The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment

 and
and
Abstract
:1. Introduction
2. Epigenetics Plays a Key Role in Th17 Cells Lineage Commitment and Plasticity
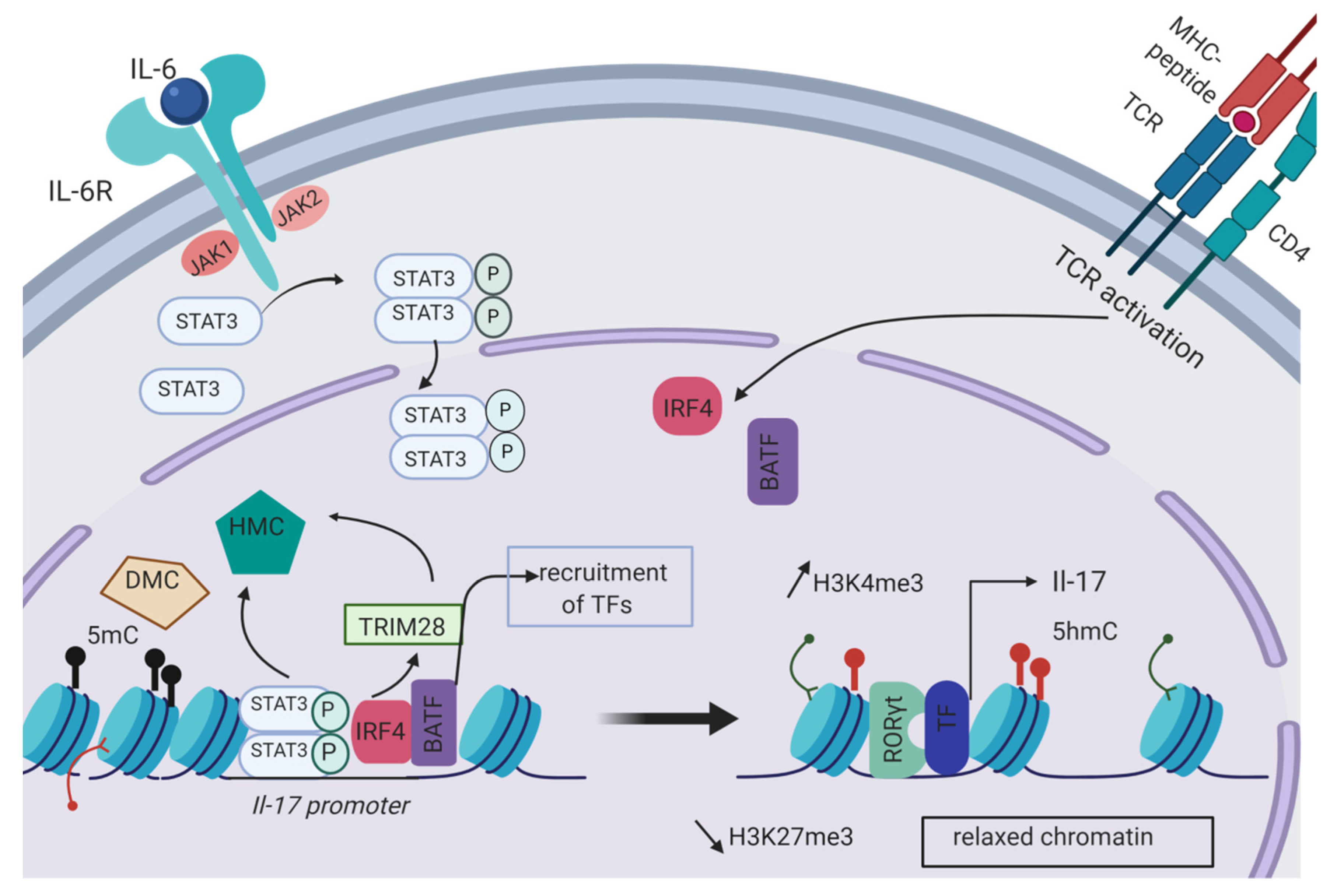
2.1. Epigenetic Initiation of the Th17 Differentiation Program
2.2. Th17 Plasticity
3. The Influence of the Tumor Microenvironment on the Epigenome of the Tumor Infiltrating Lymphocytes (TILs)
3.1. Pro-Tumoral Function of Th17 Cells
3.2. Anti-Tumoral Function of Th17 Cells
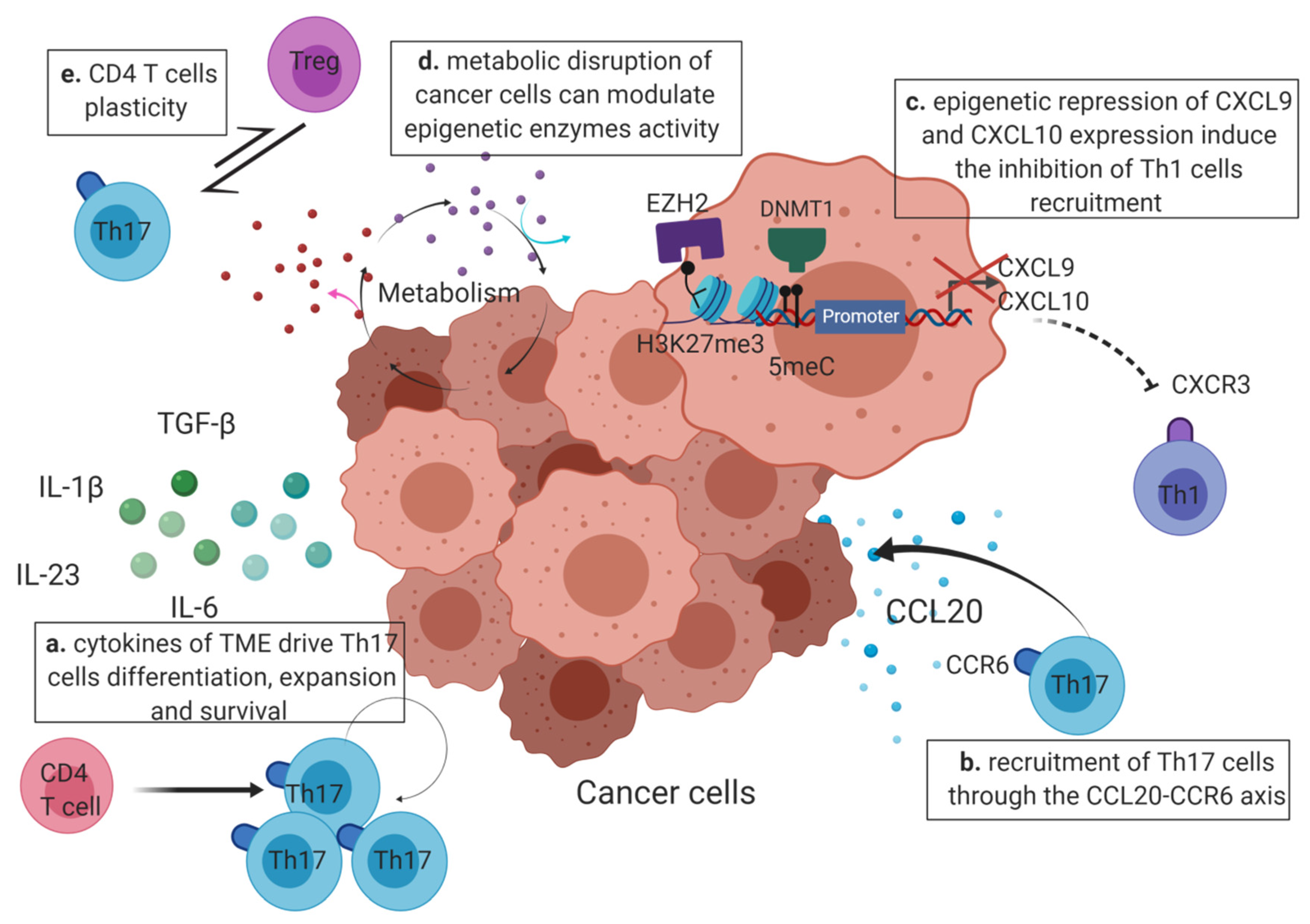
4. Epigenetic Factors Leading to Th17 Cell Predominance in the TME (Tumor Micro-Environment)
4.1. The Cytokines Produced by the TME Drive the Th17 Polarization and Expansion
4.2. Epigenetics May Enhance Th17 Recruitment or Inhibit the Recruitment of Other CD4 T Cells at the Tumor Site
4.3. Modification of the Epigenome by the Tumor Micro-Environment
4.3.1. The Hypoxic Tumor Microenvironment
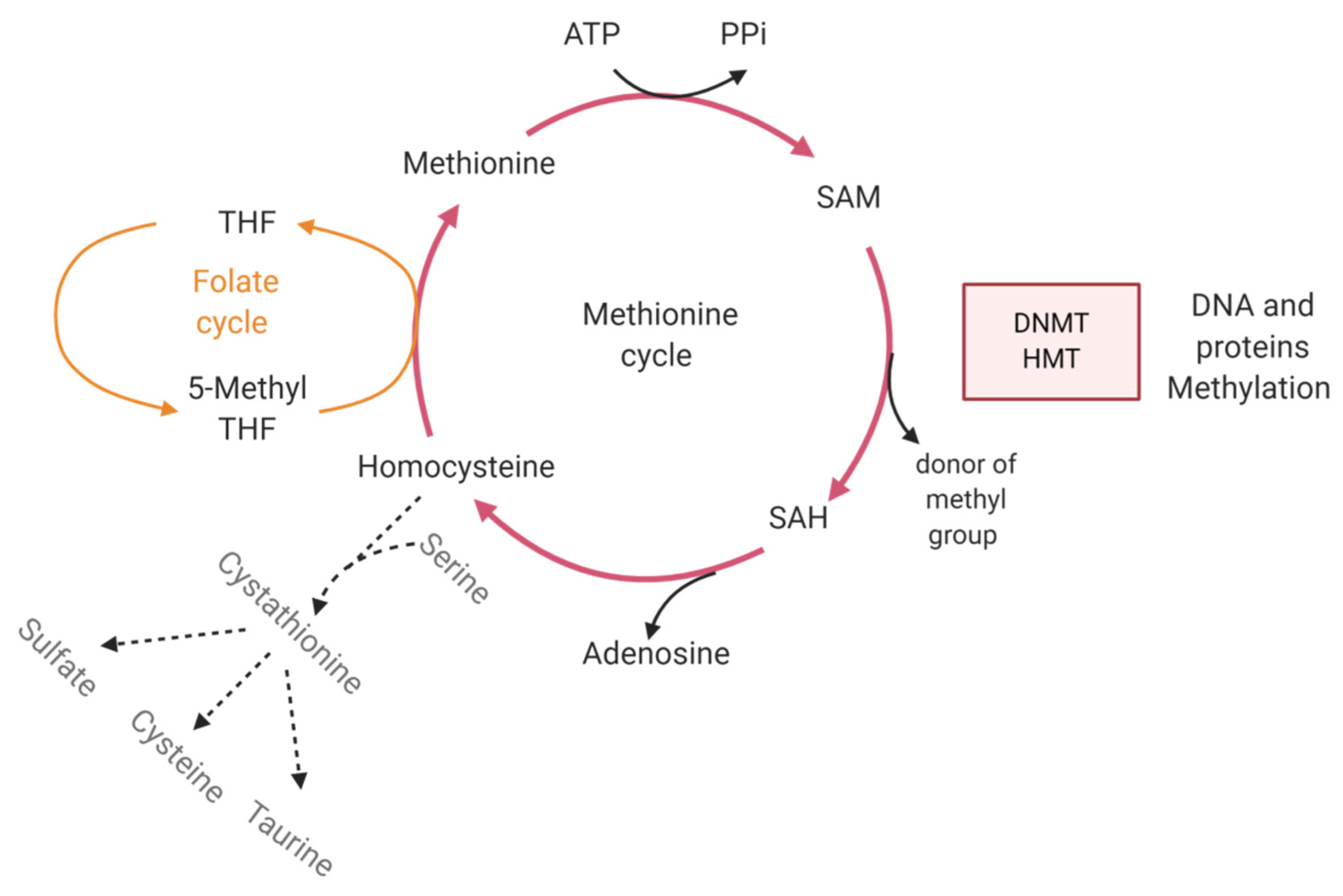
4.3.2. Metabolism
4.4. Th17 Predominance in Tumor Can Be Modulated by Plasticity
4.4.1. Treg to Th17 Cell Plasticity
4.4.2. Th17 to Th1 Cell Plasticity
5. The Promising Use of Epidrugs in Combination with Other Therapies for Anti-Tumor Treatment
5.1. Combination of Immune Checkpoint Blockade with Epidrugs
5.2. Favoring Tumor Immunogenicity with Epidrugs
5.3. Remodeling of the Tumor Micro-Environment by Epidrug Treatment
6. Epigenetic Reprogramming of CD4 T Cells to Modulate Th17 Cell Differentiation and Plasticity
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khader, S.A.; Gaffen, S.L.; Kolls, J.K. Th17 Cells at the Crossroads of Innate and Adaptive Immunity against Infectious Diseases at the Mucosa. Mucosal Immunol. 2009, 2, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct from the T Helper Type 1 and 2 Lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The Science of Change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic Tools (The Writers, The Readers and The Erasers) and Their Implications in Cancer Therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Hoefig, K.P.; Heissmeyer, V. MicroRNAs Grow up in the Immune System. Curr. Opin. Immunol. 2008, 20, 281–287. [Google Scholar] [CrossRef]
- Wei, L.; Vahedi, G.; Sun, H.-W.; Watford, W.T.; Takatori, H.; Ramos, H.L.; Takahashi, H.; Liang, J.; Gutierrez-Cruz, G.; Zang, C.; et al. Discrete Roles of STAT4 and STAT6 Transcription Factors in Tuning Epigenetic Modifications and Transcription during T Helper Cell Differentiation. Immunity 2010, 32, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Vahedi, G.; Ghoreschi, K.; Yang, X.-P.; Nakayamada, S.; Kanno, Y.; O’Shea, J.J.; Laurence, A. Helper T-Cell Differentiation and Plasticity: Insights from Epigenetics. Immunology 2011, 134, 235–245. [Google Scholar] [CrossRef]
- Lin, F.; Meng, X.; Guo, Y.; Cao, W.; Liu, W.; Xia, Q.; Hui, Z.; Chen, J.; Hong, S.; Zhang, X.; et al. Epigenetic Initiation of the TH17 Differentiation Program Is Promoted by Cxxc Finger Protein 1. Sci. Adv. 2019, 5, eaax1608. [Google Scholar] [CrossRef] [Green Version]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Muratet, M.; et al. A Validated Regulatory Network for Th17 Cell Specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, Y.; Lu, H.; Sun, S.-C.; Jin, W.; Wang, X.; Dong, C. Epigenetic Activation during T Helper 17 Cell Differentiation Is Mediated by Tripartite Motif Containing 28. Nat. Commun. 2018, 9, 1424. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.Y.; Hatfield, J.K.; Brown, M.A. Ikaros Sets the Potential for Th17 Lineage Gene Expression through Effects on Chromatin State in Early T Cell Development. J. Biol. Chem. 2013, 288, 35170–35179. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Cao, W.; Xu, L.; Chen, X.; Zhan, Y.; Yang, Q.; Liu, S.; Chen, P.; Jiang, Y.; Sun, X.; et al. The Histone H3 Lysine-27 Demethylase Jmjd3 Plays a Critical Role in Specific Regulation of Th17 Cell Differentiation. J. Mol. Cell Biol. 2015, 7, 505–516. [Google Scholar] [CrossRef]
- Baumjohann, D.; Ansel, K.M. MicroRNA-Mediated Regulation of T Helper Cell Differentiation and Plasticity. Nat. Rev. Immunol. 2013, 13, 666–678. [Google Scholar] [CrossRef]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA MiR-326 Regulates TH-17 Differentiation and Is Associated with the Pathogenesis of Multiple Sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef]
- Moisan, J.; Grenningloh, R.; Bettelli, E.; Oukka, M.; Ho, I.-C. Ets-1 Is a Negative Regulator of Th17 Differentiation. J. Exp. Med. 2007, 204, 2825–2835. [Google Scholar] [CrossRef]
- Escobar, T.M.; Kanellopoulou, C.; Kugler, D.G.; Kilaru, G.; Nguyen, C.K.; Nagarajan, V.; Bhairavabhotla, R.K.; Northrup, D.; Zahr, R.; Burr, P.; et al. MiR-155 Activates Cytokine Gene Expression in Th17 Cells by Regulating the DNA-Binding Protein Jarid2 to Relieve Polycomb-Mediated Repression. Immunity 2014, 40, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late Developmental Plasticity in the T Helper 17 Lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 Cells Give Rise to Th1 Cells That Are Required for the Pathogenesis of Colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Popp, F.C.; Soeder, Y.; Haarer, J.; Geissler, E.K.; Schlitt, H.J.; Dahlke, M.H. Conversion of Th17 into IL-17A(Neg) Regulatory T Cells: A Novel Mechanism in Prolonged Allograft Survival Promoted by Mesenchymal Stem Cell-Supported Minimized Immunosuppressive Therapy. J. Immunol. 2014, 193, 4988–4999. [Google Scholar] [CrossRef] [Green Version]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Capone, M.; Cardilicchia, E.; Frosali, F.; Querci, V.; Angeli, R.; Matucci, A.; Fambrini, M.; et al. Identification of a Novel Subset of Human Circulating Memory CD4(+) T Cells That Produce Both IL-17A and IL-4. J. Allergy Clin. Immunol. 2010, 125, 222–230.e1-4. [Google Scholar] [CrossRef]
- Zheng, W.; Flavell, R.A. The Transcription Factor GATA-3 Is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Mukasa, R.; Balasubramani, A.; Lee, Y.K.; Whitley, S.K.; Weaver, B.T.; Shibata, Y.; Crawford, G.E.; Hatton, R.D.; Weaver, C.T. Epigenetic Instability of Cytokine and Transcription Factor Gene Loci Underlies Plasticity of the T Helper 17 Cell Lineage. Immunity 2010, 32, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.-Y.; Watford, W.T.; et al. Global Mapping of H3K4me3 and H3K27me3 Reveals Specificity and Plasticity in Lineage Fate Determination of Differentiating CD4+ T Cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Kanno, Y.; Vahedi, G.; Hirahara, K.; Singleton, K.; O’Shea, J.J. Transcriptional and Epigenetic Control of T Helper Cell Specification: Molecular Mechanisms Underlying Commitment and Plasticity. Annu. Rev. Immunol. 2012, 30, 707–731. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.-H.; Floess, S.; Hagemann, S.; Deyneko, I.V.; Groebe, L.; Pezoldt, J.; Sparwasser, T.; Lochner, M.; Huehn, J. Development of a Unique Epigenetic Signature during in vivo Th17 Differentiation. Nucleic Acids Res. 2015, 43, 1537–1548. [Google Scholar] [CrossRef]
- Mazzoni, A.; Santarlasci, V.; Maggi, L.; Capone, M.; Rossi, M.C.; Querci, V.; De Palma, R.; Chang, H.-D.; Thiel, A.; Cimaz, R.; et al. Demethylation of the RORC2 and IL17A in Human CD4+ T Lymphocytes Defines Th17 Origin of Nonclassic Th1 Cells. J. Immunol. 2015, 194, 3116–3126. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Kinugasa, T.; Miyoshi, H.; Sato, K.; Yuge, K.; Ohchi, T.; Fujino, S.; Shiraiwa, S.; Katagiri, M.; Akagi, Y.; et al. A High RORγT/CD3 Ratio Is a Strong Prognostic Factor for Postoperative Survival in Advanced Colorectal Cancer: Analysis of Helper T Cell Lymphocytes (Th1, Th2, Th17 and Regulatory T Cells). Ann. Surg. Oncol. 2016, 23, 919–927. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer MiRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-P.; Yan, J.; Xu, J.; Pang, X.-H.; Chen, M.-S.; Li, L.; Wu, C.; Li, S.-P.; Zheng, L. Increased Intratumoral IL-17-Producing Cells Correlate with Poor Survival in Hepatocellular Carcinoma Patients. J. Hepatol. 2009, 50, 980–989. [Google Scholar] [CrossRef]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Bruno, T.C.; Maris, C.H.; Xu, L.; Thoburn, C.J.; DeMarzo, A.M.; Meeker, A.K.; Isaacs, W.B.; Drake, C.G. Phenotypic Analysis of Prostate-Infiltrating Lymphocytes Reveals TH17 and Treg Skewing. Clin. Cancer Res. 2008, 14, 3254–3261. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-J.; Zhou, Q.; Gu, Y.-Y.; Qin, S.-M.; Ma, W.-L.; Xin, J.-B.; Tao, X.-N.; Shi, H.-Z. Generation and Differentiation of IL-17-Producing CD4+ T Cells in Malignant Pleural Effusion. J. Immunol. 2010, 185, 6348–6354. [Google Scholar] [CrossRef] [Green Version]
- Iida, T.; Iwahashi, M.; Katsuda, M.; Ishida, K.; Nakamori, M.; Nakamura, M.; Naka, T.; Ojima, T.; Ueda, K.; Hayata, K.; et al. Tumor-Infiltrating CD4+ Th17 Cells Produce IL-17 in Tumor Microenvironment and Promote Tumor Progression in Human Gastric Cancer. Oncol. Rep. 2011, 25, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Alves, J.J.P.; De Medeiros Fernandes, T.A.A.; De Araújo, J.M.G.; Cobucci, R.N.O.; Lanza, D.C.F.; Bezerra, F.L.; Andrade, V.S.; Fernandes, J.V. Th17 Response in Patients with Cervical Cancer. Oncol. Lett. 2018, 16, 6215–6227. [Google Scholar] [CrossRef] [Green Version]
- Numasaki, M.; Fukushi, J.; Ono, M.; Narula, S.K.; Zavodny, P.J.; Kudo, T.; Robbins, P.D.; Tahara, H.; Lotze, M.T. Interleukin-17 Promotes Angiogenesis and Tumor Growth. Blood 2003, 101, 2620–2627. [Google Scholar] [CrossRef]
- Liu, J.; Duan, Y.; Cheng, X.; Chen, X.; Xie, W.; Long, H.; Lin, Z.; Zhu, B. IL-17 Is Associated with Poor Prognosis and Promotes Angiogenesis via Stimulating VEGF Production of Cancer Cells in Colorectal Carcinoma. Biochem. Biophys. Res. Commun. 2011, 407, 348–354. [Google Scholar] [CrossRef]
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 Inhibits CXCL9/10-Mediated Recruitment of CD8+ Cytotoxic T Cells and Regulatory T Cells to Colorectal Tumors. J. Immunother. Cancer 2019, 7, 324. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 Can Promote Tumor Growth through an IL-6-Stat3 Signaling Pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Végran, F.; Hichami, A.; Ladoire, S.; Derangère, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 Transcription Factors Control Th17 Cell Immunosuppressive Activity via the Regulation of Ectonucleotidase Expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The Ectonucleotidases CD39 and CD73: Novel Checkpoint Inhibitor Targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Yip, L.; Woehrle, T.; Corriden, R.; Hirsh, M.; Chen, Y.; Inoue, Y.; Ferrari, V.; Insel, P.A.; Junger, W.G. Autocrine Regulation of T-Cell Activation by ATP Release and P2X7 Receptors. FASEB J. 2009, 23, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP Inhibits the Generation and Function of Regulatory T Cells through the Activation of Purinergic P2X Receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 Promotes Tumor Development through the Induction of Tumor Promoting Microenvironments at Tumor Sites and Myeloid-Derived Suppressor Cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-Specific Th17-Polarized Cells Eradicate Large Established Melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, S.; Saez, J.J.; Fernandez, D.; Flores-Santibañez, F.; Alvarez, K.; Tejon, G.; Ruiz, P.; Maldonado, P.; Hidalgo, Y.; Manriquez, V.; et al. T Helper Type 17 Cells Contribute to Anti-Tumour Immunity and Promote the Recruitment of T Helper Type 1 Cells to the Tumour. Immunology 2013, 139, 61–71. [Google Scholar] [CrossRef]
- Ducimetière, L.; Vermeer, M.; Tugues, S. The Interplay Between Innate Lymphoid Cells and the Tumor Microenvironment. Front. Immunol. 2019, 10, 2895. [Google Scholar] [CrossRef] [Green Version]
- Loyon, R.; Jary, M.; Salomé, B.; Gomez-Cadena, A.; Galaine, J.; Kroemer, M.; Romero, P.; Trabanelli, S.; Adotévi, O.; Borg, C.; et al. Peripheral Innate Lymphoid Cells Are Increased in First Line Metastatic Colorectal Carcinoma Patients: A Negative Correlation with Th1 Immune Responses. Front. Immunol. 2019, 10, 2121. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The Paradox of Th17 Cell Functions in Tumor Immunity. Cell. Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Song, Y.; Yang, J.M. Role of Interleukin (IL)-17 and T-Helper (Th)17 Cells in Cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1–8. [Google Scholar] [CrossRef]
- Martin, F.; Apetoh, L.; Ghiringhelli, F. Controversies on the Role of Th17 in Cancer: A TGF-β-Dependent Immunosuppressive Activity? Trends Mol. Med. 2012, 18, 742–749. [Google Scholar] [CrossRef]
- Tanaka, S.; Jiang, Y.; Martinez, G.J.; Tanaka, K.; Yan, X.; Kurosaki, T.; Kaartinen, V.; Feng, X.-H.; Tian, Q.; Wang, X.; et al. Trim33 Mediates the Proinflammatory Function of Th17 Cells. J. Exp. Med. 2018, 215, 1853–1868. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-Mediated Epigenetic Reprogramming Regulates Formation of Human Pancreatic Cancer-Associated Fibroblasts. eLife 2019, 8, e50663. [Google Scholar] [CrossRef]
- Yerinde, C.; Siegmund, B.; Glauben, R.; Weidinger, C. Metabolic Control of Epigenetics and Its Role in CD8+ T Cell Differentiation and Function. Front. Immunol. 2019, 10, 2718. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX Transcriptionally and Epigenetically Programs CD8+ T Cell Exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Su, X.; Ye, J.; Hsueh, E.C.; Zhang, Y.; Hoft, D.F.; Peng, G. Tumor Microenvironments Direct the Recruitment and Expansion of Human Th17 Cells. J. Immunol. 2010, 184, 1630–1641. [Google Scholar] [CrossRef]
- Rezalotfi, A.; Ahmadian, E.; Aazami, H.; Solgi, G.; Ebrahimi, M. Gastric Cancer Stem Cells Effect on Th17/Treg Balance; A Bench to Beside Perspective. Front. Oncol. 2019, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Gu, L.; Ning, H.; Zhang, Y.; Hsueh, E.C.; Fu, M.; Hu, X.; Wei, L.; Hoft, D.F.; Liu, J. Increased Th17 Cells in the Tumor Microenvironment Is Mediated by IL-23 via Tumor-Secreted Prostaglandin E2. J. Immunol. 2013, 190, 5894–5902. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Lou, X.; He, Y. Preferential Recruitment of Th17 Cells to Cervical Cancer via CCR6-CCL20 Pathway. PLoS ONE 2015, 10, e0120855. [Google Scholar] [CrossRef]
- Yu, X.; Yuan, Z.; Yang, Z.; Chen, D.; Kim, T.; Cui, Y.; Luo, Q.; Liu, Z.; Yang, Z.; Fan, X.; et al. The Novel Long Noncoding RNA U50535 Promotes Colorectal Cancer Growth and Metastasis by Regulating CCL20. Cell Death Dis. 2018, 9, 751. [Google Scholar] [CrossRef]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential Recruitment of CCR6-Expressing Th17 Cells to Inflamed Joints via CCL20 in Rheumatoid Arthritis and Its Animal Model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Jiang, R.; Mao, C.; Shi, L.; Wang, S.; Yu, L.; Hu, Q.; Dai, D.; Xu, H. Chemokine/Chemokine Receptor Interactions Contribute to the Accumulation of Th17 Cells in Patients with Esophageal Squamous Cell Carcinoma. Hum. Immunol. 2012, 73, 1068–1072. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 Axis for Immune Activation—A Target for Novel Cancer Therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia Induces a Novel Signature of Chromatin Modifications and Global Repression of Transcription. Mutat. Res. 2008, 640, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.B.; Barton, M.C. Hypoxia-Induced and Stress-Specific Changes in Chromatin Structure and Function. Mutat. Res. 2007, 618, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volchenkov, R.; Nygaard, V.; Sener, Z.; Skålhegg, B.S. Th17 Polarization under Hypoxia Results in Increased IL-10 Production in a Pathogen-Independent Manner. Front. Immunol. 2017, 8, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic Regulation of Chromatin Modifications and Gene Expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between Epigenetics and Metabolism in Oncogenesis: Mechanisms and Therapeutic Approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Végan, F.; Humblin, E.; Boidot, R.; Rébé, C.; Derangère, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic Control of TH17 and Induced Treg Cell Balance by an Epigenetic Mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef]
- Morgillo, F.; Dallio, M.; Della Corte, C.M.; Gravina, A.G.; Viscardi, G.; Loguercio, C.; Ciardiello, F.; Federico, A. Carcinogenesis as a Result of Multiple Inflammatory and Oxidative Hits: A Comprehensive Review from Tumor Microenvironment to Gut Microbiota. Neoplasia 2018, 20, 721–733. [Google Scholar] [CrossRef]
- Bhat, M.I.; Kapila, R. Dietary Metabolites Derived from Gut Microbiota: Critical Modulators of Epigenetic Changes in Mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.H.; Yun, J.-M. Fisetin Inhibits Hyperglycemia-Induced Proinflammatory Cytokine Production by Epigenetic Mechanisms. Evid. Based Complement. Alternat. Med 2012, 2012, 639469. [Google Scholar] [CrossRef]
- Yang, S.; Wang, B.; Guan, C.; Wu, B.; Cai, C.; Wang, M.; Zhang, B.; Liu, T.; Yang, P. Foxp3+IL-17+ T Cells Promote Development of Cancer-Initiating Cells in Colorectal Cancer. J. Leukoc. Biol. 2011, 89, 85–91. [Google Scholar] [CrossRef]
- Huang, C.; Fu, Z.-X. Localization of IL-17+Foxp3+ T Cells in Esophageal Cancer. Immunol. Investig. 2011, 40, 400–412. [Google Scholar] [CrossRef]
- Kryczek, I.; Wu, K.; Zhao, E.; Wei, S.; Vatan, L.; Szeliga, W.; Huang, E.; Greenson, J.; Chang, A.; Roliński, J.; et al. IL-17+ Regulatory T Cells in the Microenvironments of Chronic Inflammation and Cancer. J. Immunol. 2011, 186, 4388–4395. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Patsoukis, N.; Petkova, V.; Boussiotis, V.A. Runx1 and Runx3 Are Involved in the Generation and Function of Highly Suppressive IL-17-Producing T Regulatory Cells. PLoS ONE 2012, 7, e45115. [Google Scholar] [CrossRef]
- Du, R.; Zhao, H.; Yan, F.; Li, H. IL-17+Foxp3+ T Cells: An Intermediate Differentiation Stage between Th17 Cells and Regulatory T Cells. J. Leukoc. Biol. 2014, 96, 39–48. [Google Scholar] [CrossRef]
- Ye, J.; Su, X.; Hsueh, E.C.; Zhang, Y.; Koenig, J.M.; Hoft, D.F.; Peng, G. Human Tumor-Infiltrating Th17 Cells Have the Capacity to Differentiate into IFN-γ+ and FOXP3+ T Cells with Potent Suppressive Function. Eur. J. Immunol. 2011, 41, 936–951. [Google Scholar] [CrossRef]
- Hamaï, A.; Pignon, P.; Raimbaud, I.; Duperrier-Amouriaux, K.; Senellart, H.; Hiret, S.; Douillard, J.-Y.; Bennouna, J.; Ayyoub, M.; Valmori, D. Human T(H)17 Immune Cells Specific for the Tumor Antigen MAGE-A3 Convert to IFN-γ-Secreting Cells as They Differentiate into Effector T Cells in vivo. Cancer Res. 2012, 72, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Peixoto, P.; Renaude, E.; Boyer-Guittaut, M.; Hervouet, E. Epigenetics, a Key Player of Immunotherapy Resistance. CDR 2018, 1, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Bretz, A.C.; Parnitzke, U.; Kronthaler, K.; Dreker, T.; Bartz, R.; Hermann, F.; Ammendola, A.; Wulff, T.; Hamm, S. Domatinostat Favors the Immunotherapy Response by Modulating the Tumor Immune Microenvironment (TIME). J. Immunother. Cancer 2019, 7, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of Histone Lysine-Specific Demethylase 1 Elicits Breast Tumor Immunity and Enhances Antitumor Efficacy of Immune Checkpoint Blockade. Oncogene 2019, 38, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Li, Y.; Pandit, H.; Li, S.; Pulliam, Z.; Zheng, Q.; Yu, Y.; Martin, R.C.G. Epigenetic Modulation Enhances Immunotherapy for Hepatocellular Carcinoma. Cell. Immunol. 2019, 336, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Collins, D.M.; Perry, A.S.; Prencipe, M. Novel Strategies for Cancer Treatment: Highlights from the 55th IACR Annual Conference. Cancers 2019, 11, 1125. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.N.H.; Gregorie, C.J.; Tomasi, T.B. Histone Deacetylase Inhibitors Induce TAP, LMP, Tapasin Genes and MHC Class I Antigen Presentation by Melanoma Cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Li, Y.; Yang, W.; Wu, H.; Li, X.; Huang, Y.; Zhou, Y.; Du, Z. Histone Deacetylase Inhibition Up-Regulates MHC Class I to Facilitate Cytotoxic T Lymphocyte-Mediated Tumor Cell Killing in Glioma Cells. J. Cancer 2019, 10, 5638–5645. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zou, J.; Wang, M.; Ding, X.; Chepelev, I.; Zhou, X.; Zhao, W.; Wei, G.; Cui, J.; Zhao, K.; et al. Critical Role of Histone Demethylase Jmjd3 in the Regulation of CD4+ T-Cell Differentiation. Nat. Commun. 2014, 5, 5780. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Zhang, M.; Wang, Y.; Fu, S.; Han, S.; Wang, L.; Wang, Q. Sirtinol Regulates the Balance of Th17/Treg to Prevent Allograft Rejection. Cell Biosci. 2017, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Chadha, S.; Wang, L.; Hancock, W.W.; Beier, U.H. Sirtuin-1 in Immunotherapy: A Janus-Headed Target. J. Leukoc. Biol. 2019, 106, 337–343. [Google Scholar] [CrossRef]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnölzer, M.; et al. SIRT1 Deacetylates RORγt and Enhances Th17 Cell Generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef]
- Akimova, T.; Xiao, H.; Liu, Y.; Bhatti, T.R.; Jiao, J.; Eruslanov, E.; Singhal, S.; Wang, L.; Han, R.; Zacharia, K.; et al. Targeting Sirtuin-1 Alleviates Experimental Autoimmune Colitis by Induction of Foxp3+ T-Regulatory Cells. Mucosal Immunol. 2014, 7, 1209–1220. [Google Scholar] [CrossRef]
- Zhang, X.; Han, S.; Kang, Y.; Guo, M.; Hong, S.; Liu, F.; Fu, S.; Wang, L.; Wang, Q.-X. SAHA, an HDAC Inhibitor, Synergizes with Tacrolimus to Prevent Murine Cardiac Allograft Rejection. Cell. Mol. Immunol. 2012, 9, 390–398. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Genes | Epigenetic Regulation | Factors Modulating the Epigenome | References | |
|---|---|---|---|---|
| Th17 differentiation | IL-17, RORγt | Histone modification complex like HAT or HMT responsible for the deposition of permissive marks | Cytokines of the TME | [9,10,12,13,14,15,16,17,60,61,62,74,75,76,77,80,81,82] |
| DNA demethylation enzymes (5hmc) | Metabolism regulates the biodisponibility of co-factors of epigenetic enzymes | |||
| miRNA | Metabolites produced by gut microbiota modulate epigenetic enzymes activity | |||
| Th17 recruitment through the CCR6-CCL20 axis | CCL20 | long non coding RNA (lncRNA-u50535) | Upregulation of lncRNA-u50535 in colorectal cancer | [64] |
| Th17 plasticity | [74,75,76,77,79,88] | |||
| - Th17 /Treg plasticity | Foxp3 | DNA methylation | Glutamate Metabolism pathway (reduction of 2 hydroxyglutarate level in Th17 cells diminish the Foxp3 promoter methylation status) | |
| - Th17/Th1 cells plasticity | IFN-γ, Tbet | Histone modification complex | Exposition of differentiated Th17 cells to another cytokinic environment |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaude, E.; Kroemer, M.; Loyon, R.; Binda, D.; Borg, C.; Guittaut, M.; Hervouet, E.; Peixoto, P. The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 1673. https://doi.org/10.3390/ijms21051673
Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, Hervouet E, Peixoto P. The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment. International Journal of Molecular Sciences. 2020; 21(5):1673. https://doi.org/10.3390/ijms21051673
Chicago/Turabian StyleRenaude, Elodie, Marie Kroemer, Romain Loyon, Delphine Binda, Christophe Borg, Michaël Guittaut, Eric Hervouet, and Paul Peixoto. 2020. "The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment" International Journal of Molecular Sciences 21, no. 5: 1673. https://doi.org/10.3390/ijms21051673