Abstract
Background: Increasingly, different heterocyclic systems have been introduced into the steroid nucleus to significantly enhance the antitumor activities of steroid molecules. However, in this study, few literature precedents describing the pyrazine heterocyclic-condensed modification to an A-ring of steroid monomers were found, although the pyrazine group is thought to be essential for the potent anticancer activity of clinically relevant drugs and natural steroid dimers. Methods and Results: Two series of novel A-ring fused steroidal pyrazines were designed and efficiently synthesized from commercially available progesterone via key α-ketoenol intermediates. Through a cell counting kit-8 cytotoxic assay of 36 derivatives for three tumor cells, 14 compounds displayed significant antiproliferative activity compared to 5-fluorouracil, especially for human prostatic tumor cells (PC-3) in vitro. Further mechanistic studies indicated that the most active compound, 12n (IC50, 0.93 μM; SI, 28.71), could induce the cell apoptosis of PC-3 cells in a dose-dependent manner and cause cell cycle arrest in the G2/M phase. The molecular docking study suggested that compound 12n fitted the active sites of cytochrome P450 17A1 (6CIZ) well. Conclusions: 12n might serve as a promising lead compound for the development of novel anticancer drugs. This facile ring-closing strategy may provide a novel and promising avenue for the cycloaddition reaction of the steroidal skeleton through α-ketoenol intermediates.
1. Introduction
Despite important advances in anticancer therapy, cancer is the second leading cause of death worldwide for humans, following cardiovascular diseases. Currently, chemotherapy has become a routine in most cancer therapy, but it is limited by the emergence of resistant cancer cells. Therefore, developing safer and more selective anticancer drugs is still an unequivocal need. Steroidal derivatives have attracted great attention due to their rigid sterane skeleton with a range of functionalization, the ability to interact with various biological targets and pathways [1], a broad spectrum of bioactivity, the specific ability to penetrate cell membranes, and so on [2,3,4]. The steroidal derivative named abiraterone has been successfully used as a commercial anti-prostate cancer drug.
Previous modifications of steroids have mainly focused on the functionalization of the C-3/6/20 position of the steroid nucleus for screening anticancer molecules (Figure 1). More than 124 compounds have significant cytotoxic effects, with IC50/GI50 values below 1 μM [5,6,7,8,9,10,11,12,13,14,15,16,17,18]. Among them, more than 52 compounds even have values below 50 nM [5,8,12,13,14,15,16,17,18]. In particular, the introduction of different heterocyclic systems into ring D, including pyri(mi)dine [15,16], thiophene, thiazole [12,13], and pyrazole [9,10,13], can significantly enhance the antitumor activities of steroid molecules, even up to nanomolar levels. This is different from the modification of the A ring, which uses the extremely potential bufalin as a starting substrate. As shown in Figure 1, there are 36 D-ring fused steroids and 57 substituted compounds linked by a side-chain which IC50 values that can be below 1 μM. However, the anticancer activity of A-ring fused steroids has scarcely been reported, expect for in 2017 (moderate antiproliferative activity) [19]. Meanwhile, few literature precedents describing the pyrazine ring-condensed modification to an A-ring of steroid monomers were also found in this study. Furthermore, the mechanism of antitumor activity for these derivatives has not been deeply investigated, especially for compounds with nanomolar IC50 values.
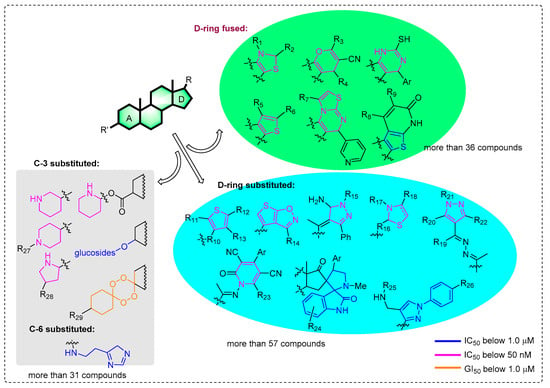
Figure 1.
Current potential anticancer steroid stuctures with the IC50/GI50 values below 1.0 μM.
In addition, the pyrazines and their derivatives are known to be very important structural motifs in various natural products, such as cephalostatins and ritterazines (Figure 2) [20,21,22], and possess extremely potent anticancer activity (sub-nanomolar levels) [23,24]. Furthermore, the pyrazine ring is also thought to be essential for their strong antitumor activity exhibited in pharmacologically active synthetic compounds (Figure 2) AZD8835 [25] and clinically relevant drugs CX5641 [26].
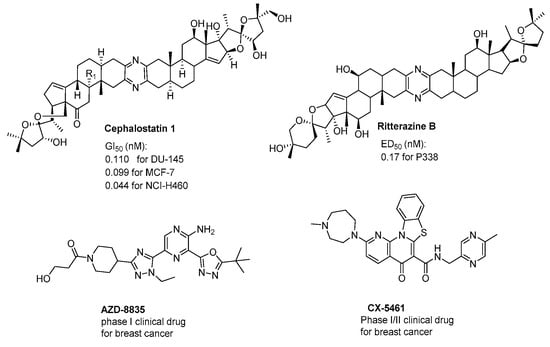
Figure 2.
The structures of anticancer agents containing a pyrazine moiety.
In view of the pharmacological importance of the steroid skeleton and pyrazine functional group in developing new biologically active steroids, we herein represent the synthesis of two series of novel steroidal A-ring fused pyrazines and their antiproliferative activity against human cancer cell lines (MCF-7, prostatic tumor cells (PC-3), and HepG2) and a human normal liver cell (THLE-2) in vitro. Our further goal was to explore their effects on the cell cycle and possible molecular mechanisms of inducing apoptosis.
2. Results
2.1. Chemistry
Thirty-six novel A-ring fused steroidal pyrazines, belonging to two different series, were designed and efficiently synthesized through four steps, with progesterone 1 as starting material. Among them, twenty-one target derivatives (5a–5f, 10a–10o), mainly bearing a benzopyrazines (quinoxalines) group, were prepared by the cyclocondensation of key intermediates containing an α-ketoenols group (2-hydroxy-3-oxo-△1,5 analog) with 1,2-diamines and substituted o-phenylenediamines (OPDs) in excellent yields (Scheme 1). The second series of steroidal A-ring fused pyrazines (12a–12o), bearing a pyrazinamides moiety, were synthesized through a ring-closing reaction of intermediate 8 and 2,3-diaminopropanamides in similar conditions (Scheme 2).
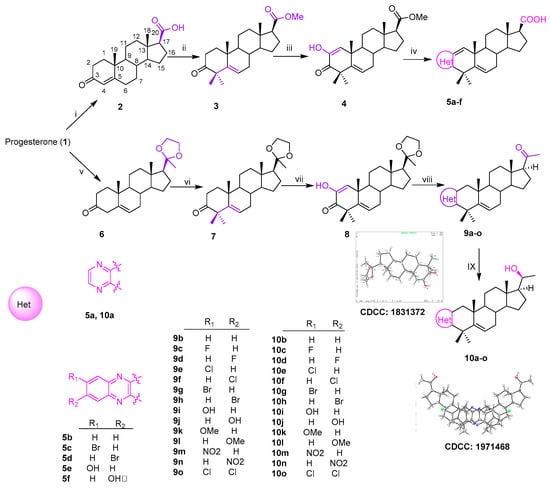
Scheme 1.
Synthesis of A-ring fused steroidal benzopyrazines.
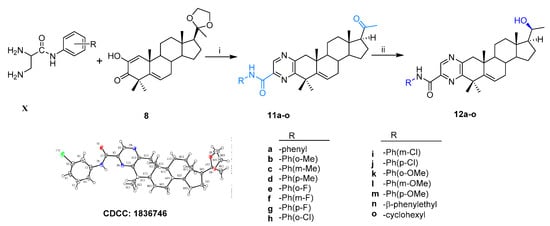
Scheme 2.
Synthesis of steroidal A-ring fused pyrazinamides. Reagents and conditions: i. (a) amine, EtOH, ρ-TSA, reflux, 85–90%; (b) 1M HCl, 95–99%; ii. NaBH4, dry MeOH/THF (v/v, 2:1), 0 °C, 85–90%.
All the structures of the newly synthesized compounds were identified by NMR and HRMS spectra. The absolute configurations of α-ketoenol intermediates 8 and 10g and the precursor of 11i were confirmed by the single crystal diffraction method using CuKα radiation and deposited with the Cambridge Crystallographic Data Centre (CCDC numbers: 1831372, 1971468 and 1836746, respectively; Scheme 1). The single-molecule structure of 10g was re-drawn by ShelXle software because of one unit containing two molecules for 10g (see section 1 of Supplementary Materials).
Reagents and conditions: i. NaOH, Br2, 76%; ii. (a) MeI, K2CO3, acetone, 40 °C, 5 h, 90%; (b) t-BuOK, t-BuOH, MeI, 60%; iii. t-BuOK, dry-THF, O2, 1 h, 69%; iv. (a) EtOH, reflux, overnight; (b) KOH, H2O, EG, reflux, 30%–67%; v. EG, (EtO)4Si, ρ-TsOH (cat.), r.t., 5 h, 95%; vi. t-BuOH, t-BuOK, 1 h, then, MeI, 50%; vii. t-BuOK, t-BuOH, THF, O2, 89%; viii. (a) amine, EtOH, ρ-TSA, reflux; (b) 1M HCl, 77%–90%; (c) NaBH4, dry MeOH/THF (v/v, 2:1), 0 °C, 80%–90%.
2.1.1. A-Ring Fused Steroidal Benzopyrazines 5a–5f and 10a–10o (Series 1)
The carboxylic acid 2 was obtained through the reaction of progesterone with bromine and sodium hydroxide [27]. The compound 2 was subsequently transformed to its corresponding methyl ester using methyl iodide [28] in acetone, and then reacted with MeI and t-BuOK in dry THF [29], which generated the 4,4-dimethyl compound 3 (Scheme 1). The resulting residue (3) was treated with an excess of t-BuOK in the t-BuOH/THF system (20:1, v/v) under an oxygen condition [30] to afford the key α-ketoenol intermediate compound 4 (Scheme 1). Target derivatives 5a–5f were achieved over two steps in excellent yields by combining 1,2-diamines or substituted OPDs with key intermediate product (4) in refluxing ethanol and then deprotecting though directly adding KOH (Scheme 1).
The compound 10a–10o was eventually prepared from key intermediate product (8) over three simple steps, as shown in Scheme Progesterone was initially selectively protected with the ethylene ketal group [31] to afford compound The subsequent reaction of 6 with t-BuOK and MeI took place in t-BuOH [32] and led to the 4,4-dimethyl compound The 4,4-dimethyl moiety was beneficial to the following C2 oxidization reaction, to afford specific 2,3-diketone product 8 under the same condition as the formation of compound 4, the configuration of which was confirmed by X-ray data (Scheme 1, CCDC: 1831372).
The compound 8 was treated with 1,2-diamines and substituted OPDs to get target compounds 9a–9b and 9g–9j, which were then converted into the corresponding target derivatives 10a–10b and 10g–10j through the removal of 20-ethylene ketal protection and reduction. This scaffold of 10g was confirmed by X-ray single crystal diffraction (CCDC: 1971468). The antitumor activities of these six compounds containing 20-OH were not obviously different from those of 5a–5f containing 20-COOH. Therefore, the remaining compounds of A-ring fused steroidal pyrazines 10c–10f and 10k–10o were obtained under the same condition. All spectral and analytical data were in agreement with the assigned structures (see section 2 of Supplementary Materials).
2.1.2. A-Ring Fused Steroidal Pyrazinamides 12a–12o (Series 2)
According to the same condition as above, the treatment of compound 8 with different substituted 2,3-diaminopropanamides (X) resulted in the formation of 11a–11o, which then changed to the corresponding target products 12a–12o (Scheme 2). The configuration of 11i bearing the ethylene ketal group was confirmed by X-ray single crystal diffraction (CCDC, 1836746). All the structures of the newly synthesized compounds were confirmed by 1H NMR, 13C NMR, and HRMS spectra (see section 2 of Supplementary Materials).
2.2. Antiproliferative Activity
All the thirty-six target derivatives (5a–5f, 10a–10o, and 12a–12o) were screened for antiproliferative activities against three human cancer cell lines (MCF-7, PC-3, and HepG2) and cytotoxicity on a normal human cell (THLE-2) at the concentration of 20 μM by using the CCK8 assay. The preliminary bioassay results are depicted in Table 1 For PC-3 cell lines, fourteen compounds exhibited equal or superior activity against the PC-3 cell line compared with the commercial drug 5-fluorouracil (5-Fu). Many compounds, such as 5d, 10d–f, 10j, 10l, 12e–f, 12h–k, and 12m, showed moderate antiproliferative activities (30% < inhibition ratios < 60%); 12g and 12n displayed remarkable activities (inhibition ratios > 60%) against PC-3 cells. However, for MCF-7 cells, many compounds, except 10l, 12g, and 12n, did not display obvious antitumor activities (inhibition ratios < 30%). In addition, for HepG2 cell lines, these compounds, except 10b, 10f, 10l, and 12n, did not display excellent antiproliferative activities (inhibition ratios < 30%). Meanwhile, most of the compounds exhibited low cytotoxicity against the THLE-2 cell line. In particular, the IC50 values of the compounds 12g and 12n were 6.88 and 0.93 μM against the PC-3 cell line, respectively, and the latter possessed a selectivity index (SI) value of up to 28.71 (Table 2).

Table 1.
Inhibitory activity of the derivatives of 5a–f, 10a–o, and 12a–o in terms of the growth of three human cancer cell lines and one human normal cell line.
Table 2.
IC50 of the derivatives 12g and 12n with PC-3 and THLE-2 cell lines.
These compounds containing a halogen atom exhibited better antitumor activity, and the products containing a substitute which was closer to the 4,4-dimethyl moiety displayed better inhibitory activity for 5b–5f and 10b–10n. Moreover, variation of the substituted position of the phenyl ring slightly affected the activities, with para ˃ ortho ˃ meta for compounds 12b–12n.
The differences in the conformation of these compounds, such as the structural rigidity or the molecular volumes, were displayed by using the Molinspiration tool (Table 3) [33]. In the first series of derivatives 10c–10o, the halogen substitute could apparently enhance the transport properties (lower topologic polar surface area (TPSA)) of compounds. In the second series of derivatives 12a–10o, the introduction of the F atom could maintain the compounds’ solubility (lower miLogP and molecular volumes). These calculated molecular properties may provide some references to compounds’ lead-like properties in prodrugs.
Table 3.
Calculated molecular properties of all the derivatives.
2.3. Morphological Detection of Cell Apoptosis
The compound 12n, which possessed the strongest activity, was selected to further assess its effects on morphological changes of PC-3 cells. After being treated with 12n at different concentrations (1.0 and 4.0 μM), the cells were stained with Giemsa stain. As shown in Figure 3a, the cells in the control (0.1% MSO) showed a regular appearance and were surrounded by abundant cytoplasm. In comparison, along with the increased concentration of 12n, the cytoplasm became deformed and clearly distinguished, and the color of the cell nucleus became deeper so that even the nuclear condensation was exhibited (Figure 3b,c). Other morphological changes, such as the loss of volume and deformation of a regular appearance, were also found. Generally speaking, these results indicated that 12n could cause the morphological change of PC-3 cells in a dose-dependent manner.
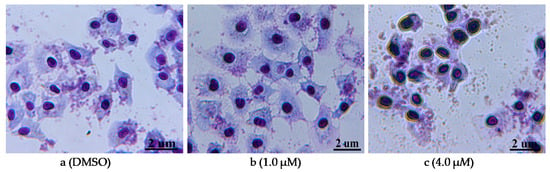
Figure 3.
Morphological changes of PC-3 cells through Giemsa staining. (a) DMSO used as the negative control; (b,c) dealt with compound 12n for 48 h at the concentration of 1.0 and 4.0 μM, respectively.
2.4. Cell Apoptosis Analysis
To clarify the effect of steroidal pyrazines on the induction of apoptosis in PC-3 cells, a flow cytometry assay with Annexin-V-FITC/PI double staining was implemented. As shown in Figure 4, the apoptotic ratios (including the early and late apoptotic ratios) of PC-3 cells treated with compound 12n increased gradually in a concentration-dependent manner (Figure 4). Moreover, the apoptosis ratios of compound 12n at different concentration gradients were found to be 8.66% (0.2 μM), 23.62% (1.0 μM), and 39.57% (5.0 μM), respectively, which were apparently higher than that of the blank control group (5.77%) (Table 4). These results indicated that 12n induced the apoptosis of PC-3 cells in a dose-dependent manner.
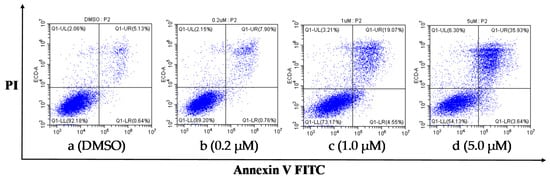
Figure 4.
Compound 12n induced apoptosis in PC-3 cells after 48 h through treatment with 12n at different concentrations (0.2, 1.0, and 5.0 μM).
Table 4.
The percentage of cell apoptosis of PC3 cells after 48 h through treatment with 12n at different concentrations (0.2, 1.0, and 5.0 μM).
2.5. Cell Cycle Assay
To further clarify the effects of A-ring fused steroidal pyrazines on cell growth, the cell cycle distribution of PC-3 cells was investigated through treatment with compound 12n at concentration gradients for 48 h (Figure 5 and Table 5). Treatment with 12n at various concentrations of 0.2, 1.0, and 5.0 μM resulted in a significant accumulation of 16.09%, 46.19%, and 44.27% of cells in the G2/M phase (control group, 15.69%). When the concentration of 12n was 8 μM, the percentage of cells in the G1 phase increased to 49.62% and G2/M phase increased to 44.27%, whilst exhibiting a substantial decrease of the S phase. These results suggested that derivative 12n could cause obvious G2/M phase arrest in a dose-dependent manner.
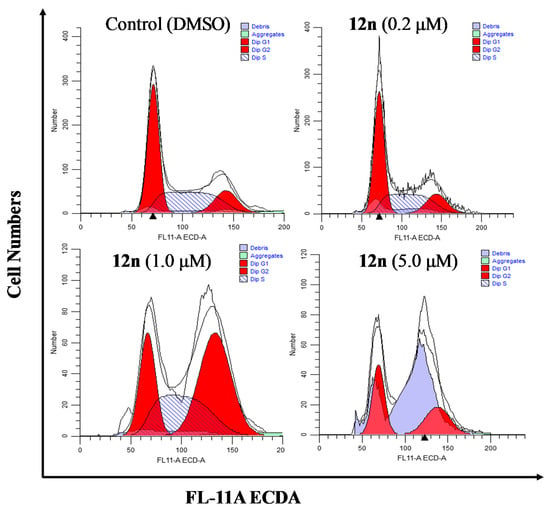
Figure 5.
Effects of compound 12n on the cell cycle distribution of PC-3 cells after treatment for 48 h.
Table 5.
Distribution of the cell cycle after treatment with 12n in PC-3 cells.
2.6. The Molecular Docking
Potential molecular targets for the most active compound, 12n, were screened by rapid molecular docking against more than ten proteins using the MOE software (Table 6). The cytochromes P450 17A1/19A1 (CYP17A1/CYP17A1) have a strong correlation with cell apoptosis, because most of them have an Abiraterone-specific active site. As can be seen in Table 6, the CYP17A1 proteins (except 3S7S) have stronger binding abilities than the reference drug Abiraterone acetate (−8.91 kcal/mol). Furthermore, the binding ability of 6CIZ proteins with 12n was the strongest (−11.72 kcal/mol). This protein could be used to test its binding abilities with various steroidal pyrazines.
Table 6.
Possible cancer-associated protein targets for compound 12n screened by fast docking against the protein data bank (PDB) database.
As can be seen (Table 7), 36 steroidal pyrazines were further checked by comparisons with Abiraterone acetate used as a commercial anticancer drug. Successful docking indicated that most of the designed compounds had research significance, especially compounds 12a–12n, which had virtual ligand screening (VLS) score values lower than −10.0 kcal/mol (Abiraterone acetate, −8.91). In addition, among the compounds, 12n exhibited the lowest score value (−11.87). Steroidal pyrazinamides had lower scores than benzopyrazine derivatives 10a–o, suggesting better anticancer activity. Substituents close to 4,4-dimethyl had lower scores than those located further away from 4,4-dimethyl. The tendency of docking scores was consistent with that of antiproliferative activities of compounds (Table 1).
Table 7.
Binding affinity scores of the compounds 5a−f, 10a−o, and 12a−o with 6CIZ.
Furthermore, the molecular interactions of the most active compound, 19, with 6CIZ were displayed in detail by using the MOE software. The hypothetical 3D binding modes of compound 12n and the reference ligands with 6CIZ are presented in Figure 6a. Compound 12n was predicted to adopt a similar binding space and orientation as Abiraterone. A similar analysis suggests that 12n could interact strongly with CYP17A1 family proteins (6CIZ). In addition, the two-dimensional (2D) interaction diagram for compound 12n with 6CIZ is shown in Figure 6b. The hydrogen bond in this complex is the most notable: the pyrizine ring, β-phenethylamine ring, and 20-OH group could form a hydrogen bond with Ala 302, Gly 444, and Arg of 6CIZ, respectively.
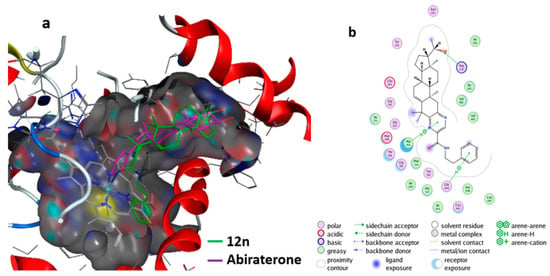
Figure 6.
Interaction of the compound 12n with the CYP17A1 (6CIZ) receptor: (a) docking of compound 12n (green) with 6CIZ and Abiraterone (purple) was used as the reference; (b) interaction of compound 12n with 6CIZ.
The molecule of compound 12n could also hydrophobically interact with amino acid residues Ala302, Phe114, Val483, and Val482 (Figure 6b). The pyrizine group played the key role in determining the inhibitory properties of compound 12, because it not only resulted in the formation of the H-bond with Ala302, but also interacted with Ala302 through hydrophobic and van der Waals contacts [34]. The most active compound, 12n, fitted the active sites of CYP17A1 (6CIZ) well. These docking results further proved the bioactivity and pharmacological potential of compound 12n against PC-3 cells. These ligand-based screening results could be used as a guide for the deep optimization of pre-drugs.
2.7. Preliminary Structure–Activity Relationship
Generally, combining the CCK-8 assay and molecular docking screening results, a preliminary structure–activity relationship could be obtained. The halogen substituents were beneficial to improving the antitumor activity. For benzopyrazine derivatives 10a–o, substituents close to 4,4-dimethyl were better than those located further away from 4,4-dimethyl. The modification using pyrazinamides was more conducive to anticancer activity than that using benzopyrazines.
3. Discussion
The formation of α-ketoenols (2-hydroxy-3-oxo-△1,5 analogs) was the step bottleneck in the whole synthetic strategy. The desired A-ring fused steroidal pyrazines could be obtained from compound 3 or 7 in quite low yields, without the α-ketoenol intermediates (4 or 8), even in harsh reaction conditions (sulfur and refluxing morpholine) [30]. However, the corresponding α-ketoenols might be prepared in complex reaction conditions, according to previous reports [35,36], such as in dry toluene at −25 °C for 1.5–4 h with t-BuOK (1.5 equiv.) and (3 equiv.) 18-crown-6 under dry O2, and easily accompanied by a byproduct through oxygenation at CFinally, the introduction of 4,4-dimethyl could not only maintain the activity of target compounds, but also be favorable for the efficient formation of α-ketoenols at room temperature. In addition, the 4,4-dimethyl group was widespread in sterols derived from the natural lanostane or cycloartane skeletons and was also included in triterpenoids which showed broad spectrum antitumor activity [37].
Through modifications of the steroidal D-ring and its C-20 side-chain, more than 35 steroid monomers with nanomolar IC50 values below 50 nM were synthesized by Banday’s group in 2010 [8] and by Mohareb’s group in 2012, 2013, and 2014 [12,13,14,15,16]. Additionally, about nine steroid monomers were reported by Ma’s group [17] that had less than 50 nM levels through 3-nitrogen-containing esterificated at the C-3 position based on special bufalin, which originally had excellent antitumor activity. However, the mechanism of their antitumor activity for steroid monomer derivatives still has not been deeply investigated, especially for compounds with nanomolar IC50 values. Only a few preliminary mechanisms were reported by Shi et al. and Liu et al. for potential synthetic derivatives with IC50 values below 1 μM. An androsten-17-(1’,3’,4’)-pyrazole (IC50 0.53 μM) could cause Hela cell line apoptosis and arrest the cell cycle in the S phase [10]. Another steroidal spiro-oxindole (IC50 0.71 μM) arrested the cell cycle in the G2/M phase [6]. Furthermore, for ring-condensed derivatives, the steroidal ring A/D-fused arylpyrazoles displayed moderate anticancer activity (4.0–5.0 μM) that might cause cell cycle arrest in the G2/M phase for T47D cells and MDA-MB-231cells, respectively [19]. In this work, compound 12n could also cause cell cycle arrest in the G2/M phase.
Generally, there are currently two major apoptotic pathways used by cytotoxic drugs: one that begins with the ligation of death receptors and subsequently activates the initiator caspase-8, and another that involves the “activation” of the mitochondria pathway. For natural steroid monomers like α-solanine and α-chaconine, several studies have found that they could suppress the migration and invasion of cancer cells through the inhibition of PI3K/Akt and MAPK signaling pathways and also suppress metastasis by declining the NF-κB activity [11]. However, for steroid dimers, the ED50 /IC50 values could be below 1 μM for 20 natural cephalostatins, 19 ritterazines, and only 1 [1,2,4] triazolo [1,5-α] pyrimidine-based phenyl-linked steroid dimer [38,39]. These mechanical studies of cephalostatin 1 and the synthetic steroid dimer suggested that they could induce apoptosis, probably through the mitochondrial pathway, which was a distinctive feature of dimeric steroids and different from that of steroid monomers [23,39]. In summary, the anticancer mechanism still requires further research for satisfying the development of novel anticancer drugs, especially for synthetic fused derivatives with IC50 values below 1 μM.
4. Materials and Methods
4.1. General
All chemicals and solvents were purchased from commercial sources (Chron Chemical Co., Ltd., Chengdu, China; Aladdin Industrial Co., Ltd., Shanghai, China) and used as received, except for THF and t-BuOH, which were distillated when needed. The 1H-NMR and 13C-NMR spectra were investigated using an Avance spectrometer (Bruker, Billerica, MA, USA) at 500 and 125 MHz. Chemical shifts were measured relative to the residual solvent peaks of CDCl3 and DMSO-d6, with tetramethylsilane (TMS) as the internal standard (Aladdin, Shanghai, China). High-resolution mass spectroscopy was undertaken using an AB SCIEX Triple TOF 5600+ spectrometer (AB SCIEX, Boston, MA, USA). Melting points were measured through capillary melting apparatus and uncorrected (SGW X-4, Shanghai, China). The column chromatography and thin-layer chromatography (TLC) were implemented on a silica gel 100–200 mesh and silica GF254 with ultraviolet detection purchased from Qingdao Marine Chemical Company (China). Four cell lines (MCF-7, PC-3, HepG2, and THLE-2) were obtained from the ATCC (Manassas, VA, USA) and were cultured using Ham’s F-12K (Wisent, Nanjing, China), DMEM medium (Hyclone Laboratories Inc., Logan, Utah, USA) and 10% FBS (Gibco&Invitrogen, Carlsbad, CA, USA). The optical density (OD) values were recorded using a multimode plate reader (PerkinElmer VICTOR Nivo, Turku, Finland). The biological samples were processed and analyzed by flow cytometry (CytoFLEX, California, United States). Molecular docking experiments were implemented using the MOE software (CCG, Quebec, Canada).
4.1.1. Synthesis of 4,4-dimethyl-3-oxo-pregnen-5-ene-17-carboxylate (3)
The carboxylic acid compound 2 could be easily obtained and recrystallized from acetone, according to a previously described method [27]. The esterification reaction of 2 was also carried out as previously described [28]. The 4,4-dimethylsteroid 3 could be prepared following the reported method [29]. The 1H and 13C NMR spectra of compounds 2 and 3 are available in the section 2.2 of Supplementary Materials.
4.1.2. Synthesis of 4,4-dimethyl-2-hydroxy-3-oxo-pregnen-1,5-diene-17β-carboxylate (4)
To a solution of 3 (7.2 g, 20.0 mmol) in t-BuOH (200 mL) and THF (5.0 mL), t-BuOK (4.50 g, 40.00 mmol) was added, and the mixture was stirred at 40 °C under O2 for 40 min. After the THF was removed, the residue was replenished with brine solution (80 mL). The mixture was extracted with EtOA, and the organic layer was washed with saturated NaCl solution, dried over Na2SO4, and concentrated. Then, the crude solid was purified through silica gel column chromatography (PE/EA, 10/1, v/v), to afford the α-ketoenol 4 (5.2 g, 69%) as a white solid [30]. m.p. 152−154 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.27 (d, J = 8.8 Hz, 1H, OH), 6.00 (d, J = 8.8 Hz, 1H, H1), 5.57 (t, J = 3.5 Hz, 1H, H6), 3.60 (s, 3H, COOCH3), 2.37 (t, J = 9.3 Hz, 1H), 2.23 – 2.07 (m, 2H), 1.99 (td, J = 13.2, 6.6 Hz, 3H), 1.75 – 1.60 (m, 6H,), 1.29 (s, 3H), 1.24 (s, 6H, 2 × CH3), 1.21 (s, 2H), 0.63 (d, J = 4.0 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO) δ 198.05, 173.50, 144.06, 143.89, 122.71, 120.78, 55.61, 54.13, 51.05, 48.10, 47.55, 43.33, 37.80, 37.41, 31.05, 30.79, 30.65, 24.40, 23.79, 23.27, 23.22, 20.37, 13.23; HRMS (ESI) m/z calcd for C23H32O4 [M + H]+ 373.2373, found 373.2357.
4.1.3. General Procedure for the Preparation of Target Compounds 5a–5f
To a solution of 4 (372.2 mg, 1.0 mmol) in anhydrous EtOH (20 mL), anhydrous magnesium sulfate (0.5 g, 4 eq.) and ethylene diamine (2 eq.) were added. The mixture was heated to reflux for 8 h and then filtered, and the filtrate was concentrated to provide the crude white solid. The residue was purified by flash column chromatography on silica gel (petroleum ether/EtOAc 3:1, v/v) to afford the crude product, which was used for the next reaction, without further purification. Immediately, the crude product was dissolved in ethylene glycol (10.0 mL), an aqueous solution of KOH (1.0 mL, 40% m/v) was added, and the resulting suspension was heated at 130 °C under reflux for 20 min until a clear red solution was obtained [29]. After being cooled down to r.t., saturated brine (20 mL) and HCl 10% (10 mL) were added to adjust the pH to 3, and the precipitate was extracted with EtOAc. The organic layer was washed with saturated brine, dried over anhydrous Na2SO4, and evaporated in vacuo. The crude product was purified by column chromatography on silica (petroleum ether/EtOAc gradient, 3:1 to 1:1), to yield the desired A-ring fused steroidal pyrazine 5a. White solid; yield: 67%; m.p. 270–292 °C. 1H NMR (500 MHz, DMSO-d6) δ 11.93 (s, 1H, COOH), 8.48 (d, J = 2.4 Hz, 1H, pyrazine), 8.34 (d, J = 2.6 Hz, 1H, pyrazine), 5.81 (dt, J = 7.1, 3.4 Hz, 1H, H6), 2.97 (d, J = 15.6 Hz, 1H, H1), 2.64 (d, J = 15.6 Hz, 1H, H1), 2.43 (d, J = 14.9 Hz, 1H), 2.30 (t, J = 9.1 Hz, 1H), 2.15 (dt, J = 17.3, 5.2 Hz, 1H), 2.09 – 1.93 (m, 2H), 1.77 – 1.63 (m, 4H), 1.54 (s, 3H, CH3), 1.52 (d, J = 3.2 Hz, 1H), 1.27 (s, 3H, CH3), 1.25 – 1.17 (m, 4H), 0.70 (s, 3H, CH3), 0.65 (s, 3H, CH3); 13C NMR (126 MHz, DMSO) δ 174.65, 158.32, 150.32, 148.64, 142.61, 141.48, 120.09, 55.56, 54.46, 48.20, 43.13, 43.00, 40.86, 37.63, 37.19, 33.34, 31.56, 31.33, 31.08, 24.03, 23.26, 20.48, 20.20, 13.HRMS (ESI) m/z calcd for C24H32N2O2 [M + H]+ 381.2536, found 381.2538.
Target derivatives 5b–5f were achieved using the same protocols by combining o-phenylenediamine (OPD) or substituted OPD with key intermediate product (4), but without anhydrous MgSO4, in refluxing ethanol, and then deprotecting through adding KOH, with 50%–70% yields for two steps. Those novel derivatives were identified by 1H NMR, 13C NMR, and HRMS(ESI) spectra (see section 2.3 and 4 in Supplementary Materials).
4.1.4. Synthesis of 4,4-dimethyl-2-hydroxy-3-oxo-pregnen-1,5-diene-20-ethylene ketal (8)
Progesterone 20-ethylene ketal (6) could be prepared according to the reported methods [31]. Then 4,4-dimethyl-3-oxo-pregnen-1,5-diene-20-ethylene ketal (7) was synthesized following the reported methods [32], and without HCl aqueous solution to adjust the pH, in case of losing the 20-ethylene ketal. These novel compounds 6 and 7 were identified by 1H NMR, 13C NMR, and HRMS(ESI) spectra (see section 2.4, 2.5 and 4 in Supplementary Materials).
The α-ketoenol product 8 was achieved using the above procedure of compound The configuration of compound 8 was confirmed by X-ray data (Scheme 1, CCDC: 1831372). White solid; yield: 65%; m.p. 148−150°C; 1H NMR (500 MHz, DMSO-d6) δ 8.32 (s, 1H, OH), 6.03 (s, 1H, H1), 5.59 (d, J = 4.6 Hz, 1H, H6), 3.97 – 3.87 (m, 2H, -OCH2CH2O-), 3.82 (dt, J = 17.7, 7.0 Hz, 2H, -OCH2CH2O-), 2.16 – 2.05 (m, 2H, H7), 1.77 – 1.50 (m, 8H), 1.32 (s, 3H, CH3), 1.27 (s, 3H, CH3), 1.23 (s, 6H, 2 × CH3), 1.20 – 1.02 (m, 4H), 0.78 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 199.40, 143.84, 142.83, 123.11, 122.10, 111.86, 65.19, 63.22, 58.00, 57.06, 48.23, 47.25, 41.75, 39.19, 38.47, 32.08, 31.43, 30.55, 24.59, 23.97, 23.62, 23.20, 23.01, 20.77, 13.01; HRMS (ESI) m/z: calcd for C25H36O4 [M + H]+ 401.2686, found 401.2678.
4.1.5. General Procedure for the Conversion of 8 to 9a–9o and 10a–10o
To a solution of 8 (400 mg, 1.0 mmol,) in dry EtOH (10 mL), anhydrous magnesium sulfate (0.5 g, 4 eq.) and ethylene diamine (2 eq.) were added, and the mixture was heated to reflux for 8 h. The mixture was filtered and the filtrate was concentrated to give a white mixture. The residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc 2:1, v/v) to afford a white solid, which was used for the next reaction, without further purification. The crude product was then dissolved in MeOH/DCM (5 mL, 4:1, v/v), three drops of 3 M HCl were added, and the clear solution was stirred for 10 min at r.t. After being adjusted to pH = 7 by NaHCO3, the solvents were removed and the residue was purified by column chromatography (PE:EA = 3:1), to afford 9a. Furthermore, to a solution of 9a in dry MeOH/THF (10 mL, 2:1, v/v), NaBH4 (80 mg) at 0 °C was added portion-wise, and the resulting mixture was stirred at 0 °C for 15 min. The reaction was quenched (saturated aqueous NH4Cl, 10 mL), the solvent was evaporated, and the residue was dissolved in water and EtOAc (40 mL, 1:1, v/v). The organic layer was extracted with EtOAc (30 mL × 3), dried over anhydrous Na2SO4, and evaporated in vacuo. The crude product was purified by silica column chromatography (DCM/MT = 20:1), to afford compound 10a, white solid, yield: 67%; m.p. 150–152 °C; 1H NMR (500 MHz, Chloroform-d) δ 8.42 (d, J = 2.7 Hz, 1H, pyrazine), 8.27 (d, J = 2.6 Hz, 1H, pyrazine), 5.79 (dd, J = 5.3, 2.5 Hz, 1H, H6), 3.75 (dt, J = 12.0, 5.8 Hz, 1H, H20), 3.10 (d, J = 15.7 Hz, 1H, H1), 2.64 (d, J = 15.6 Hz, 1H, H1), 2.29 – 2.11 (m, 2H), 1.79 – 1.66 (m, 4H), 1.61 (s, 3H, CH3), 1.53 (td, J = 13.1, 3.9 Hz, 2H), 1.38 (d, J = 9.0 Hz, 1H), 1.33 (s, 3H, CH3), 1.25 (p, J = 4.7 Hz, 3H), 1.21 (d, J = 8.5 Hz, 2H), 1.16 (d, J = 6.1 Hz, 3H, CH3), 0.80 (s, 6H, 2 × CH3); 13C NMR (125 MHz, CDCl3) δ 159.76, 151.36, 149.14, 142.65, 141.31, 120.77, 70.64, 58.55, 56.48, 49.06, 43.99, 42.46, 41.41, 39.99, 37.86, 33.69, 32.11, 31.86, 31.44, 25.84, 24.63, 23.88, 21.12, 20.56, 12.58; HRMS (ESI) m/z calcd for C25H36N2O [M + H]+ 381.2900, found 381.2885.
Compounds 9b–9o and 10b–10o were synthesized similarly using the above procedure, but without MgSOCollectively, the compound 8 was treated with o-phenylenediamine (OPD, 1.5 eq.) and substituted OPD (1.5 eq.) to get the A-ring fused steroidal pyrazines 9b–9o (24–71% yields for two steps), which were then converted into the corresponding target derivatives 10b-o through the removal of ketal protection and reduction (70–89%). Furthermore, this scaffold 10g was confirmed by X-ray single crystal diffraction (CCDC: 1971468). Those novel compounds were identified by 1H NMR, 13C NMR, and HRMS(ESI) spectra (see section 2.6 and 4 Supplementary Materials).
4.1.6. General Procedure for the Conversion of 8 to 11a–11o and 12a–12o
The 2,3-diaminopropanamides (X) could be prepared by using the standard coupling reagent (DIPEA/HBTU), according to published literature [40] and were then used directly to trigger A-ring condensation with α-ketoenols intermediate Furthermore, the ring-closing reaction was carried out in the same condition as that of compound 10b, and resulted in the formation of 11a–11o (31–64% for two steps), which then changed to the corresponding target products 12a–12o in 75–96% yields (Scheme 2). This scaffold of 11i containing the ethylene ketal group was identified by X-ray single crystal diffraction (CCDC, 1836746, see Scheme 2). Those novel compounds were identified by 1H NMR, 13C NMR, and HRMS(ESI) spectra (see section 2.7 and 4 in Supplementary Materials).
4.2. The X-ray Structure of Three Representative Compounds
X-ray-quality crystal of compounds 8 and 10g was obtained from pure acetone after 3 days and gained from DCM/MT (1:1) after 5 days. X-ray-quality crystal of the compound 11i containing the ethylene ketal protecting group was obtained from cyclohexane/acetone (3:1) after 3 days. A suitable crystal was selected on a SuperNova, Dual, Cu at zero, AtlasS2 diffractometer (Rigaku Corporation, Tokyo, Japan). The crystal was kept at 100.00(10) K during data collection. The atomic coordinates have been deposited at the Cambridge Crystallographic Data Center (CCDC).
4.3. In Vitro Cytotoxicity
Thirty-six novel steroidal A-ring fused pyrazine hybrids were evaluated by Cell Counting Kit-8 (CCK-8) assays [41], to evaluate their in vitro antiproliferative activities on three human cancer cell lines and one normal cell line: MCF-7 (breast carcinoma), PC-3 (prostate carcinoma), HepG2 (liver hepatocellular carcinoma), and THLE-2 (normal lung cell), with 5-fluorouracil as the positive control.
Cells (3 × 103 cells per well) seeded into 96-well plates (Costar, Shanghai, China) were incubated with Ham’s F-12K containing various concentrations of steroidal pyrazines (5a–5f, 10a–10o, and 12a–12o) for 48 h at 37 °C and 5% COThen, 10 μL of CCK-8 reagent (Beyotime, Shanghai, China) dissolved in PBS (Phosphate buffered saline) was added per well, and continuously incubated at 37 °C for 1 h. Finally, the absorbance of samples taken from each well was measured on a microplate reader at 450 nm. The antiproliferative activities were expressed as inhibitory activity and IC50 values calculated using GraphPad Software 5.0 (GraphPad Software, San Diego, CA, USA), which were the mean values of three independent tests and are shown in Table 1 and Table 2, respectively. The formula used for cell inhibitory rates was (1 - sample absorption/vehicle absorption) × 100%.
4.4. Calculation of the Molecular Properties of All Derivatives
The molecular properties were calculated using the Molinspiration tool (http://www.molinspiration.com) [33].
4.5. Giemsa Staining for the Detection of Morphological Features of Apoptosis
Giemsa stain was used to assess the morphological changes of PC-3 cells, according to the standard Giemsa procedure established by the supplier (Solarbio, Beijng, China). Briefly, PC-3 cells were plated at 3 × 103 cells/well into a 96-well plates. PC-3 cells after treatment with 12n for 48 h were fixed with methanol (75%) for 10 min at room temperature. Then, the cells were stained with Giemsa (diluted with phosphate buffer), observed, and photographed under the microscope at 200 × (Olympus Corporation, Tokyo, Japan ) [42]. PC-3 cell lines that did not deal with the compound 12n were used as the control.
4.6. Cell Apoptosis Assay
The cell apoptosis was evaluated with Annexin V-FITC/PI dual staining (Beyotime, Shanghai, China), as previously described [43]. PC-3 cells (1 × 106 cells per well) seeded into 6-well plates (Costar, Shanghai, China) were incubated with Ham’s F-12K containing various concentrations (0.2, 1.0, and 5.0 μM) of compound 12n for 48 h. The biological samples were analyzed using flow cytometry (CytoFLEX, CA, USA).
4.7. Cell Cycle Assay
The cell cycle was examined by PI single staining (Beyotime, Shanghai, China), as previously described [43]. PC-3 cells (1 × 106 cells per well) seeded into 6-well plates were incubated with Ham’s F-12K containing various concentrations (0.2, 1.0, and 5.0 μM) of 12n for 48 h. The samples were analyzed using flow cytometry for DNA content (CytoFLEX, California, United States).
4.8. Molecular Docking Study
The X-ray structures and data of protein targets were obtained from the protein data bank (PDB) (www.rcsb.org): CYP17A1 (17a-hydroxylase/C17,20-lyase, PDB ID: 6CIZ, 6CHI, 6CIR, 5UYS, 4NKZ, 4NKX, 4NKV, 4NKW, and 3RUK) and CYP19A1 (aromatase, PDB ID: 3S7S).
All designed compound structures were energy-minimized using the ChemdBio software and saved in an sdf format. Molecular docking was implemented with the default settings using the MOE software, referring to the published reports [34,44,45]. The numbers of placement poses were 20 for flash docking and 300 for molecular interaction.
5. Conclusions
In summary, 36 novel steroidal A-ring fused pyrazines, belonging to two different series, were efficiently prepared with key α-ketoenol intermediates. This facile ring-closing strategy may provide a novel and promising avenue for the cycloaddition reaction of the steroidal skeleton. The most active compound, 12n, had a lower IC50 value (0.93 μM) and a higher SI value (28.71) compared to human prostatic cells (PC-3). Furthermore, 12n could induce apoptosis of PC-3 cells and cause cell cycle arrest in the G2/M phase. The molecular docking study suggested that 12n could be used as an inhibitor of CYP17A1 (6CIZ). Collectively, 12n may serve as a new candidate for anticancer drug discovery. In addition, the anticancer mechanism still requires further research for satisfying the development of novel anticancer drugs, especially for synthetic fused steroidal derivatives.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/5/1665/s1. The following are available online: The NMR spectra of all compounds and the HRMS spectra of representative compounds.
Author Contributions
S.W. conceived and conducted this work, and wrote the manuscript; N.L. re-elucidated the compound structures via the new HRMS data and revised the manuscript; X.Y. and H.Q. provided help in the HRMS analysis; J.W. designed the project and revised the final paper; all authors approved the final version of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (Grant No. 81773603).
Acknowledgments
We acknowledge the Life Science Large Research Core Services of the university, and also acknowledge Hongli Zhang for providing help during the NMR analysis. We acknowledge Guixai Li from Changzhi Medical College for providing help with the CytExpert software for flow cytometry analysis. We also acknowledge Jingjing Zhang from the School of Life Science of Zhengzhou University for providing help with the MOE molecular docking.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| OPDs | o-Phenylenediamines |
| CCDC | Cambridge crystallographic data centre |
| EG | Ethylene glycol |
| (EtO)4Si | Tetraethyl orthosilicate |
| CCK8 | Cell counting kit-8 |
| MOE | Molecular operating environment |
| CYP17A1 | Cytochrome P450 17A1 |
| miLogP | LogP calculated by Molinspiration tool |
| TPSA | Topologic polar surface area |
References
- Salvador, J.A.R.; Carvalho, J.F.S.; Neves, M.A.C.; Silvestre, S.M.; Leitao, A.J.; Silva, M.M.C.; Melo, M. Anticancer steroids: linking natural and semi-synthetic compounds. Nat. Prod. Rep. 2013, 30, 324–374. [Google Scholar] [CrossRef]
- Baji, Á.; Gyovai, A.; Wölfling, J.; Minorics, R.; Ocsovszki, I.; Zupkó, I.; Frank, É. Microwave-assisted one-pot synthesis of steroid–quinoline hybrids and an evaluation of their antiproliferative activities on gynecological cancer cell lines. Rsc Adv. 2016, 6, 27501–27516. [Google Scholar] [CrossRef]
- Corona-Diaz, A.; Garcia-Merinos, J.P.; Ochoa, M.E.; Del Rio, R.E.; Santillan, R.; Rojas-Lima, S.; Morzycki, J.W.; Lopez, Y. TiCl4 catalyzed cleavage of (25R)-22-oxo-23-spiroketals. Synthesis of sapogenins with furostanol and pyranone E rings on the side chain. Steroids 2019, 152, 108488. [Google Scholar] [CrossRef]
- Kiss, A.; Wolfling, J.; Mernyak, E.; Frank, E.; Benke, Z.; Ashkan Senobar Tahaei, S.; Zupko, I.; Maho, S.; Schneider, G. Stereocontrolled synthesis of the four possible 3-methoxy and 3-benzyloxy-16-triazolyl-methyl-estra-17-ol hybrids and their antiproliferative activities. Steroids 2019, 108500. [Google Scholar] [CrossRef]
- Monier, M.; El-Mekabaty, A.; Abdel-Latif, D.; Dogru Mert, B.; Elattar, K.M. Heterocyclic steroids: Efficient routes for annulation of pentacyclic steroidal pyrimidines. Steroids 2019, 108548. [Google Scholar] [CrossRef]
- Yu, B.; Shi, X.-J.; Qi, P.-P.; Yu, D.-Q.; Liu, H.-M. Design, synthesis and biological evaluation of novel steroidal spiro-oxindoles as potent antiproliferative agents. J. Steroid Biochem. Mol. Biol. 2014, 141, 121–134. [Google Scholar] [CrossRef]
- Banday, A.H.; Mir, B.P.; Lone, I.H.; Suri, K.A.; Kumar, H.M.S. Studies on novel D-ring substituted steroidal pyrazolines as potential anticancer agents. Steroids 75, 805–809. [CrossRef]
- Banday, A.H.; Shameem, S.A.; Gupta, B.D.; Kumar, H.M.S. D-ring substituted 1,2,3-triazolyl 20-keto pregnenanes as potential anticancer agents: Synthesis and biological evaluation. Steroids 75, 801–804. [CrossRef]
- Li, J.; Zhao, X.; Li, L.; Yuan, Z.; Tan, F.; Shi, B.; Zhang, J. Design, synthesis and cytotoxic activity of a novel series of steroidal phenylpyrazoles. Steroids 2016, 107, 45–54. [Google Scholar] [CrossRef]
- Li, J.; Huo, H.; Guo, R.; Liu, B.; Li, L.; Dan, W.; Xiao, X.; Zhang, J.; Shi, B. Facile and efficient access to Androsten-17-(1’,3’,4’)-pyrazoles and Androst-17beta-(1’,3’,4’)-pyrazoles via Vilsmeier reagents, and their antiproliferative activity evaluation in vitro. Eur. J. Med. Chem. 2017, 130, 1–14. [Google Scholar] [CrossRef]
- Jiang, Q.W.; Chen, M.W.; Cheng, K.J.; Yu, P.Z.; Wei, X.; Shi, Z. Therapeutic potential of steroidal alkaloids in cancer and other diseases. Med. Res. Rev. 2016, 36, 119–143. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Al-Omran, F. Reaction of pregnenolone with cyanoacetylhydrazine: Novel synthesis of hydrazide-hydrazone, pyrazole, pyridine, thiazole, thiophene derivatives and their cytotoxicity evaluations. Steroids 2012a, 77, 1551–1559. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Wardakhan, W.W.; Elmegeed, G.A.; Ashour, R.M.S. Heterocyclizations of pregnenolone: Novel synthesis of thiosemicarbazone, thiophene, thiazole, thieno 2,3-b pyridine derivatives and their cytotoxicity evaluations. Steroids 2012b, 77, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; El-Sayed, N.N.E.; Abdelaziz, M.A. The Knoevenagel reactions of pregnenolone with cyanomethylene reagents: synthesis of thiophene, thieno 2,3-b pyridine, thieno 3,2-d isoxazole derivatives of pregnenolone and their in vitro cytotoxicity towards tumor and normal cell lines. Steroids 2013, 78, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Abbas, N.S.; Abdelaziz, M.A. Heterocyclic ring extension of androstenedione: Synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids 2014a, 86, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Al-Omran, F.; Azzam, R.A. Heterocyclic ring extension of estrone: Synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids 2014b, 84, 46–56. [Google Scholar] [CrossRef]
- Ma, B.A.; Xiao, Z.Y.; Chen, Y.J.; Lei, M.; Meng, Y.H.; Guo, D.A.; Liu, X.; Hu, L.H. Synthesis and structure-activity relationships study of cytotoxic bufalin 3-nitrogen-containing-ester derivatives. Steroids 2013, 78, 508–512. [Google Scholar] [CrossRef]
- Terzić, N.; Opsenica, D.; Milić, D.; Tinant, B.; Smith, K.S.; Milhous, W.K.; Šolaja, B.A. Deoxycholic acid-derived tetraoxane antimalarials and antiproliferatives. J. Med.Chem. 2007, 50, 5118–5127. [Google Scholar] [CrossRef]
- Baji, A.; Kovacs, F.; Motyan, G.; Schneider, G.; Wolfling, J.; Sinka, I.; Zupko, I.; Ocsovszki, I.; Frank, E. Investigation of pH and substituent effects on the distribution ratio of novel steroidal ring D- and A-fused arylpyrazole regioisomers and evaluation of their cell-growth inhibitory effects in vitro. Steroids 2017, 126, 35–49. [Google Scholar] [CrossRef]
- Pettit, G.R.; Inoue, M.; Kamano, Y.; Herald, D.L.; Arm, C.; Dufresne, C.; Christie, N.D.; Schmidt, J.M.; Doubek, D.L.; Krupa, T.S. Isolation and structure of the powerful cell growth inhibitor cephalostatin. J. Am. Chem. Soc. 1988, 110, 2006–2007. [Google Scholar] [CrossRef]
- LaCour, T.G.; Guo, C.X.; Bhandaru, S.; Boyd, M.R.; Fuchs, P.L. Interphylal product splicing: The first total syntheses of cephalostatin 1, the north hemisphere of ritterazine G, and the highly active hybrid analogue, ritterostatin G(N)1(N). J. Am. Chem. Soc. 1998, 120, 692–707. [Google Scholar] [CrossRef]
- Nahar, L.; Sarker, S.D.; Turner, A.B.L.; Nahar, S.D. A review on synthetic and natural steroid dimers 1997–2006. Curr. Med. Chem 2007, 14, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Dirsch, V.M.; Müller, I.M.; Eichhorst, S.T.; Pettit, G.R.; Kamano, Y.; Inoue, M.; Xu, J.P.; Ichihara, Y.; Wanner, G.; Vollmar, A.M. Cephalostatin 1 selectively triggers the release of Smac/DIABLO and subsequent apoptosis that is characterized by an increased density of the mitochondrial matrix. Cancer Res. 2003, 63, 8869–8876. [Google Scholar] [PubMed]
- Pettit, G.R.; Xu, J.P.; Chapuis, J.C.; Melody, N. The cephalostatins. Isolation, structure, and cancer cell growth inhibition of cephalostatin. J. Nat. Prod. 2015, 78, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Delouvrie, B.; Ellston, R.; Fitzek, M.; Germain, H.; Green, S.; Hancox, U.; Harris, C.S.; Hudson, K.; et al. Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl)pyrazin-2-yl)-1-ethyl-1,2,4 -triazol-3-yl)piperidin-1-yl)-3-hydroxypropan-1-one (AZD8835): A potent and selective inhibitor of PI3Kalpha and PI3Kdelta for the treatment of cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5155–5162. [Google Scholar] [CrossRef] [PubMed]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the first direct and selective inhibitor of rna polymerase i, for cancer therapeutics. Acs Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef]
- Lao, K.; Sun, J.; Wang, C.; Lyu, W.; Zhou, B.; Zhao, R.; Xu, Q.; You, Q.; Xiang, H. Design, synthesis and biological evaluation of novel androsta-3,5-diene-3-carboxylic acid derivatives as inhibitors of 5α-reductase type 1 and 2. Steroids 2017, 124, 29–34. [Google Scholar] [CrossRef]
- Annand, J.R.; Bruno, P.A.; Mapp, A.K.; Schindler, C.S. Synthesis and biological evaluation of pharbinilic acid and derivatives as NF-kappaB pathway inhibitors. Chem. Commun. 2015, 51, 8990–8993. [Google Scholar] [CrossRef]
- Alonso, F.; Cirigliano, A.M.; Davola, M.E.; Cabrera, G.M.; Garcia Linares, G.E.; Labriola, C.; Barquero, A.A.; Ramirez, J.A. Multicomponent synthesis of 4,4-dimethyl sterol analogues and their effect on eukaryotic cells. Steroids 2014, 84, 1–6. [Google Scholar] [CrossRef]
- Fu, H.J.; Zhou, Y.R.; Bao, B.H.; Jia, M.X.; Zhao, Y.; Zhang, L.; Li, J.X.; He, H.L.; Zhou, X.M. Tryptophan hydroxylase 1 (Tph-1)-targeted bone anabolic agents for osteoporosis. J Med Chem 2014, 57, 4692–4709. [Google Scholar] [CrossRef]
- Zolottsev, V.A.; Zavarzin, I.V.; Shirinyan, V.Z.; Levina, I.S. Synthesis of E- and Z-isomeric progesterone 3-O-methyloximes. Russ. Chem. Bull. 2013, 62, 2086–2087. [Google Scholar] [CrossRef]
- Mao, S.W.; Chen, H.; Yu, L.F.; Lv, F.; Xing, Y.J.; Liu, T.; Xie, J.; Tang, J.; Yi, Z.; Yang, F. Novel 3,4-seco bile acid diamides as selective anticancer proliferation and migration agents. Eur. J. Med. Chem. 2016, 122, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Available online: www.molinspiration.com (accessed on 9 January 2020).
- Tolmacheva, I.A.; Nazarov, A.V.; Eroshenko, D.V.; Grishko, V.V. Synthesis, cytotoxic evaluation, and molecular docking studies of the semi-synthetic “triterpenoid-steroid” hybrids. Steroids 2018, 140, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Frimer, A.A.; Gilinsky-Sharon, P.; Hameiri, J.; Aljadeheff, G. Superoxide, tert-butoxide, and hydroxide-mediated autoxidation of 3-oxo-Δ4 steroids in aprotic media. J. Org. Chem. 1982, 47, 2818–2819. [Google Scholar] [CrossRef]
- Frimer, A.A.; Hameiri-Buch, J.; Ripshtos, S.; Gilinsky-Sharon, P. A facile two-step high-yield approach to 2-oxasteroids. Tetrahedron 1986, 42, 5693–5706. [Google Scholar] [CrossRef]
- Salvador, J.A.; Moreira, V.M.; Goncalves, B.M.; Leal, A.S.; Jing, Y. Ursane-type pentacyclic triterpenoids as useful platforms to discover anticancer drugs. Nat. Prod. Rep. 2012, 29, 1463–1479. [Google Scholar] [CrossRef]
- Lutfun Nahar, S.D.S. Steroid dimers: Chemistry and applications in drug design and delivery; John Wiley & Sons, Ltd: Leicester, UK, 2012; pp. 1–422. [Google Scholar] [CrossRef]
- Yu, B.; Shi, X.-J.; Zheng, Y.-F.; Fang, Y.; Zhang, E.; Yu, D.-Q.; Liu, H.-M. A novel [1,2,4] triazolo [1,5-α] pyrimidine-based phenyl-linked steroid dimer: Synthesis and its cytotoxic activity. Eur. J. Med. Chem. 2013, 69, 323–330. [Google Scholar] [CrossRef]
- Islam, M.K.; Kim, S.; Kim, H.K.; Kim, Y.H.; Lee, Y.M.; Choi, G.; Baek, A.R.; Sung, B.K.; Kim, M.; Cho, A.E.; et al. Synthesis and evaluation of Manganese(II)-based ethylenediaminetetraacetic acid-ethoxybenzyl conjugate as a highly stable hepatobiliary magnetic resonance imaging contrast agent. Bioconjug. Chem. 2018, 29, 3614–3625. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Mirza, M.B.; Elkady, A.I.; Al-Attar, A.M.; Syed, F.Q.; Mohammed, F.A.; Hakeem, K.R. Induction of apoptosis and cell cycle arrest by ethyl acetate fraction of Phoenix dactylifera L. (Ajwa dates) in prostate cancer cells. J. Ethnopharmacol. 2018, 218, 35–44. [Google Scholar] [CrossRef]
- Gu, H.L.; Li, N.; Dai, J.K.; Xi, Y.X.; Wang, S.J.; Wang, J.R. Synthesis and in vitro antitumor activity of novel bivalent beta-carboline-3-carboxylic acid derivatives with DNA as a potential target. Int. J. Mol. Sci. 2018, 19, 3179. [Google Scholar] [CrossRef]
- Song, Y.L.; Tian, C.P.; Yan, W.; Jiang, L.H.; Shen, L.Q. Design, synthesis and antitumor activity of steroidal pyridine derivatives based on molecular docking. Steroids 2019, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, A.R.; Petri, E.T.; Klisurić, O.R.; Ćelić, A.S.; Jakimov, D.S.; Djurendić, E.A.; Gaši, K.M.P.; Sakač, M.N. Synthesis and anticancer cell potential of steroidal 16,17-seco-16,17a-dinitriles: Identification of a selective inhibitor of hormone-independent breast cancer cells. Bioorganic Med. Chem. 2015, 23, 703–711. [Google Scholar] [CrossRef] [PubMed]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).