Integration of Rap1 and Calcium Signaling



Abstract
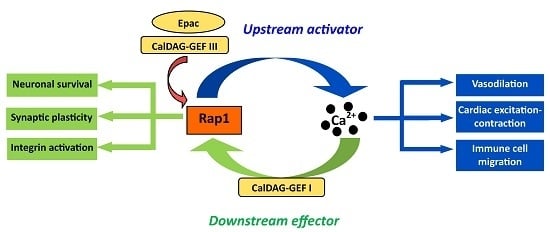
1. Discovery, Early and Classical Functions of Rap1: Ras Antagonism, Integrin Activation
2. Posttranslational Modifications and Cellular Localization of Rap1
3. Ca2+ Signaling and Rap1
4. Rap1 Activators in Integration of Ca2+ Signaling
4.1. CalDAG-GEFs
4.2. Epac
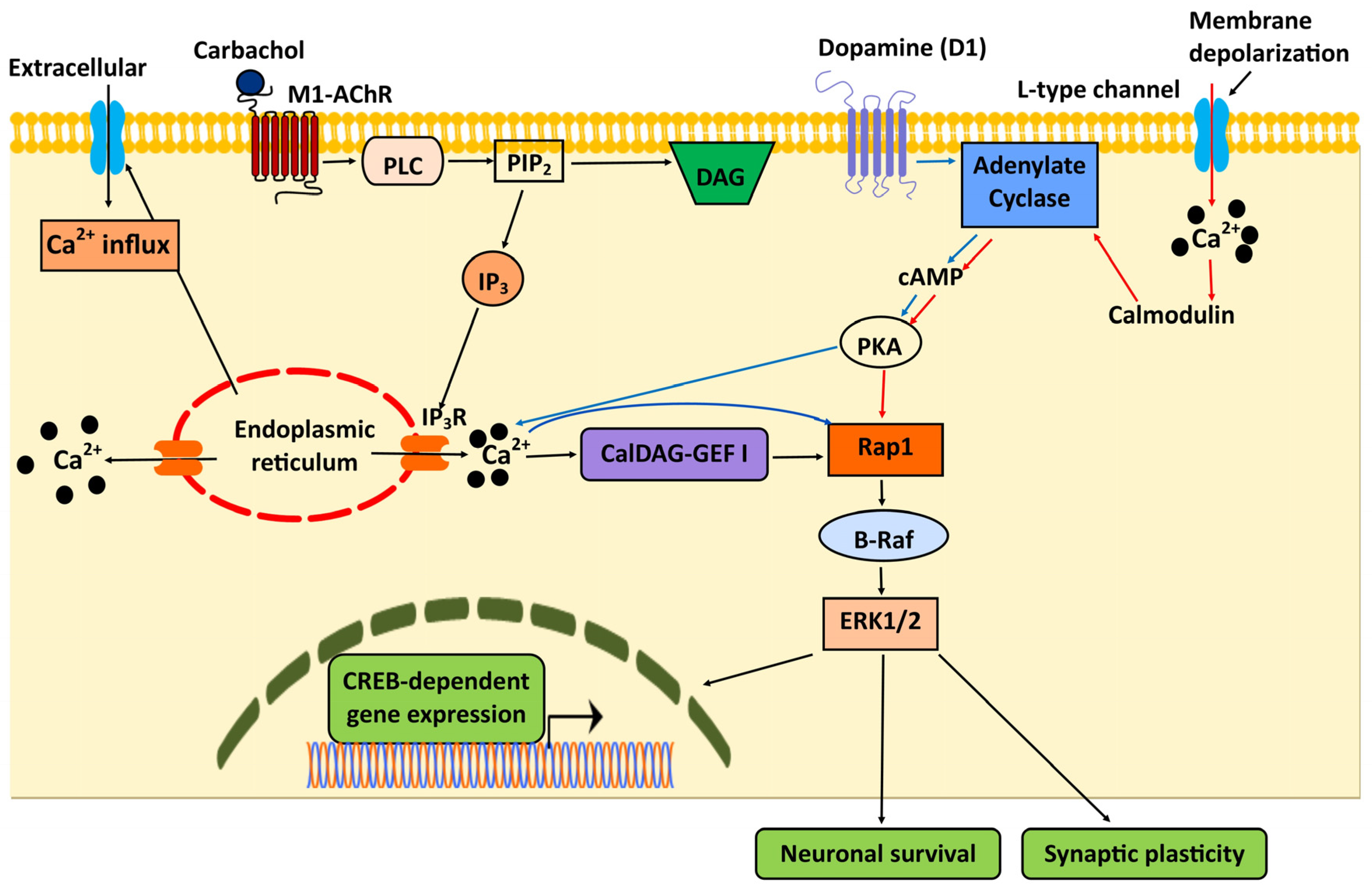
5. Integration of Rap1 and Ca2+ Signaling in the Central Nervous System (CNS)
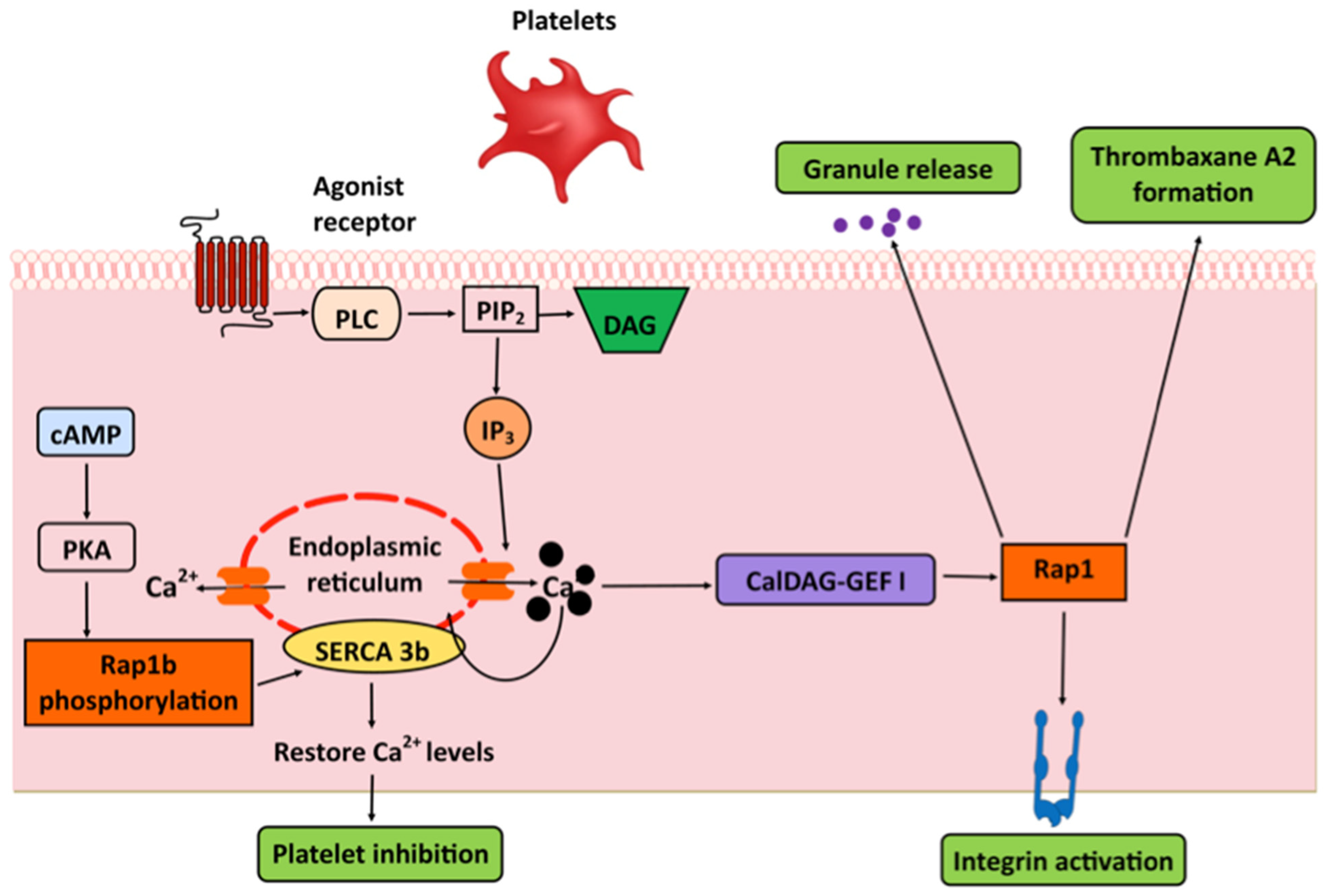
6. Platelets: Integrins and SERCA
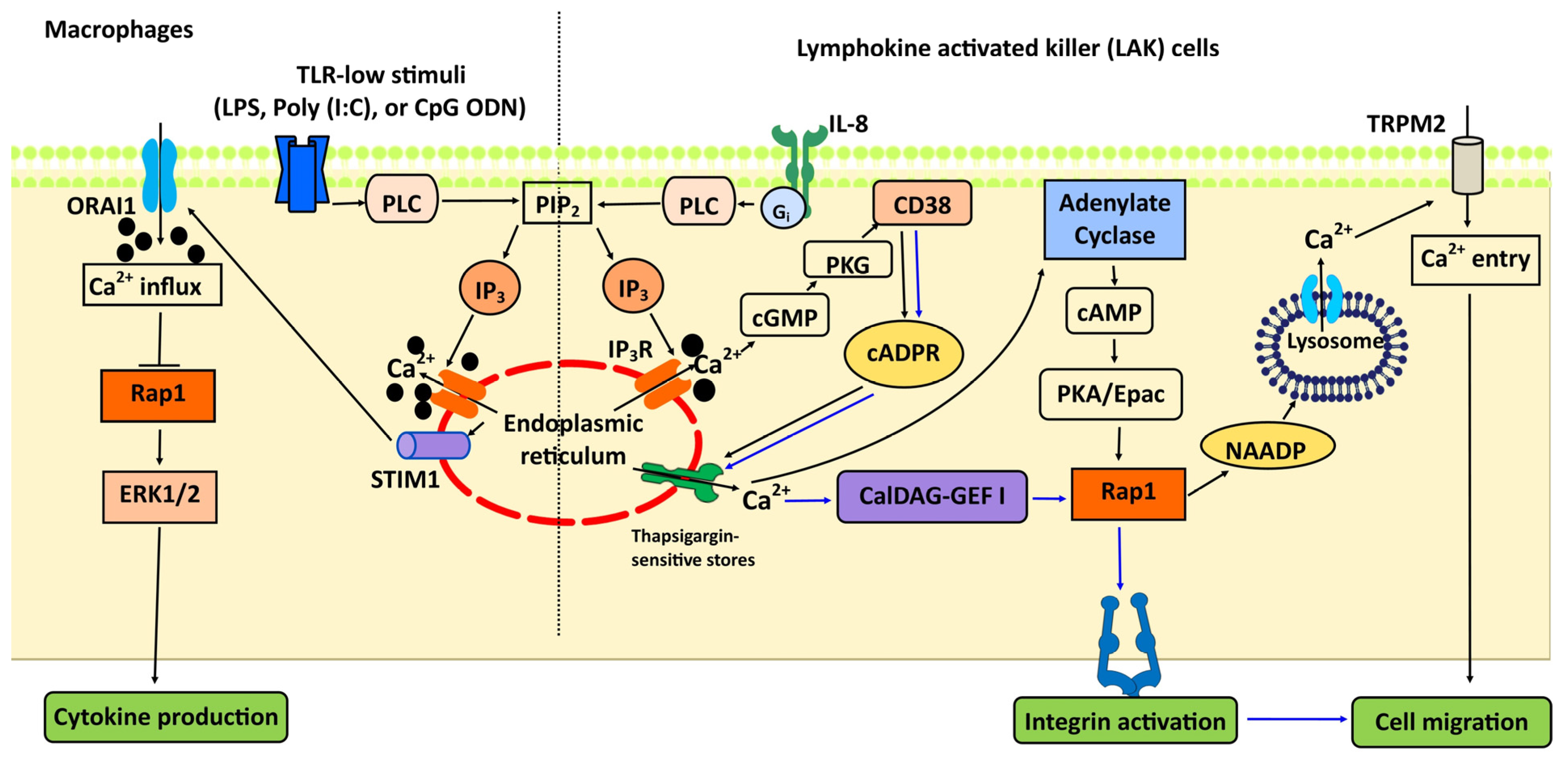
7. Rap1 and Ca2+ Signaling in the Immune System: TLR, Integrins, and Chemotaxis
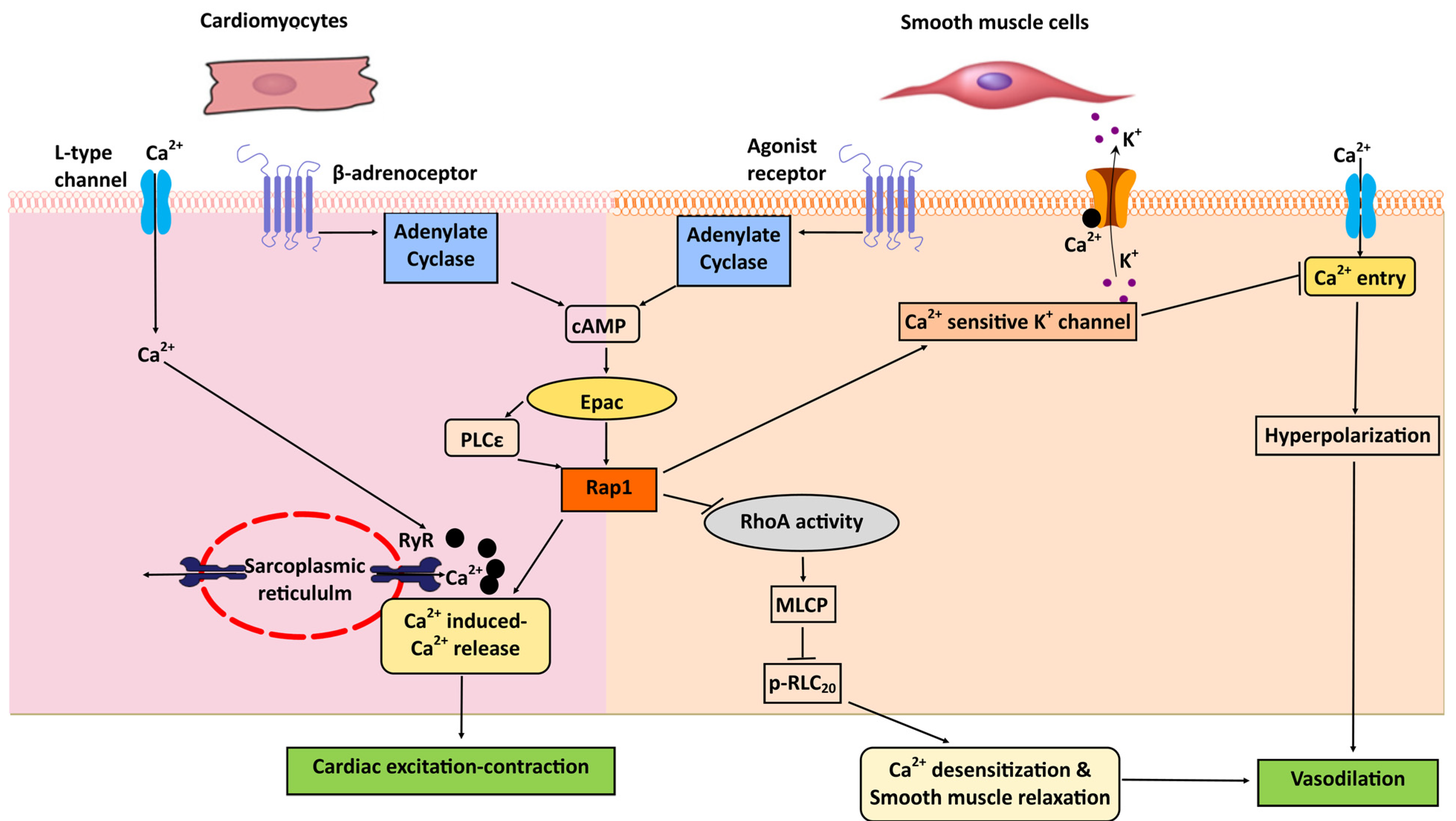
8. Heart: Excitation-Contraction Coupling; Cardiac Hypertrophy
9. Vascular Smooth Muscle Cells: Vasorelaxation
10. Endothelium: NO, Vasorelaxation, Vasoreactivity
11. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ca2+ | Calcium |
| CalDAG-GEF | Ca2+ and DAG-activated GEF |
| cAMP | cyclic adenosine monophosphate |
| CICR | Ca2+-induced Ca2+ release |
| CLL | chronic lymphocytic leukemia |
| CNS | central nervous system |
| CREB | cAMP-response element binding protein |
| eNOS | endothelial nitric oxide synthase |
| Epac | exchange protein activated by cAMP |
| ERK | extracellular signal-regulated kinase-1 |
| GAPs | GTPase-activating proteins |
| GEFs | guanine nucleotide exchange factors |
| GPCR | G protein coupled receptor |
| IP3 | inositol 1,4,5-trisphosphate |
| MAPK | mitogen-activated protein kinase |
| NO | nitric oxide |
| PDEs | phosphodiesterases |
| PIP | phosphatidyl inositol phosphate |
| PKA | protein kinase A |
| PLC | phospholipase C |
| REM | Ras exchange motif |
| RIAM | Rap1-GTP-interacting adaptor molecule |
| RLC20 | myosin regulatory light chain |
| RyR | ryanodine receptor |
| SERCA | sarcoendoplasmic reticulum Ca2+-ATPase |
| TKR | tyrosine kinase receptors |
| TLR | Toll-like receptor |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
References
- Noda, M.; Kitayama, H.; Matsuzaki, T.; Sugimoto, Y.; Okayama, H.; Bassin, R.H.; Ikawa, Y. Detection of genes with a potential for suppressing the transformed phenotype associated with activated ras genes. Proc. Natl. Acad. Sci. USA 1989, 86, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, H.; Sugimoto, Y.; Matsuzaki, T.; Ikawa, Y.; Noda, M. A ras-related gene with transformation suppressor activity. Cell 1989, 56, 77–84. [Google Scholar] [CrossRef]
- Pizon, V.; Lerosey, I.; Chardin, P.; Tavitian, A. Nucleotide sequence of a human cDNA encoding a ras-related protein (rap1B). Nucleic Acids Res. 1988, 16, 7719. [Google Scholar] [CrossRef] [PubMed]
- Pizon, V.; Chardin, P.; Lerosey, I.; Olofsson, B.; Tavitian, A. Human cDNAs rap1 and rap2 homologous to the Drosophila gene Dras3 encode proteins closely related to ras in the ‘effector’ region. Oncogene 1988, 3, 201–204. [Google Scholar]
- Bos, J.L. All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J. 1998, 17, 6776–6782. [Google Scholar] [CrossRef] [PubMed]
- Milburn, M.V.; Tong, L.; deVos, A.M.; Brunger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef]
- Noguchi, H.; Ikegami, T.; Nagadoi, A.; Kamatari, Y.O.; Park, S.Y.; Tame, J.R.; Unzai, S. The structure and conformational switching of Rap1B. Biochem. Biophys. Res. Commun. 2015, 462, 46–51. [Google Scholar] [CrossRef]
- van den Berghe, N.; Cool, R.H.; Wittinghofer, A. Discriminatory residues in Ras and Rap for guanine nucleotide exchange factor recognition. J. Biol. Chem. 1999, 274, 11078–11085. [Google Scholar] [CrossRef]
- Raaijmakers, J.H.; Bos, J.L. Specificity in Ras and Rap signaling. J. Biol. Chem. 2009, 284, 10995–10999. [Google Scholar] [CrossRef]
- Boettner, B.; Van Aelst, L. Control of cell adhesion dynamics by Rap1 signaling. Curr. Opin. Cell Biol. 2009, 21, 684–693. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M. Rap1 in endothelial biology. Curr. Opin. Hematol. 2017, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Karbstein, K. Role of GTPases in ribosome assembly. Biopolymers 2007, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jamroz-Wisniewska, A.; Beltowski, J. Protein isoprenylation. Postepy Biochem. 2004, 50, 316–329. [Google Scholar]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, D.L.; Peterson, S.N.; Ostrowski, M.C.; Lapetina, E.G. Cyclic AMP-dependent activation of Rap1b. J. Biol. Chem. 1995, 270, 10373–10376. [Google Scholar] [CrossRef]
- Altschuler, D.; Lapetina, E.G. Mutational analysis of the cAMP-dependent protein kinase-mediated phosphorylation site of Rap1b. J. Biol. Chem. 1993, 268, 7527–7531. [Google Scholar]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Stork, P.J. Phosphorylation of Rap1 by cAMP-dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by cAMP. J. Biol. Chem. 2017, 292, 1449–1461. [Google Scholar] [CrossRef]
- Ntantie, E.; Gonyo, P.; Lorimer, E.L.; Hauser, A.D.; Schuld, N.; McAllister, D.; Kalyanaraman, B.; Dwinell, M.B.; Auchampach, J.A.; Williams, C.L. An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering. Sci. Signal 2013, 6, ra39. [Google Scholar] [CrossRef]
- Wilson, J.M.; Prokop, J.W.; Lorimer, E.; Ntantie, E.; Williams, C.L. Differences in the Phosphorylation-Dependent Regulation of Prenylation of Rap1A and Rap1B. J. Mol. Biol. 2016, 428, 4929–4945. [Google Scholar] [CrossRef]
- Epstein, P.M. Different phosphodiesterases (PDEs) regulate distinct phosphoproteomes during cAMP signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 7741–7743. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.N.; Ovens, J.D.; Huston, E.; Umana, M.B.; Wilson, L.S.; Netherton, S.J.; Lynch, M.J.; Baillie, G.S.; Houslay, M.D.; Maurice, D.H. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J. Biol. Chem. 2010, 285, 33614–33622. [Google Scholar] [CrossRef] [PubMed]
- Nancy, V.; Callebaut, I.; El Marjou, A.; de Gunzburg, J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 2002, 277, 15076–15084. [Google Scholar] [CrossRef] [PubMed]
- Dumbacher, M.; Van Dooren, T.; Princen, K.; De Witte, K.; Farinelli, M.; Lievens, S.; Tavernier, J.; Dehaen, W.; Wera, S.; Winderickx, J.; et al. Modifying Rap1-signalling by targeting Pde6δ is neuroprotective in models of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 50. [Google Scholar] [CrossRef]
- Sakurai, A.; Fukuhara, S.; Yamagishi, A.; Sako, K.; Kamioka, Y.; Masuda, M.; Nakaoka, Y.; Mochizuki, N. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell 2006, 17, 966–976. [Google Scholar] [CrossRef]
- Beranger, F.; Goud, B.; Tavitian, A.; de Gunzburg, J. Association of the Ras-antagonistic Rap1/Krev-1 proteins with the Golgi complex. Proc. Natl. Acad. Sci. USA 1991, 88, 1606–1610. [Google Scholar] [CrossRef]
- Pizon, V.; Desjardins, M.; Bucci, C.; Parton, R.G.; Zerial, M. Association of Rap1a and Rap1b proteins with late endocytic/phagocytic compartments and Rap2a with the Golgi complex. J. Cell Sci. 1994, 107 Pt 6, 1661–1670. [Google Scholar]
- Maridonneau-Parini, I.; de Gunzburg, J. Association of rap1 and rap2 proteins with the specific granules of human neutrophils. Translocation to the plasma membrane during cell activation. J. Biol. Chem. 1992, 267, 6396–6402. [Google Scholar]
- Berger, G.; Quarck, R.; Tenza, D.; Levy-Toledano, S.; de Gunzburg, J.; Cramer, E.M. Ultrastructural localization of the small GTP-binding protein Rap1 in human platelets and megakaryocytes. Br. J. Haematol. 1994, 88, 372–382. [Google Scholar] [CrossRef]
- Wang, Z.; Dillon, T.J.; Pokala, V.; Mishra, S.; Labudda, K.; Hunter, B.; Stork, P.J. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 2006, 26, 2130–2145. [Google Scholar] [CrossRef]
- Sarker, M.; Goliaei, A.; Golesi, F.; Poggi, M.; Cook, A.; Khan, M.A.I.; Temple, B.R.; Stefanini, L.; Canault, M.; Bergmeier, W.; et al. Subcellular localization of Rap1 GTPase activator CalDAG-GEFI is orchestrated by interaction of its atypical C1 domain with membrane phosphoinositides. J. Thromb. Haemost. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid Redox Signal 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Olszak, I.T.; Poznansky, M.C.; Evans, R.H.; Olson, D.; Kos, C.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 2000, 105, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, W.S.; Giuliano, S.; Kulkarni, S.; Dopheide, S.M.; Harper, I.S.; Jackson, S.P. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J. Cell Biol. 2003, 160, 1151–1161. [Google Scholar] [CrossRef]
- Amberg, G.C.; Navedo, M.F. Calcium dynamics in vascular smooth muscle. Microcirculation 2013, 20, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bootman, M.D.; Lipp, P.; Berridge, M.J. The organisation and functions of local Ca2+signals. J. Cell Sci. 2001, 114, 2213–2222. [Google Scholar]
- Werry, T.D.; Wilkinson, G.F.; Willars, G.B. Mechanisms of cross-talk between G-protein-coupled receptors resulting in enhanced release of intracellular Ca2+. Biochem. J. 2003, 374 Pt 2, 281–296. [Google Scholar] [CrossRef]
- Putney, J.W., Jr.; Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S. Mechanisms of capacitative calcium entry. J. Cell Sci. 2001, 114 Pt 12, 2223–2229. [Google Scholar]
- Franke, B.; Akkerman, J.W.; Bos, J.L. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 1997, 16, 252–259. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.J.; Ingham, R.J.; Bos, J.L.; Kurosaki, T.; Gold, M.R. Activation of the Rap1 GTPase by the B cell antigen receptor. J. Biol. Chem. 1998, 273, 29218–29223. [Google Scholar] [CrossRef] [PubMed]
- Zwartkruis, F.J.; Wolthuis, R.M.; Nabben, N.M.; Franke, B.; Bos, J.L. Extracellular signal-regulated activation of Rap1 fails to interfere in Ras effector signalling. EMBO J. 1998, 17, 5905–5912. [Google Scholar] [CrossRef] [PubMed]
- Ebinu, J.O.; Bottorff, D.A.; Chan, E.Y.; Stang, S.L.; Dunn, R.J.; Stone, J.C. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science 1998, 280, 1082–1086. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Toki, S.; Canales, J.J.; Harlan, P.; Blumenstiel, J.P.; Chen, E.J.; Bany, I.A.; Mochizuki, N.; Ashbacher, A.; et al. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 13278–13283. [Google Scholar] [CrossRef]
- Clyde-Smith, J.; Silins, G.; Gartside, M.; Grimmond, S.; Etheridge, M.; Apolloni, A.; Hayward, N.; Hancock, J.F. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 2000, 275, 32260–32267. [Google Scholar] [CrossRef]
- Teixeira, C.; Stang, S.L.; Zheng, Y.; Beswick, N.S.; Stone, J.C. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 2003, 102, 1414–1420. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Wong, G.W.; Krilis, S.A.; Madhusudhan, M.S.; Sali, A.; Stevens, R.L. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 2002, 277, 25756–25774. [Google Scholar]
- Crittenden, J.R.; Bergmeier, W.; Zhang, Y.; Piffath, C.L.; Liang, Y.; Wagner, D.D.; Housman, D.E.; Graybiel, A.M. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004, 10, 982–986. [Google Scholar] [CrossRef]
- Reuther, G.W.; Lambert, Q.T.; Rebhun, J.F.; Caligiuri, M.A.; Quilliam, L.A.; Der, C.J. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J. Biol. Chem. 2002, 277, 30508–30514. [Google Scholar] [CrossRef]
- Roose, J.P.; Mollenauer, M.; Gupta, V.A.; Stone, J.; Weiss, A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell. Biol. 2005, 25, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Limnander, A.; Depeille, P.; Freedman, T.S.; Liou, J.; Leitges, M.; Kurosaki, T.; Roose, J.P.; Weiss, A. STIM1, PKC-delta and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nat. Immunol. 2011, 12, 425–433. [Google Scholar] [CrossRef]
- Yang, H.-C.; Liang, Y.-J.; Wu, Y.-L.; Chung, C.-M.; Chiang, K.-M.; Ho, H.-Y.; Ting, C.-T.; Lin, T.-H.; Sheu, S.-H.; Tsai, W.-C.; et al. Genome-wide association study of young-onset hypertension in the Han Chinese population of Taiwan. PLoS ONE 2009, 4, e5459. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Anderson, A.L.; Hidaka, M.; Swetenburg, R.L.; Patterson, C.; Stanford, W.L.; Bautch, V.L. A vascular gene trap screen defines RasGRP3 as an angiogenesis-regulated gene required for the endothelial response to phorbol esters. Mol. Cell. Biol. 2004, 24, 10515–10528. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.A.; Deng, W.; Ren, J.; Li, R.; Sondek, J.; Bergmeier, W. Calcium-induced structural rearrangements release autoinhibition in the Rap-GEF CalDAG-GEFI. J. Biol. Chem. 2018, 293, 8521–8529. [Google Scholar] [CrossRef]
- Walker, S.A.; Cullen, P.J.; Taylor, J.A.; Lockyer, P.J. Control of Ras cycling by Ca2+. Febs Lett. 2003, 546, 6–10. [Google Scholar] [CrossRef]
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.C. Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef]
- Bos, J.L.; de Rooij, J.; Reedquist, K.A. Rap1 signalling: Adhering to new models. Nat. Rev. Mol. Cell Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef]
- Yamashita, S.; Mochizuki, N.; Ohba, Y.; Tobiume, M.; Okada, Y.; Sawa, H.; Nagashima, K.; Matsuda, M. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J. Biol. Chem. 2000, 275, 25488–25493. [Google Scholar] [CrossRef]
- Lorenzo, P.S.; Beheshti, M.; Pettit, G.R.; Stone, J.C.; Blumberg, P.M. The Guanine Nucleotide Exchange Factor RasGRP Is a High-Affinity Target for Diacylglycerol and Phorbol Esters. Mol. Pharmacol. 2000, 57, 840–846. [Google Scholar] [PubMed]
- Lorenzo, P.S.; Kung, J.W.; Bottorff, D.A.; Garfield, S.H.; Stone, J.C.; Blumberg, P.M. Phorbol Esters Modulate the Ras Exchange Factor RasGRP3. Cancer Res. 2001, 61, 943–949. [Google Scholar] [PubMed]
- Johnson, J.E.; Goulding, R.E.; Ding, Z.; Partovi, A.; Anthony, K.V.; Beaulieu, N.; Tazmini, G.; Cornell, R.B.; Kay, R.J. Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem. J. 2007, 406, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, W.G.R., III; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, Q.; Zhao, Y.; Schwarz, B.J.; Stallone, J.N.; Heaps, C.L.; Han, G. Activation of G protein-coupled estrogen receptor 1 induces coronary artery relaxation via Epac/Rap1-mediated inhibition of RhoA/Rho kinase pathway in parallel with PKA. PLoS ONE 2017, 12, e0173085. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A Family of cAMP-Binding Proteins that Directly Activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef]
- Holz, G.G.; Kang, G.; Harbeck, M.; Roe, M.W.; Chepurny, O.G. Cell physiology of cAMP sensor Epac. J. Physiol. 2006, 577 Pt 1, 5–15. [Google Scholar] [CrossRef]
- Rehmann, H.; Arias-Palomo, E.; Hadders, M.A.; Schwede, F.; Llorca, O.; Bos, J.L. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 2008, 455, 124–127. [Google Scholar] [CrossRef]
- de Rooij, J.; Rehmann, H.; van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J. Biol. Chem. 2000, 275, 20829–20836. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Morel, E.; Dominguez-Rodriguez, A.; Llach, A.; Lezoualc’h, F.; Benitah, J.P.; Gomez, A.M. Epac in cardiac calcium signaling. J. Mol. Cell. Cardiol. 2013, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Bare, D.J.; Galice, S.; Shannon, T.R.; Bers, D.M. β-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell. Cardiol. 2017, 108, 8–16. [Google Scholar] [CrossRef]
- Lezcano, N.; Mariángelo, J.I.E.; Vittone, L.; Wehrens, X.H.T.; Said, M.; Mundiña-Weilenmann, C. Early effects of Epac depend on the fine-tuning of the sarcoplasmic reticulum Ca2+ handling in cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 114, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Joseph, J.W.; Chepurny, O.G.; Monaco, M.; Wheeler, M.B.; Bos, J.L.; Schwede, F.; Genieser, H.-G.; Holz, G.G. Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J. Biol. Chem. 2003, 278, 8279–8285. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Chepurny, O.G.; Rindler, M.J.; Collis, L.; Chepurny, Z.; Li, W.-H.; Harbeck, M.; Roe, M.W.; Holz, G.G. A cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in mouse pancreatic beta cells. J. Physiol. 2005, 566, 173–188. [Google Scholar] [CrossRef]
- Pratt, E.P.; Salyer, A.E.; Guerra, M.L.; Hockerman, G.H. Ca2+ influx through L-type Ca2+ channels and Ca2+-induced Ca2+ release regulate cAMP accumulation and Epac1-dependent ERK 1/2 activation in INS-1 cells. Mol. Cell. Endocrinol. 2016, 419, 60–71. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zanassi, P.; Paolillo, M.; Feliciello, A.; Avvedimento, E.V.; Gallo, V.; Schinelli, S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 2001, 276, 11487–11495. [Google Scholar] [CrossRef]
- Morozov, A.; Muzzio, I.A.; Bourtchouladze, R.; Van-Strien, N.; Lapidus, K.; Yin, D.; Winder, D.G.; Adams, J.P.; Sweatt, J.D.; Kandel, E.R. Rap1 Couples cAMP Signaling to a Distinct Pool of p42/44MAPK Regulating Excitability, Synaptic Plasticity, Learning, and Memory. Neuron 2003, 39, 309–325. [Google Scholar] [CrossRef]
- Grewal, S.S.; Horgan, A.M.; York, R.D.; Withers, G.S.; Banker, G.A.; Stork, P.J. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J. Biol. Chem. 2000, 275, 3722–3728. [Google Scholar] [CrossRef]
- Subramanian, J.; Dye, L.; Morozov, A. Rap1 signaling prevents L-type calcium channel-dependent neurotransmitter release. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 7245–7252. [Google Scholar] [CrossRef]
- Guo, F.F.; Kumahara, E.; Saffen, D. A CalDAG-GEFI/Rap1/B-Raf cassette couples M (1) muscarinic acetylcholine receptors to the activation of ERK1/2. J. Biol. Chem. 2001, 276, 25568–25581. [Google Scholar] [CrossRef]
- Kim, J.; Wei, D.-S.; Hoffman, D.A. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J. Physiol. 2005, 569, 41–57. [Google Scholar] [CrossRef]
- Grewal, S.S.; Fass, D.M.; Yao, H.; Ellig, C.L.; Goodman, R.H.; Stork, P.J. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J. Biol. Chem. 2000, 275, 34433–34441. [Google Scholar] [CrossRef] [PubMed]
- Ster, J.; De Bock, F.; Guerineau, N.C.; Janossy, A.; Barrere-Lemaire, S.; Bos, J.L.; Bockaert, J.; Fagni, L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 2519–2524. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Bergmeier, W. CalDAG-GEFI and platelet activation. Platelets 2010, 21, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lim, C.J.; Watanabe, N.; Soriani, A.; Ratnikov, B.; Calderwood, D.A.; Puzon-McLaughlin, W.; Lafuente, E.M.; Boussiotis, V.A.; Shattil, S.J.; et al. Reconstructing and Deconstructing Agonist-Induced Activation of Integrin αIIbβ3. Curr. Biol. 2006, 16, 1796–1806. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef]
- Calderwood, D.A. The Rap1-RIAM pathway prefers b2 integrins. Blood 2015, 126, 2658–2659. [Google Scholar] [CrossRef]
- Stritt, S.; Wolf, K.; Lorenz, V.; Vogtle, T.; Gupta, S.; Bosl, M.R.; Nieswandt, B. Rap1-GTP-interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood 2015, 125, 219–222. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M.; Smyth, S.S.; Schoenwaelder, S.M.; Fischer, T.H.; White, G.C. 2nd, Rap1b is required for normal platelet function and hemostasis in mice. J. Clin. Investig. 2005, 115, 680–687. [Google Scholar] [CrossRef]
- Canault, M.; Ghalloussi, D.; Grosdidier, C.; Guinier, M.; Perret, C.; Chelghoum, N.; Germain, M.; Raslova, H.; Peiretti, F.; Morange, P.E.; et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J. Exp. Med. 2014, 211, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Stefanini, L. Novel molecules in calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Roden, R.C.; Bergmeier, W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 2009, 114, 2506–2514. [Google Scholar] [CrossRef]
- Magnier, C.; Corvazier, E.; Aumont, M.C.; Le Jemtel, T.H.; Enouf, J. Relationship between Rap1 protein phosphorylation and regulation of Ca2+ transport in platelets: A new approach. Biochem. J. 1995, 310 Pt 2, 469–475. [Google Scholar] [CrossRef][Green Version]
- Lacabaratz-Porret, C.; Corvazier, E.; Kovacs, T.; Bobe, R.; Bredoux, R.; Launay, S.; Papp, B.; Enouf, J. Platelet sarco/endoplasmic reticulum Ca2+ ATPase isoform 3b and Rap 1b: Interrelation and regulation in physiopathology. Biochem. J. 1998, 332, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Bobe, R.; Dally, S.; Chaabane, C.; Corvazier, E.; Polidano, E.; Bredoux, R.; Enouf, J. Platelet Ca2+ ATPases: Identification and regulation in hypertension. Curr. Hypertens. Rev. 2010, 6, 155–165. [Google Scholar] [CrossRef]
- Jeyaraj, S.C.; Unger, N.T.; Chotani, M.A. Rap1 GTPases: An emerging role in the cardiovasculature. Life Sci. 2011, 88, 645–652. [Google Scholar] [CrossRef]
- Ghandour, H.; Cullere, X.; Alvarez, A.; Luscinskas, F.W.; Mayadas, T.N. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood 2007, 110, 3682–3690. [Google Scholar] [CrossRef]
- Bergmeier, W.; Goerge, T.; Wang, H.W.; Crittenden, J.R.; Baldwin, A.C.; Cifuni, S.M.; Housman, D.E.; Graybiel, A.M.; Wagner, D.D. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Investig. 2007, 117, 1699–1707. [Google Scholar] [CrossRef]
- Lee, H.S.; Lim, C.J.; Puzon-McLaughlin, W.; Shattil, S.J.; Ginsberg, M.H. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J. Biol. Chem. 2009, 284, 5119–5127. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Kim, C.; Ginsberg, M.H. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016, 128, 479–487. [Google Scholar] [CrossRef]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Lowell, C.A. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 2009, 27, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Stadtmann, A.; Brinkhaus, L.; Mueller, H.; Rossaint, J.; Bolomini-Vittori, M.; Bergmeier, W.; Van Aken, H.; Wagner, D.D.; Laudanna, C.; Ley, K.; et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 2011, 41, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- Carbo, C.; Duerschmied, D.; Goerge, T.; Hattori, H.; Sakai, J.; Cifuni, S.M.; White Ii, G.C.; Chrzanowska-Wodnicka, M.; Luo, H.R.; Wagner, D.D. Integrin-independent role of CalDAG-GEFI in neutrophil chemotaxis. J. Leukoc. Biol. 2010, 88, 313–319. [Google Scholar] [CrossRef]
- Pasvolsky, R.; Feigelson, S.W.; Kilic, S.S.; Simon, A.J.; Tal-Lapidot, G.; Grabovsky, V.; Crittenden, J.R.; Amariglio, N.; Safran, M.; Graybiel, A.M.; et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J. Exp. Med. 2007, 204, 1571–1582. [Google Scholar] [CrossRef]
- Kilic, S.S.; Etzioni, A. The clinical spectrum of leukocyte adhesion deficiency (LAD) III due to defective CalDAG-GEF1. J. Clin. Immunol. 2009, 29, 117–122. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Van Bruggen, R.; Kamerbeek, N.; Tool, A.T.J.; Hicsonmez, G.; Gurgey, A.; Karow, A.; Verhoeven, A.J.; Seeger, K.; Sanal, Ö.; et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood 2007, 109, 3529–3537. [Google Scholar] [CrossRef]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef]
- Abram, C.L.; Lowell, C.A. Leukocyte adhesion deficiency syndrome: A controversy solved. Immunol. Cell Biol. 2009, 87, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Mele, S.; Devereux, S.; Pepper, A.G.; Infante, E.; Ridley, A.J. Calcium-RasGRP2-Rap1 signaling mediates CD38-induced migration of chronic lymphocytic leukemia cells. Blood Adv. 2018, 2, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Graeff, R.; Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca2+ signaling pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Rah, S.Y.; Mushtaq, M.; Nam, T.S.; Kim, S.H.; Kim, U.H. Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells. J. Biol. Chem. 2010, 285, 21877–21887. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.; Gartner, K.; Scheuermann, U.; Zoeller, T.; Hantzschmann, J.; Over, B.; Foermer, S.; Heeg, K.; Bekeredjian-Ding, I. Ca2+-related signaling events influence TLR9-induced IL-10 secretion in human B cells. Eur. J. Immunol. 2014, 44, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Chen, T.; Yang, M.; Wang, L.; Yu, Z.; Xie, B.; Qian, C.; Xu, S.; Li, N.; Cao, X.; et al. Extracellular calcium elicits feedforward regulation of the Toll-like receptor-triggered innate immune response. Cell Mol. Immunol. 2017, 14, 180–191. [Google Scholar] [CrossRef]
- Tang, S.; Chen, T.; Yu, Z.; Zhu, X.; Yang, M.; Xie, B.; Li, N.; Cao, X.; Wang, J. RasGRP3 limits Toll-like receptor-triggered inflammatory response in macrophages by activating Rap1 small GTPase. Nat. Commun. 2014, 5, 4657. [Google Scholar] [CrossRef]
- Fujita, T.; Umemura, M.; Yokoyama, U.; Okumura, S.; Ishikawa, Y. The role of Epac in the heart. Cell Mol. Life Sci. Cmls 2017, 74, 591–606. [Google Scholar] [CrossRef]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Schmidt, M.; Evellin, S.; Weernink, P.A.; von Dorp, F.; Rehmann, H.; Lomasney, J.W.; Jakobs, K.H. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat. Cell Biol. 2001, 3, 1020–1024. [Google Scholar] [CrossRef]
- Oestreich, E.A.; Malik, S.; Goonasekera, S.A.; Blaxall, B.C.; Kelley, G.G.; Dirksen, R.T.; Smrcka, A.V. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J. Biol. Chem. 2009, 284, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, E.A.; Wang, H.; Malik, S.; Kaproth-Joslin, K.A.; Blaxall, B.C.; Kelley, G.G.; Dirksen, R.T.; Smrcka, A.V. Epac-mediated activation of phospholipase C (epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 2007, 282, 5488–5495. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Marcantoni, A.; Gastineau, M.; Birkedal, R.; Rochais, F.; Garnier, A.; Lompre, A.M.; Vandecasteele, G.; Lezoualc’h, F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 2005, 97, 1296–1304. [Google Scholar] [CrossRef]
- Metrich, M.; Laurent, A.C.; Breckler, M.; Duquesnes, N.; Hmitou, I.; Courillau, D.; Blondeau, J.P.; Crozatier, B.; Lezoualc’h, F.; Morel, E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell. Signal. 2010, 22, 1459–1468. [Google Scholar] [CrossRef]
- Dodge-Kafka, K.L.; Kapiloff, M.S. The mAKAP signaling complex: Integration of cAMP, calcium, and MAP kinase signaling pathways. Eur. J. Cell Biol. 2006, 85, 593–602. [Google Scholar] [CrossRef]
- Zieba, B.J.; Artamonov, M.V.; Jin, L.; Momotani, K.; Ho, R.; Franke, A.S.; Neppl, R.L.; Stevenson, A.S.; Khromov, A.S.; Chrzanowska-Wodnicka, M.; et al. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J. Biol. Chem. 2011, 286, 16681–16692. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Maarsingh, H.; Elzinga, C.R.; Schuur, J.; Menzen, M.; Halayko, A.J.; Meurs, H.; Schmidt, M. Epac as a novel effector of airway smooth muscle relaxation. J. Cell. Mol. Med. 2011, 15, 1551–1563. [Google Scholar] [CrossRef]
- Lakshmikanthan, S.; Zieba Bartosz, J.; Ge, Z.-D.; Momotani, K.; Zheng, X.; Lund, H.; Artamonov Mykhaylo, V.; Maas Jason, E.; Szabo, A.; Zhang David, X.; et al. Rap1b in Smooth Muscle and Endothelium Is Required for Maintenance of Vascular Tone and Normal Blood Pressure. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1486–1494. [Google Scholar] [CrossRef]
- Roberts, O.L.; Kamishima, T.; Barrett-Jolley, R.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery. J. Physiol. 2013, 591, 5107–5123. [Google Scholar] [CrossRef]
- Pannekoek, W.-J.; Post, A.; Bos, J.L. Rap1 signaling in endothelial barrier control. Cell Adhes. Migr. 2014, 8, 100–107. [Google Scholar] [CrossRef]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, M.R.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. Febs Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Fu, P.; Wu, T.; Dubrovskyi, O.; Sarich, N.; Poroyko, V.; Birukov, K.G. Afadin controls p120-catenin-ZO-1 interactions leading to endothelial barrier enhancement by oxidized phospholipids. J. Cell. Physiol. 2012, 227, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Tian, X.; Tian, Y.; Higginbotham, K.; Birukov, K.G. Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol. Biol. Cell 2013, 24, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Pannekoek, W.-J.; Vliem, M.J.; Bos, J.L. Multiple Rap1 effectors control Epac1-mediated tightening of endothelial junctions. Small GTPases 2018, 1–8. [Google Scholar] [CrossRef]
- Birukova, A.A.; Burdette, D.; Moldobaeva, N.; Xing, J.; Fu, P.; Birukov, K.G. Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc. Res. 2010, 79, 128–138. [Google Scholar] [CrossRef]
- Birukova, A.A.; Meng, F.; Tian, Y.; Meliton, A.; Sarich, N.; Quilliam, L.A.; Birukov, K.G. Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 778–791. [Google Scholar] [CrossRef]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflamm. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Satoh, K.; Ichihara, K.; Landon, E.J.; Inagami, T.; Tang, H. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors block calcium-dependent tyrosine kinase Pyk2 activation by angiotensin II in vascular endothelial cells. involvement of geranylgeranylation of small G protein Rap1. J. Biol. Chem. 2001, 276, 15761–15767. [Google Scholar] [CrossRef]
- Ohtsu, H.; Suzuki, H.; Nakashima, H.; Dhobale, S.; Frank, G.D.; Motley, E.D.; Eguchi, S. Angiotensin II signal transduction through small GTP-binding proteins: Mechanism and significance in vascular smooth muscle cells. Hypertension 2006, 48, 534–540. [Google Scholar] [CrossRef]
- Townsley, M.I. Permeability and calcium signaling in lung endothelium: Unpack the box. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; White, G.C.; Quilliam, L.A.; Whitehead, K.J. Small GTPase Rap1 is essential for mouse development and formation of functional vasculature. PLoS ONE 2015, 10, e0145689. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanthan, S.; Zheng, X.; Nishijima, Y.; Sobczak, M.; Szabo, A.; Vasquez-Vivar, J.; Zhang, D.X.; Chrzanowska-Wodnicka, M. Rap1 promotes endothelial mechanosensing complex formation, NO release and normal endothelial function. EMBO Rep. 2015, 16, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanthan, S.; Sobczak, M.; Li Calzi, S.; Shaw, L.; Grant, M.B.; Chrzanowska-Wodnicka, M. Rap1B promotes VEGF-induced endothelial permeability and is required for dynamic regulation of the endothelial barrier. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117 Pt 12, 2427–2429. [Google Scholar] [CrossRef]
- O’Shaughnessy, E.C.; Stone, O.J.; LaFosse, P.K.; Azoitei, M.L.; Tsygankov, D.; Heddleston, J.M.; Legant, W.R.; Wittchen, E.S.; Burridge, K.; Elston, T.C.; et al. Software for lattice light-sheet imaging of FRET biosensors, illustrated with a new Rap1 biosensor. J. Cell Biol. 2019, 218, 3153–3160. [Google Scholar] [CrossRef]
- Busse, R.; Mulsch, A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. Febs Lett. 1990, 265, 133–136. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef]
- Michel, T.; Feron, O. Nitric oxide synthases: Which, where, how, and why? J. Clin. Investig. 1997, 100, 2146–2152. [Google Scholar] [CrossRef]
- Lin, S.; Fagan, K.A.; Li, K.X.; Shaul, P.W.; Cooper, D.M.; Rodman, D.M. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 2000, 275, 17979–17985. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Name of the Protein | Molecular Structure Domains | Protein Length | GEF Activity |
|---|---|---|---|---|
| RAPGEF3 | Epac1 |  | 881 | Rap1, Rap2 |
| RAPGEF4 | Epac2 |  | 1011 | Rap2 |
| RASGRP2 | CalDAG GEF-I |  | 609 | Rap1a>N-Ras |
| RASGRP | CalDAG GEF-II |  | 797 | H-Ras, R-Ras |
| RASGRP3 | CalDAG GEF-III |  | 689 | H-Ras, R-Ras, M-Ras, Rap1a, Rap2a |
| RASGRP4 | CalDAG GEF-IV |  | 673 | H-Ras |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosuru, R.; Chrzanowska, M. Integration of Rap1 and Calcium Signaling. Int. J. Mol. Sci. 2020, 21, 1616. https://doi.org/10.3390/ijms21051616
Kosuru R, Chrzanowska M. Integration of Rap1 and Calcium Signaling. International Journal of Molecular Sciences. 2020; 21(5):1616. https://doi.org/10.3390/ijms21051616
Chicago/Turabian StyleKosuru, Ramoji, and Magdalena Chrzanowska. 2020. "Integration of Rap1 and Calcium Signaling" International Journal of Molecular Sciences 21, no. 5: 1616. https://doi.org/10.3390/ijms21051616
APA StyleKosuru, R., & Chrzanowska, M. (2020). Integration of Rap1 and Calcium Signaling. International Journal of Molecular Sciences, 21(5), 1616. https://doi.org/10.3390/ijms21051616