroom spines are not stable, leading to impaired synaptic plasticity.
In addition to these critical physiological functions, an excess of Ca2+ entry—or Ca2+ overload—leads to neuron cell death. For instance, excess of glutamate and/or K+ in the extracellular medium, two conditions that may concur during stroke, overactivate both NMDARs and voltage-gated Ca2+ channels leading to an excess of Ca2+ entry. This excess is managed in healthy neurons by activating energy consuming Ca2+ pumps that clear Ca2+ loads efficiently but slowly, intracellular Ca2+ buffers, and mitochondria.
Mitochondria act as sinks of Ca
2+ loads that quickly and efficiently remove Ca
2+ from intracellular Ca
2+ hot spots without consuming energy. This is possible because of the mitochondrial Ca
2+ uniporter (MCU), a Ca
2+-activated Ca
2+ channel that transports Ca
2+ efficiently down the huge mitochondrial membrane potential (∆Ψm) [
10,
11]. ∆Ψm is negative inside the mitochondrial matrix and is the electromotive force for mitochondrial Ca
2+ uptake. Formerly, it was believed that mitochondria simply act as Ca
2+ stores. However, more recently, it has been established that the Ca
2+ taken up by mitochondria exits back to the cytosol in exchange for Na
+ through the mitochondrial Na
+/Ca
2+ exchanger (NCLX), or in exchange for H
+ through the mitochondrial H
+/Ca
2+ exchanger. However, if Ca
2+ signals are exceedingly larger and/or more sustained than certain values, the mitochondrial extrusion mechanisms are not able to extrude Ca
2+ at a consistent rate, leading to mitochondrial Ca
2+ overload. This process induces the opening of the mitochondrial permeability transition pore (mPTP), a channel complex located at the interface between the inner and the outer mitochondrial membranes [
12], resulting in the release of cytochrome c and other pro-apoptotic proteins to the cytosol. This is a point of no return for apoptosome activation irreversibly leading to apoptotic cell death. This process is also favored by an excess of reactive oxygen species (ROS), loss of mitochondrial membrane potential, and low ATP levels [
13]. Accordingly, all the mechanisms that promote excessive Ca
2+ entry, Ca
2+ release, and/or lack of intracellular Ca
2+ buffering may contribute to Ca
2+-dependent cell death or increased susceptibility to cell death. This scenario is often favored in different pathophysiological situations, such as excitotoxicity, neuroinflammation, aging, and/or neurodegenerative disorders, where some neurotoxins involved in Ca
2+ overload may accumulate, including glutamate, aspartate, K
+, glycine, and amyloid β peptide species.
Additionally, an increased firing of neurons has also been linked to aging in dopaminergic neurons, caused by a remodeling of voltage-gated Ca
2+ channels [
14]. Moreover, aging also depends on the depletion of intracellular Ca
2+ buffers in specific brain regions. For example, loss of calbindin with aging has been suggested to contribute to the increased susceptibility to Ca
2+ overload in hippocampal neurons in AD [
15].
4. Effects of Neurotoxins in Short-Term and Long-Term Cultures of Rat Hippocampal Neurons
The effects of several neurotoxins, such as the glutamate receptor agonist NMDA or the amyloid β peptide, have been tested in short-term and long-term cultures of rat hippocampal neurons resembling young and aged neurons respectively, and their toxic effects on cell death and Ca2+ signaling among others have been compared between the two groups.
NMDA did not induce cell death in hippocampal neurons cultured for only two days in vitro (2 DIV), considered young neurons. After 8 DIV, NMDA slightly but significantly increased the rate of neuronal apoptosis from about 5% to 10%. However, this effect dramatically increased at 13 to 21 DIV, considered aged neurons, in which NMDA exposure increased apoptosis from about 12% to 60–80%. Thus, long-term cultured neurons reflecting aged neurons were highly sensitive to excitotoxicity [
22].
Interestingly, the rises in intracellular Ca
2+ concentrations induced by NMDA were very small or nearly negligible at 2 DIV, significantly increased at 8 DIV, and became exceedingly large at 13 to 21 DIV, establishing a correlation between the NMDA-induced Ca
2+ increase and the apoptosis rate. The Ca
2+ rises induced by plasma membrane depolarization with high K
+ concentration medium also increased with culture time but the difference was smaller [
22]. These results suggest that in vitro aging is associated with an enhanced expression of NMDARs. Data obtained using semi-quantitative immunofluorescence indicated that in vitro aging is associated with an increased expression of NR1 and NR2A subunits of the NMDAR, and decreased expression of NR2B subunit [
22]. Interestingly, this pattern of changes resembles the pattern reported in in vivo aging [
21].
Accordingly, enhanced Ca
2+ entry mediated by an increased expression of NMDARs could be involved in the enhanced susceptibility to neuronal damage in aged neurons. This possibility is supported by additional evidence showing that NMDA does not promote mitochondrial Ca
2+ uptake in short-term cultured rat hippocampal neurons [
22]. However, at 8 DIV, and particularly at >13 DIV, NMDA promotes large increases in mitochondrial Ca
2+ concentration ([Ca
2+]
mit). Consistently, inhibiting the driving force for mitochondrial Ca
2+ uptake (loss of ∆Ψm) with low concentrations of mitochondrial uncouplers, such as FCCP, or several NSAIDs acting as mild mitochondrial uncouplers, prevented NMDA-induced mitochondrial Ca
2+ uptake and neuron cell death [
22]. Taking this into account, we can conclude that aging is associated with enhanced susceptibility to cell death due to Ca
2+ channel remodeling, at least in vitro. This remodeling depends on changes in the expression of NMDAR subunits, enhanced Ca
2+ entry and mitochondrial Ca
2+ overload. In addition, inhibiting this mitochondrial Ca
2+ overload protects from neuronal cell death. Interestingly, this mechanism may explain the anti-inflammatory-independent mechanism of neuroprotection afforded by low concentrations of NSAIDs [
23].
AD is the neurodegenerative disorder most commonly associated with age, which implies neuronal cell death and progressive cognitive impairment. Even though it is one of the most prevalent dementias, the direct cause of AD is still unknown. It has been proposed that soluble low molecular weight aggregates (oligomers) of the amyloid β peptide (Aβo) are the main neurotoxin in AD. Aβo promote Ca
2+ entry into the cytosol and mitochondrial Ca
2+ uptake in rat cerebellar granule cells and hippocampal neurons leading to mitochondrial Ca
2+ overload and apoptosis [
24]. These effects have been partially attributed to activation of NMDA receptors by Aβo [
25]. However, it has been claimed that some of these results are related to glutamate contamination of the media in which the synthetic Aβo were prepared [
26,
27]. Nevertheless, further results using Aβo prepared in media devoid of glutamate receptor agonists confirmed that Aβo promote Ca
2+ entry and mitochondrial Ca
2+ overload in primary cultures of both rat cerebellar neurons and hippocampal neurons [
24,
27].
Aβ itself has been hypothesized to form Ca
2+-permeable pores or non-specific ion channels in the plasma membrane [
28,
29]. On the other hand, presenilins (PSs), the catalytic core of γ-secretase which participates in the amyloid precursor protein (APP) processing, have been proposed to be an ER Ca
2+-leak channel [
30], independently of their catalytic activity in the γ-secretase. It has been shown that mutations in PSs can disturb Ca
2+ release through the leak channels, leading to an increase of Ca
2+ in the ER and increasing the vulnerability to neurodegeneration [
31]. Therefore, Aβ pores and/or Aβ-activated NMDA receptors, together with mutant PS channels may all lead to severe general Ca
2+ overload in genetic cases of AD.
Interestingly, Aβo had no effect on apoptosis in short-term cultures of rat hippocampal neurons, but started to promote cell death at 8 DIV and, after longer culture periods (>13 DIV), oligomers caused apoptosis in more than 40% of the neurons as assessed by fluorescence imaging [
32]. Consistently, Aβo induced no rise in cytosolic Ca
2+ concentration in young neurons, but started to increase cytosolic Ca
2+ at 8 DIV, and the effects increased further in aged neurons at >13 DIV. These effects were observed only in neurons but not in glial cells extracted from rat hippocampi, as demonstrated by simultaneous Ca
2+ imaging and immunofluorescence in the same individual cells. Moreover, Aβo promoted no rise in mitochondrial Ca
2+ concentration in short-term cultures of rat hippocampal neurons. However, Aβo induced a small rise in mitochondrial Ca
2+ at 8 DIV, and the effects increased rather dramatically in aged neurons at >13 DIV [
32]. In addition, Aβo promoted cytochrome c release only in in vitro aged neurons but not in the short-term neurons.
Interestingly, inhibition of mitochondrial Ca
2+ uptake using low concentrations of mitochondrial uncouplers or several NSAIDs prevented apoptosis induced by Aβo without having any effect on cytosolic Ca
2+ [
32]. The effects of NSAIDs are mimicked by R enantiomers, like R-flurbiprofen, that lack anti-inflammatory activity, indicating that neuroprotection relates to NSAID’s ability to inhibit mitochondrial Ca
2+ overload rather than to their anti-inflammatory activity. However, it must be emphasized that neuroprotection is only achieved at low NSAID concentrations that depolarize mitochondria only partially. At large concentrations, NSAIDs collapse mitochondrial potential and induce apoptosis even in the absence of Aβo [
32]. These data indicate that the vulnerability of rat hippocampal neurons to Aβo depends strongly on the time of culture related to the phenotypic age of the neurons. They also point to mitochondrial Ca
2+ as a key player in the Aβo-induced neurotoxicity. In addition, they provide strong evidence that NSAIDs may protect against Aβo toxicity and perhaps AD by preventing the associated mitochondrial Ca
2+ overload.
Unfortunately, latest trials with NSAIDs aimed at preventing or treating AD have given rather pessimistic results. In the last trial carried out in cognitively intact individuals at risk (INTREPAD), the sustained treatment with naproxen sodium increased the frequency of adverse health effects but did not reduce apparent progression of pre-symptomatic AD [
33]. Further research is required to ascertain discrepancies with previous studies.
AD has also been related to neuroinflammation. The AD brain shows increased levels of pro-inflammatory factors, such as pro-inflammatory cytokines, complement components, and proteases, which are recognized by different receptors in glial cells and neurons. Neuroinflammation comprises a set of cellular and molecular responses, and recent evidence suggests that Ca
2+ signaling is also involved in this regard. Specifically, toll-like receptors (TLRs) are transmembrane pattern-recognition receptors of the innate immune system that recognize several pathogen-derived and tissue damage-related ligands. It has been proposed that TLR signaling may contribute to age-related neurodegenerative diseases, including AD, suggesting a possible interplay between inflammation and Aβo in AD [
34]. Consistent with this view, it has been recently shown that lipopolysacharide (LPS), an agonist of TLR4, increases cytosolic Ca
2+ and promotes apoptosis in long-term cultures of rat hippocampal neurons but not in short-term cultured neurons [
35]. Both, the increase in cytosolic Ca
2+ and neuronal apoptosis induced by LPS were prevented by the TLR4 antagonist CAY10614. Interestingly, hippocampal neurons express TLR4 receptors and the expression level increases with time in culture consistently with the rise in TLR4 in aging brains [
36]. Furthermore, chronic exposure of aged hippocampal cultures to Aβo further increased TLR4 expression and enhanced cytosolic Ca
2+ rises and apoptosis induced by LPS. These results suggest that in vitro aging increases the susceptibility of rat hippocampal neurons to LPS-induced damage, and these effects seem to be related to changes in the TLR4 expression [
35].
Altogether, these data suggest that rat hippocampal neurons in long-term culture may be a good model to investigate age-related changes in susceptibility to neurotoxins related to excitotoxicity, AD, and neuroinflammation.
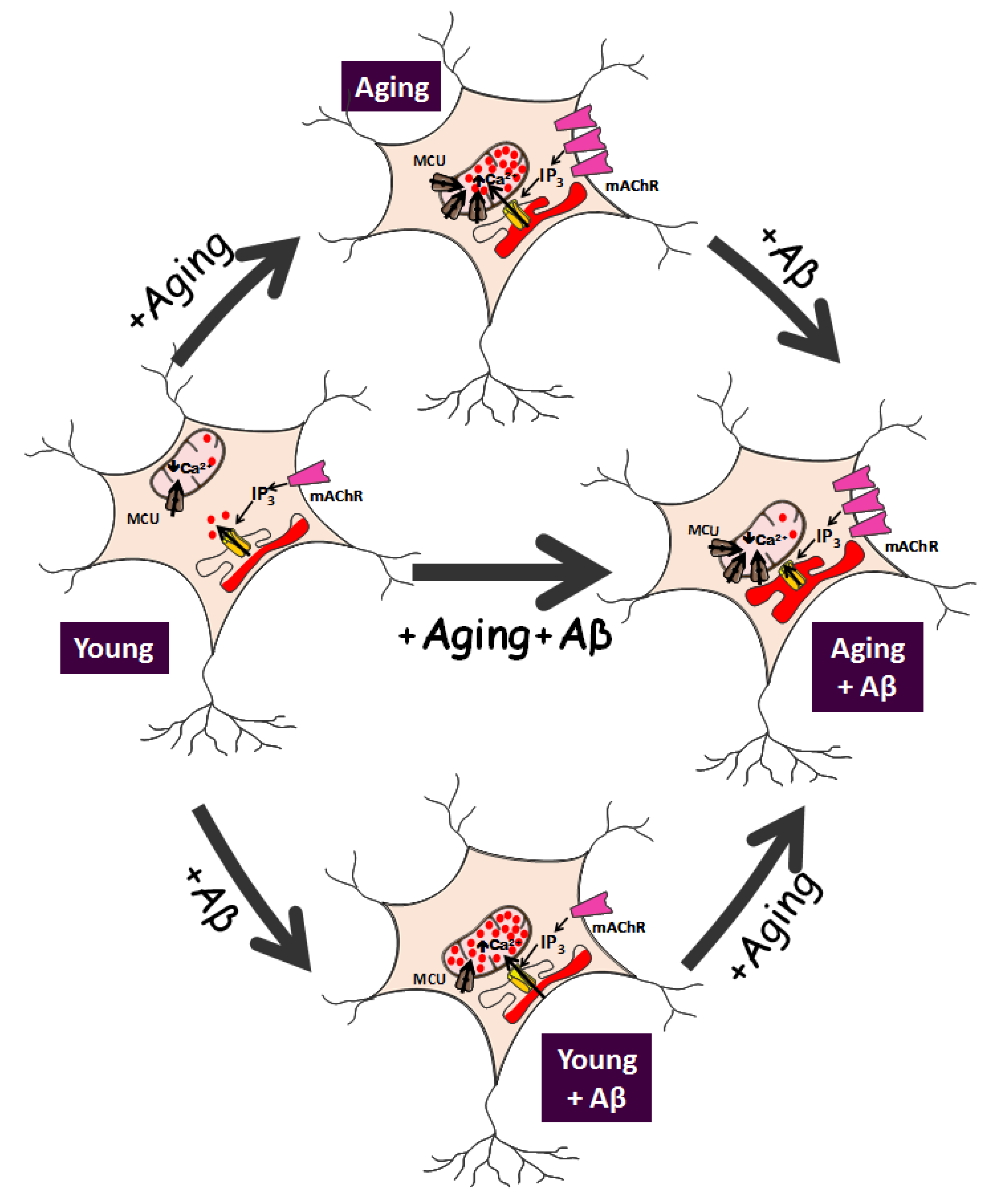
5. Remodeling of Intracellular Ca2+ and Store-Operated Ca2+ Entry (SOCE) in In Vitro Aged Neurons
Data suggest that long-term cultured neurons resume many of the characteristics of aged neurons, at least regarding susceptibility to neuron damage and changes in expression of receptors targeted by neurotoxins related to age-dependent neurodegenerative processes and Ca2+ signaling. Therefore, changes in intracellular Ca2+ homeostasis (Ca2+ remodeling) could be involved in the aging process. These changes have recently been explored in the model of a long-term culture of rat hippocampal neurons.
One of the characteristics of cultured hippocampal neurons is that they connect each other forming neural networks. This is evidenced by their ability to display spontaneous and synchronous intracellular Ca
2+ oscillations after a few days in vitro, when neurons express mature NMDARs [
36,
37]. These oscillations can be abolished by tetrodotoxin, indicating they are dependent on synaptic network communication among neurons. However, after several weeks in vitro, the neuronal cultures lose this ability and show increased resting levels of intracellular Ca
2+ concentration relative to the short-term cultures instead [
38]. Chronic exposure of rat hippocampal neurons to Aβo for 24 h has no effect on synchronic cytosolic Ca
2+ oscillations. Intriguingly, in aged neurons, Aβo treatment resumes synchronic oscillations [
38]. Ca
2+ hyperactivity has also been shown in in vivo mouse models of cerebral amyloidosis [
39]. The functional consequences of this effect are not clear, but they provide evidence that aged neurons are sensitive to Aβo whereas young neurons are not.
SOCE is an important Ca
2+ entry pathway activated after Ca
2+ release from the ER induced by physiological agonists such as acetylcholine or glutamate activating metabotropic receptors. SOCE is involved in many different physiological functions in most cell types [
40]. In neurons, the physiological role of SOCE is not well known. Recent data provided by several laboratories indicate that SOCE could be critical for the stabilization of mushroom spines involved in memory storage. Importantly, mushroom spines’ stability is downregulated in age-related neurodegenerative disorders, such as AD and Parkinson’s disease [
36]. At the molecular level, SOCE is activated by the interaction of an ER Ca
2+ sensor, the stromal interacting molecular 1 (STIM1), with a plasma membrane channel, Orai1. Other isoforms of these proteins, including STIM2, Orai2, and Orai3, can also be involved. Rat hippocampal neurons display SOCE and express both Orai1 and STIM1, the classic molecular players involved in SOCE. Interestingly, the amplitude of SOCE in rat hippocampal neurons decreases significantly with culture time [
37]. Specifically, the rise in cytosolic Ca
2+ induced by extracellular Ca
2+ addition to cultured neurons pre-treated with thapsigargin (commonly used to deplete intracellular Ca
2+ stores) is easily recorded in short-term cultures of rat hippocampal neurons, and decreases significantly in long-term cultures [
37]. Loss of SOCE is associated with a decreased expression of both Orai1 and STIM1 in aged neurons. Interestingly, exposure to Aβo has no effect on SOCE in the short-cultured ones. However, Aβo treatment decreases SOCE to even lower levels in the long-term cultures [
38]. Accordingly, it is conceivable that a loss of SOCE in aged neurons may contribute to a cognitive decline in the elderly, particularly in excess of Aβo, a scenario observed in AD. Notice that Aβo may be present in the brain of patients for decades; however, damage is only evident as they age, suggesting the interesting possibility that it is the combination of Aβo excess and the age-associated remodeling of intracellular Ca
2+ homeostasis which is harmful for neurons. If this is confirmed, then the reversal of Ca
2+ remodeling could be an efficient novel approach for AD prevention.
6. Remodeling of Ca2+ Store Content and Ca2+ Release in In Vitro Aged Neurons
As stated above, mitochondrial Ca
2+ uptake is critically involved in neuronal apoptosis. Due to the low Ca
2+-affinity of the MCU, mitochondria take up Ca
2+ from microdomains, which exhibit high Ca
2+ concentrations and are typically formed at the inner mouth of plasma membrane Ca
2+ channels [
41]. In addition, mitochondria also sense high Ca
2+ domains formed nearby IP
3 receptor channels in the ER, particularly those located at the interface between the ER and mitochondria, at the mitochondria-associated membranes, the so-called MAMs [
42]. Accordingly, the Ca
2+ content of intracellular Ca
2+ stores, the extent of Ca
2+ release though the IP
3 receptors, and the resulting Ca
2+ uptake by mitochondria are key parameters of intracellular Ca
2+ homeostasis in neurons. Recent data indicate that in vitro aging is associated with changes in several parameters consistently with an age-related remodeling of intracellular Ca
2+ homeostasis [
37,
38] [
Figure 1]. Specifically, Ca
2+ store content increases with time in culture. This has been shown by the size of the rise in cytosolic Ca
2+ induced by low concentrations of the Ca
2+ ionophore ionomycin that release Ca
2+ from intracellular stores [
37]. Consistently, caffeine (an agonist of ryanodine receptors) and acetylcholine, that promotes IP
3 synthesis and the ensuing activation of IP
3 receptors, induce a Ca
2+ release from the ER that is significantly enhanced in aged cultures compared to young cultures [
37]. Therefore, ER Ca
2+ release is enhanced in aging neurons. This may be a logic consequence of enhanced resting intracellular Ca
2+ levels in aged cells as mentioned above, but this may be also mediated by changes in the expression of IP
3 receptors. In fact, immunofluorescence imaging using specific antibodies against different IP
3 receptor subunits suggests that the expression of all three IP
3 receptor subunits (IP
3R1, IP
3R2, and IP
3R3) is enhanced in aged neurons relative to young neurons [
38]. This provides an additional mechanism for enhanced ER Ca
2+ release in aged neurons. Interestingly, it has also been reported that Aβo treatment enhances further Ca
2+ store content and Ca
2+ release induced by acetylcholine or caffeine. Again, these effects are only observed in aged cultures of rat hippocampal neurons but not in young cultures [
38]. Therefore, in vitro aging increases Ca
2+ store content and Ca
2+ release from intracellular stores, and these effects are further potentiated by Aβo treatment (
Figure 1).
7. Remodeling of Ca2+ Transfer from ER to Mitochondria Induced by Aβ and Aging
A functional consequence of enhanced Ca
2+ store content and Ca
2+ release in aged neurons could be enhanced Ca
2+ transfer from ER to mitochondria. The ER–mitochondria coupling is gaining momentum particularly because of the discovery of MAMs, specialized junctions of interaction between the ER and mitochondria [
42]. Ca
2+ transfer from ER to mitochondria has been investigated also longitudinally in cultures of rat hippocampal neurons.
Specifically, Calvo-Rodriguez et al. [
37] showed no or little Ca
2+ transfer from ER to mitochondria in young cultures of rat hippocampal neurons. When stimulating hippocampal neurons with acetylcholine to release Ca
2+ from the ER, the rise in cytosolic Ca
2+ was similar in young cultures in normal conditions or in presence of a mitochondrial uncoupler to prevent mitochondrial Ca
2+ uptake [
37]. However, in aged cultures, the rise in cytosolic Ca
2+ induced by acetylcholine increased dramatically if mitochondria were not able to take up Ca
2+ (following pre-treatment with the mitochondrial uncoupler). The above evidence indicates that, in contrast to young neurons, most of the Ca
2+ released by the ER is taken by the surrounding mitochondria in the aged neurons [
37]. This view is further supported by direct measurements of mitochondrial Ca
2+ uptake using bioluminescence imaging of individual neurons transfected with mitochondria-targeted aequorin [
43]. In this sense, Ca
2+ release after acetylcholine stimulation induced no or very little mitochondrial Ca
2+ uptake in young cultures. On the contrary, it induced large mitochondrial Ca
2+ uptake in the aged cultures [
37]. Therefore, in vitro neuronal aging is associated with a deep remodeling of the coupling ER–mitochondria that favors Ca
2+ transfer from the ER to mitochondria [
Figure 1].
Several mechanisms could contribute to the enhanced transfer of Ca
2+ from ER to mitochondria in the aged neurons. The first one has been mentioned above: increased ER Ca
2+ content together with enhanced ER Ca
2+ release. Changes in expression of acetylcholine, IP
3, and ryanodine receptors could contribute to these changes. As stated above, immunofluorescence of cultured neurons suggests upregulation of all three IP
3 receptor subunits in long-term cultures [
38]. Second, evidence from confocal microscopy imaging analysis of tagger ER and mitochondria suggests that ER–mitochondria colocalization is enhanced in long-term cultures of rat hippocampal neurons [
38], which could also contribute to the enhanced Ca
2+ transfer between these two organelles.
Mitochondria may also contribute to the aging-induced enhanced Ca
2+ transfer ER–mitochondria. As stated above, mitochondrial Ca
2+ uptake depends on MCU and ∆Ψm. Recent evidence indicates that MCU expression increases with time in culture, suggesting that MCU is upregulated in aged hippocampal neurons [
37]. These data may explain the increased mitochondrial Ca
2+ uptake in aged cultures described above. However, since Ca
2+ release from the ER is also enhanced in aged neurons, the specific contribution of each mechanism cannot be distinguished. This was analyzed in detail using permeabilized neurons subjected to a clamped cytosolic Ca
2+ concentration. In this experimental setting, mitochondrial Ca
2+ uptake was paradoxically lower in aged neurons compared to young neurons. This cannot be attributed to a decreased MCU expression. Instead, it could be explained by the partial loss of ∆Ψm reported in aged neurons [
37]. Therefore, these data suggest that long-term cultured hippocampal neurons display an enhanced ER–mitochondria Ca
2+ transfer mediated by several mechanisms mostly related to enhanced ER Ca
2+ release and increased ER–mitochondria colocalization in aged neurons in culture [
Figure 1].
What are the functional consequences of enhanced Ca2+ transfer from ER to mitochondria in aged neurons? It may favor the activation of Ca2+-dependent dehydrogenases involved in the Krebs cycle, thus supporting ATP synthesis in metabolically compromised aged neurons. However, this improvement of energy production increases the risk of mitochondrial Ca2+ overload and apoptosis. In consequence, the remodeling of intracellular Ca2+ homeostasis observed in aged neurons in vitro may contribute to explain some of the characteristics of brain aging, including cognitive decline and enhanced susceptibility to neuron damage.
Ca
2+ transfer from ER to mitochondria has also been investigated in the context of AD, in cultures of rat hippocampal neurons treated with Aβo. Evidence indicates that a 24 h treatment with Aβo increases the ER–mitochondria Ca
2+ transfer. This effect is observed in short-term cultures of rat hippocampal neurons [
Figure 1]. Remarkably, this is the only effect induced by Aβo in young neurons. It is important to bear in mind that, as stated above, Aβo have no effect on resting cytosolic Ca
2+, SOCE, Ca
2+ store content or ER Ca
2+ release in young cultures. Therefore, Aβo-enhanced Ca
2+ transfer from ER to mitochondria cannot be attributed to possible effects on Ca
2+ store content, Ca
2+ release, or changes in expression of MCU since Aβo treatment in young neurons does not influence any of the above mentioned parameters. However, Aβo treatment also increases ER–mitochondria colocalization in young cultures. Therefore, these data imply that Aβo increase ER–mitochondria Ca
2+ transfer most likely by promoting the colocalization of these two organelles. In fact, it has been proposed that modulation of the ER–mitochondria interface could be “the” physiological role of Aβo [
38]. This function may be beneficial in young neurons by improving the coupling ATP synthesis rate to energy demands after cell stimulation. In fact, Aβo decrease partially mitochondrial membrane potential in young neurons consistently with enhanced respiration and metabolic activity [
38]. Nevertheless, Aβo effects are not deleterious to young neurons as evidenced by the lack of apoptosis induced by Aβo. The reason for this effect might be that, despite increased Aβo-induced ER–mitochondria colocalization, mitochondrial Ca
2+ overload is limited in this scenario by the low Ca
2+ store content, the small Ca
2+ release, and the low expression levels of IP
3 receptors and MCU.
It is not entirely clear how a peptide such as Aβ could mechanistically affect the intracellular localization of ER–mitochondria and increase their interaction. Area-Gomez et al. first found increased ER–mitochondrial contacts in AD patient fibroblasts and PS-mutant cells [
44]. Additionally, they recently proposed that C99 (a terminal fragment generated after the cleavage by β-secretase of the amyloid precursor protein (APP) that is then cleaved by γ-secretase to produce Aβ) is found in the MAMs, and its localization is increased in AD, causing mitochondrial dysfunction. Importantly, uncleaved C99 in the MAMs induces both physical and functional enhancement of ER–mitochondrial connections [
45]. Along these lines, by using pure mitochondrial fractions, Ankarcrona’s group recently proposed that Aβ peptides are generated in the MAMs, which may disturb ER, mitochondria, and ER–mitochondria contact site function if the peptide is produced in excess [
46].
Aβo effects on apoptosis may be quite different in the aging scenario. Aged neurons do display enhanced Ca
2+ store content, large Ca
2+ release after stimulation, and increased expression of IP
3 receptors and MCU. Surprisingly, in aged neurons treated with Aβo, Ca
2+ transfer from ERto mitochondria is rather abolished [
38]. This effect is observed despite that ER–mitochondria colocalization is enhanced in aged cells, and increased even further after Aβo treatment [
Figure 1]. However, as stated above, mitochondrial membrane potential is quite low in aged cells, and further decreased by Aβo treatment. This scenario of very low mitochondrial membrane potential, ER Ca
2+ overload, and enhanced ER–mitochondrial coupling may favor failure of mitochondrial Ca
2+ homeostasis and mPTP opening, leading to apoptosis [
38]. Further research is required to confirm this evidence in vivo. However, if this is true, then the combination of age-related, intracellular Ca
2+ remodeling with the physiological effect of Aβo increasing ER–mitochondria coupling may be critically involved in the catastrophic consequences of neuronal damage in AD. Some of the evidences reviewed here should be confirmed in in vivo aging. However, the preparation of long-term cultures of rat hippocampal neurons as a model of aging [
37,
38] is worthy of consideration when it comes to investigating functional and molecular changes related to aging. Finally, we would like to emphasize that in this context, the pharmacological modulation of Ca
2+ fluxes, particularly at the ER–mitochondria interface, should be considered further for fighting cognitive decline and neurodegenerative disorders in the elderly.

 ,
, {kind=link}
