Amino Acids 563–566 of the Na+/H+ Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity



Abstract
:1. Introduction
2. Results
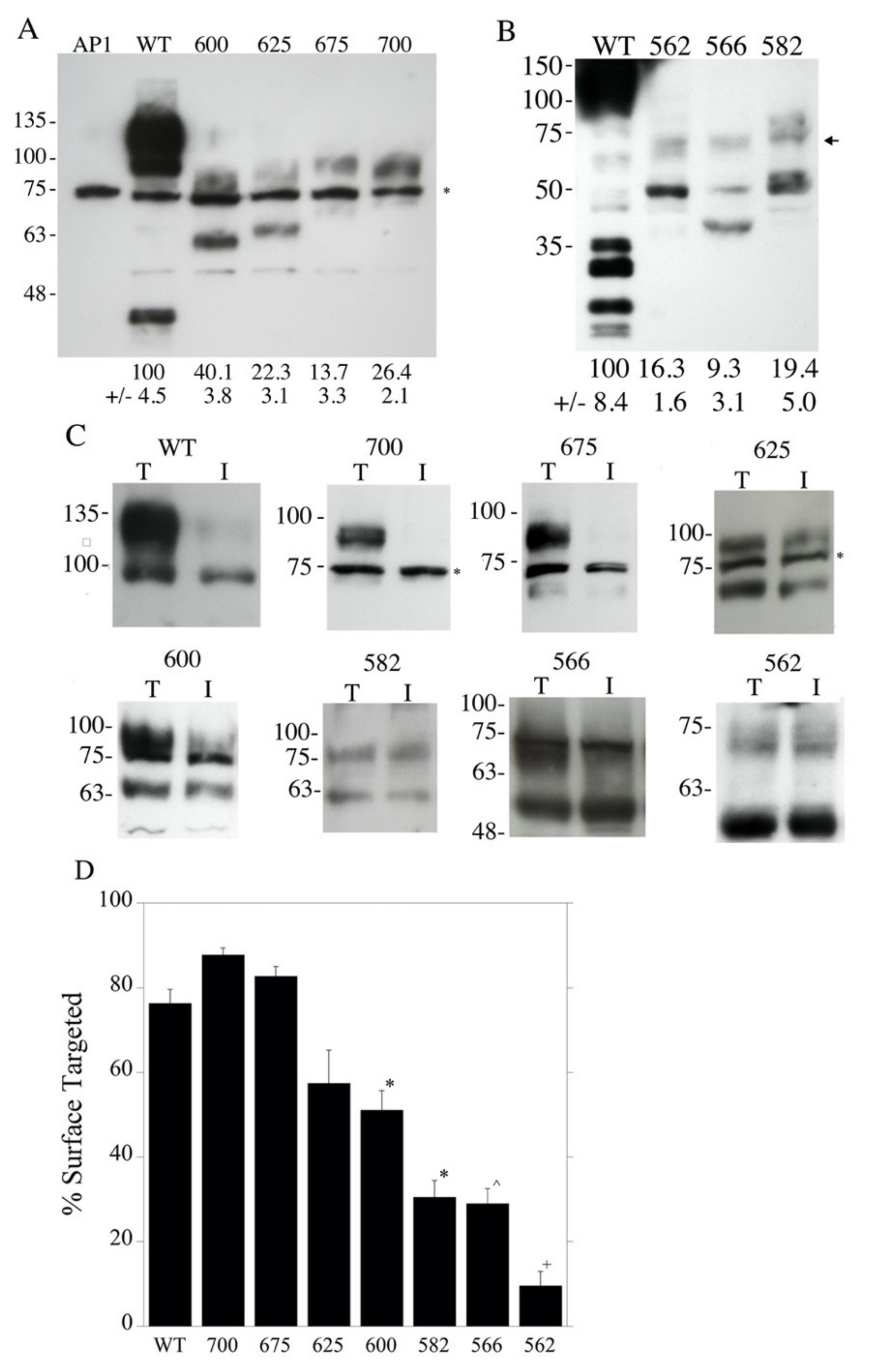
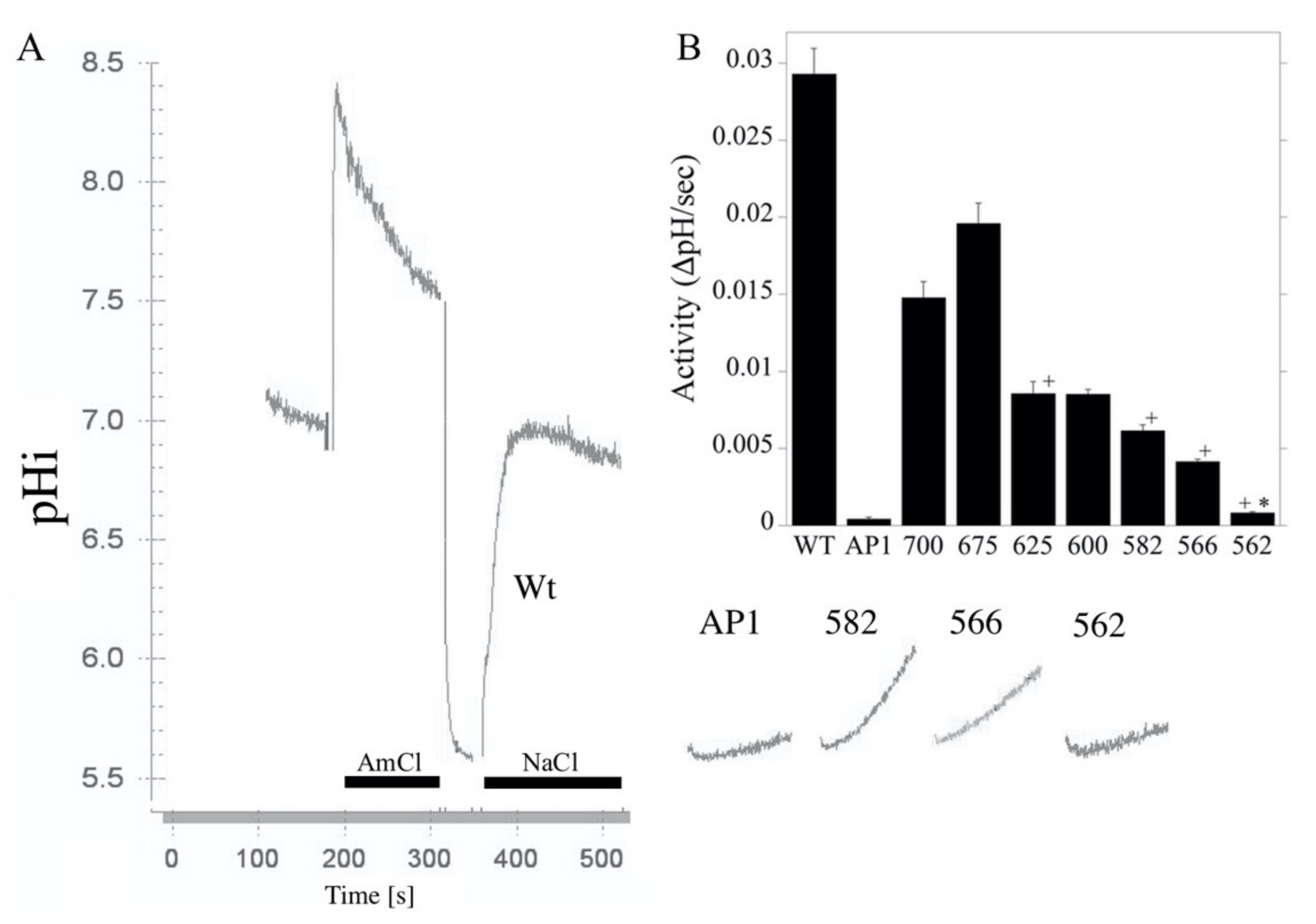
2.1. Construction and Characterization of NHE Mutants
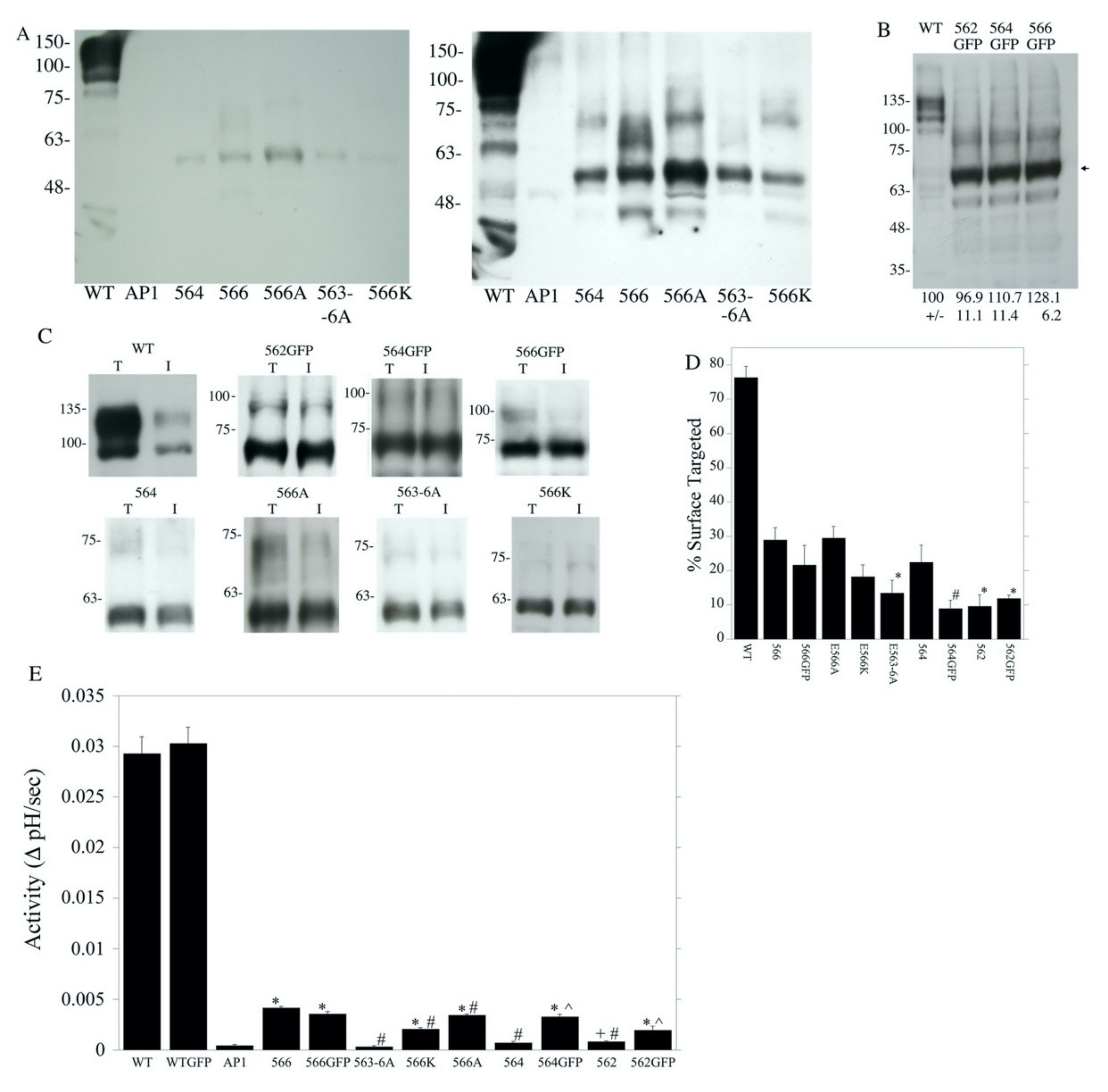
2.2. Characterization of Amino Acids 563–566 of the Cytosolic C-Terminal Tail
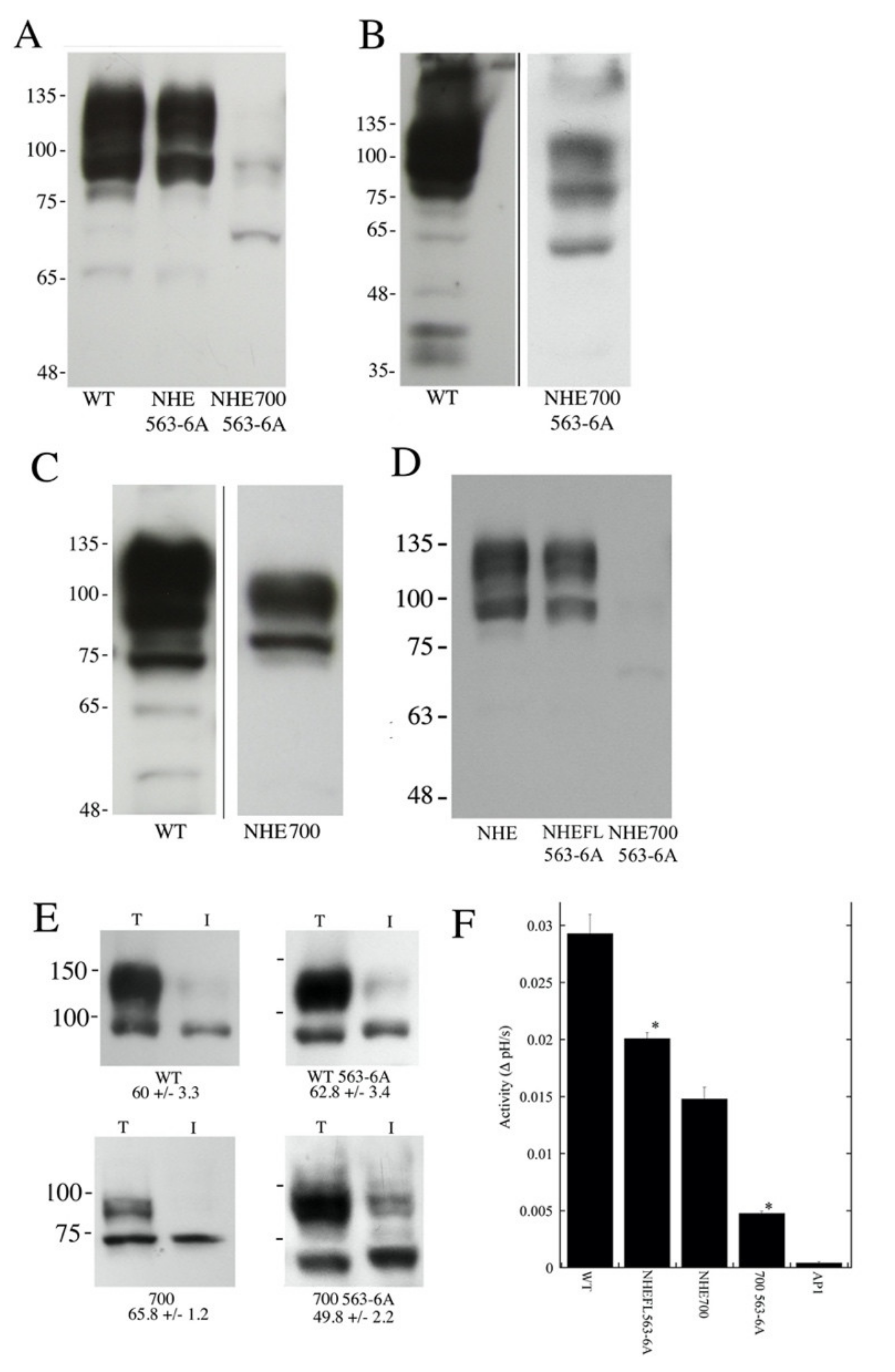
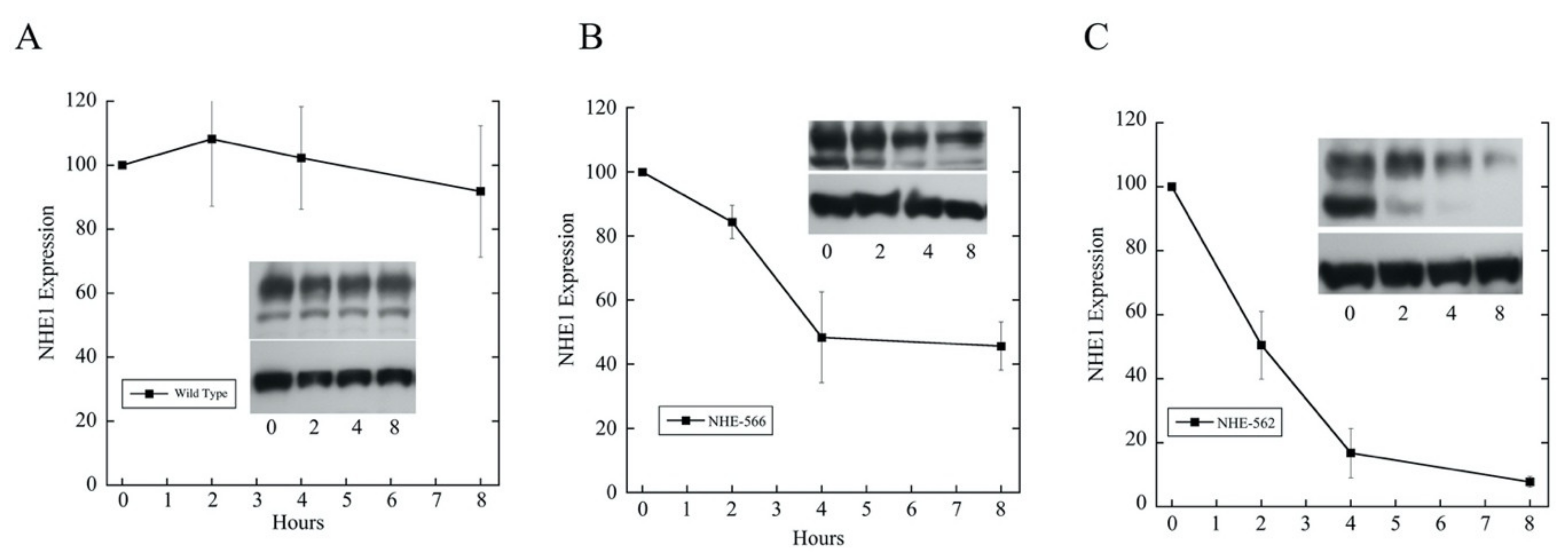
2.3. Effects of Mutations of Amino Acids 563–566 on Longer NHE1 Proteins
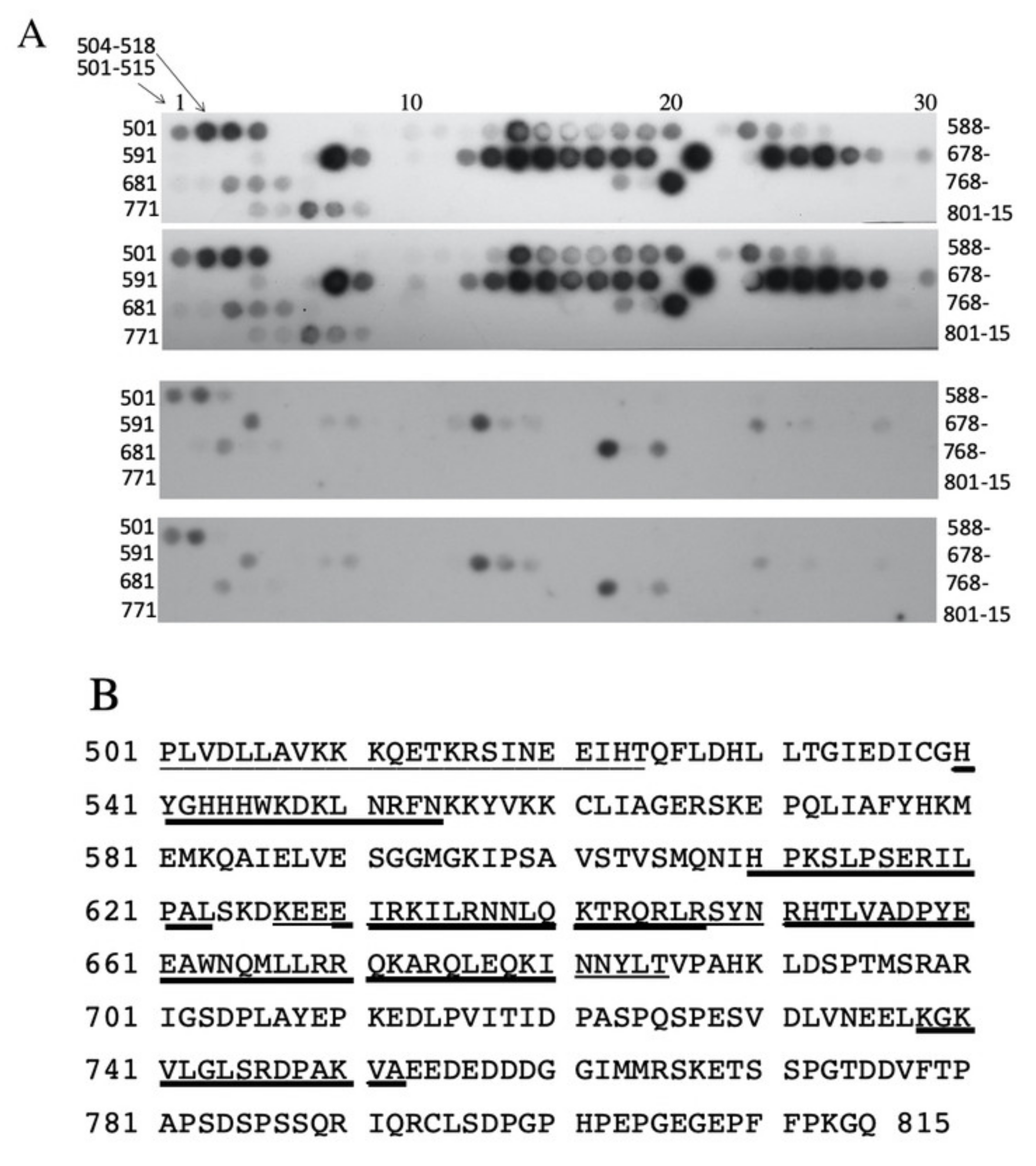
2.4. Examination of Amino Acids 562–568 Binding to the C-Terminal Tail Region
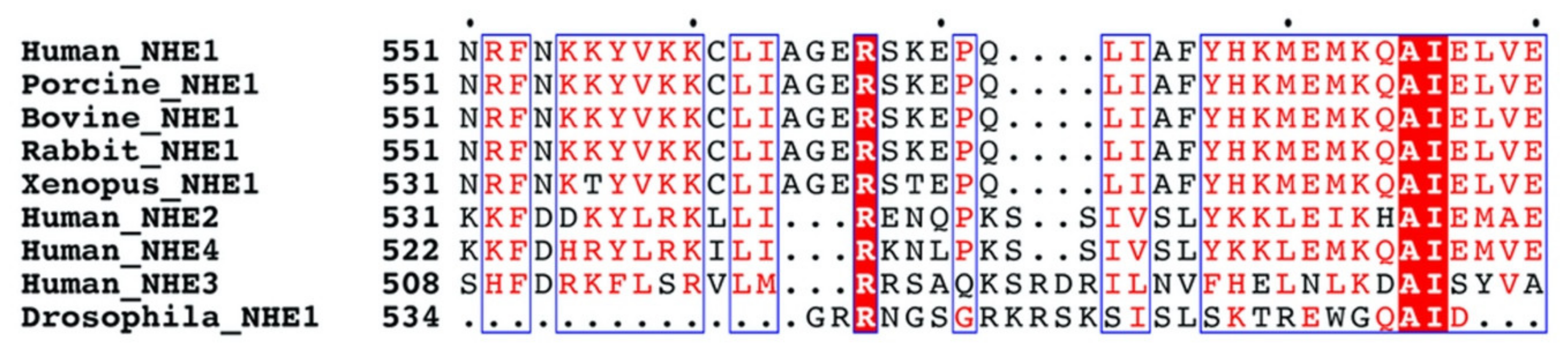
2.5. Multiple Sequence Alignment of NHE1
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Stable Transfection
4.3. Mutant Plasmid Construction
4.4. Cell Surface Expression
4.5. SDS-PAGE and Immunoblotting
4.6. Protein Degradation of Wild Type and Mutant NHE1 Proteins
4.7. Peptide Blot
4.8. Intracellular pH Measurement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| ERM | ezrin, radixin and moesin |
| GFP | green fluorescent protein |
| NHE1 | Na+/H+ exchanger isoform 1 |
| pHi | intracellular pH |
References
- Fliegel, L. The Na+/H+ exchanger isoform 1. Int. J. Biochem. Cell Biol. 2005, 37, 33–37. [Google Scholar] [CrossRef]
- Hendus-Altenburger, R.; Kragelund, B.B.; Pedersen, S.F. Structural dynamics and regulation of the mammalian SLC9A family of Na(+)/H(+) exchangers. Curr. Top. Membr. 2014, 73, 69–148. [Google Scholar] [PubMed]
- Orlowski, J.; Kandasamy, R.A.; Shull, G.E. Molecular cloning of putative members of the Na+/H+ exchanger gene family. J. Biol. Chem. 1992, 267, 9331–9339. [Google Scholar] [PubMed]
- Takaichi, K.; Wang, D.; Balkovetz, D.F.; Warnock, D.G. Cloning, sequencing, and expression of Na+/H+ antiporter cDNAs from human tissues. Am. J. Physiol. 1992, 262, C1069–C1076. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, J.; Grinstein, S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004, 447, 549–565. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, T.; Park, E.S.; Yang, S.; Jeong, D.; Choi, Y.; Rho, J. NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival [corrected]. Biochem. Biophys. Res. Commun. 2008, 369, 320–326. [Google Scholar] [CrossRef]
- Karmazyn, M.; Gan, T.; Humphreys, R.A.; Yoshida, H.; Kusumoto, K. The myocardial Na+-H+ exchange. Structure, regulation, and its role in heart disease. Circ. Res. 1999, 85, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Fliegel, L. Na(+)/H(+) exchanger-mediated hydrogen ion extrusion as a carcinogenic signal in triple-negative breast cancer etiopathogenesis and prospects for its inhibition in therapeutics. Semin. Cancer Biol. 2017, 43, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130100. [Google Scholar] [CrossRef] [Green Version]
- Malo, M.E.; Fliegel, L. Physiological role and regulation of the Na+/H+ exchanger. Can. J. Physiol. Pharmacol. 2006, 84, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Guissart, C.; Li, X.; Leheup, B.; Drouot, N.; Montaut-Verient, B.; Raffo, E.; Jonveaux, P.; Roux, A.F.; Claustres, M.; Fliegel, L.; et al. Mutation of SLC9A1, encoding the major Na+/H+ exchanger, causes ataxia-deafness Lichtenstein-Knorr syndrome. Hum. Mol. Genet. 2015, 24, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fliegel, L. A novel human mutation in the SLC9A1 gene results in abolition of Na+/H+ exchanger activity. PLoS ONE 2015, 10, e0119453. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Augustine, A.; Chen, S.; Fliegel, L. Stop Codon Polymorphisms in the Human SLC9A1 Gene Disrupt or Compromise Na+/H+ Exchanger Function. PLoS ONE 2016, 11, e0162902. [Google Scholar] [CrossRef] [Green Version]
- Haworth, R.S.; Frohlich, O.; Fliegel, L. Multiple carbohydrate moieties on the Na+/H+ exchanger. Biochem. J. 1993, 289, 637–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauble, N.; Cavalie, A.; Zimmermann, R.; Jung, M. Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels (Austin) 2014, 8, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, S.; Pang, T.; Su, X.; Shigekawa, M. Second mutations rescue point mutant of the Na(+)/H(+) exchanger NHE1 showing defective surface expression. FEBS Lett. 2000, 487, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Silva, N.L.C.L.; Haworth, R.S.; Singh, D.; Fliegel, L. The Carboxyl-terminal region of the Na+/H+ exchanger interacts with mammalian heat shock protein. Biochemistry 1995, 34, 10412–10420. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Counillon, L. The SLC9A-C Mammalian Na(+)/H(+) Exchanger Family: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef]
- Gouw, M.; Michael, S.; Samano-Sanchez, H.; Kumar, M.; Zeke, A.; Lang, B.; Bely, B.; Chemes, L.B.; Davey, N.E.; Deng, Z.; et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018, 46, D428–D434. [Google Scholar] [CrossRef]
- Wakabayashi, S.; Bertrand, B.; Ikeda, T.; Pouyssegur, J.; Shigekawa, M. Mutation of calmodulin-binding site renders the Na+/H+ exchanger (NHE1) highly H(+)-sensitive and Ca2+ regulation-defective. J. Biol. Chem. 1994, 269, 13710–13715. [Google Scholar]
- Hisamitsu, T.; Pang, T.; Shigekawa, M.; Wakabayashi, S. Dimeric interaction between the cytoplasmic domains of the Na+/H+ exchanger NHE1 revealed by symmetrical intermolecular cross-linking and selective co-immunoprecipitation. Biochemistry 2004, 43, 11135–11143. [Google Scholar] [CrossRef] [PubMed]
- Shimada-Shimizu, N.; Hisamitsu, T.; Nakamura, T.Y.; Wakabayashi, S. Evidence that Na+/H+ exchanger 1 is an ATP-binding protein. FEBS J. 2013, 280, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Denker, S.P.; Huang, D.C.; Orlowski, J.; Furthmayr, H.; Barber, D.L. Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H(+) translocation. Mol. Cell 2000, 6, 1425–1436. [Google Scholar] [CrossRef]
- Aharonovitz, O.; Zaun, H.C.; Balla, T.; York, J.D.; Orlowski, J.; Grinstein, S. Intracellular pH regulation by Na(+)/H(+) exchange requires phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 2000, 150, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ding, J.; Liu, Y.; Brix, B.J.; Fliegel, L. Functional analysis of acidic amino acids in the cytosolic tail of the Na+/H+ exchanger. Biochemistry 2004, 43, 16477–16486. [Google Scholar] [CrossRef]
- Ding, J.; Rainey, J.K.; Xu, C.; Sykes, B.D.; Fliegel, L. Structural and functional characterization of transmembrane segment VII of the Na+/H+ exchanger isoform 1. J. Biol. Chem. 2006, 281, 29817–29829. [Google Scholar] [CrossRef] [Green Version]
- Slepkov, E.R.; Chow, S.; Lemieux, M.J.; Fliegel, L. Proline residues in transmembrane segment IV are critical for activity, expression and targeting of the Na+/H+ exchanger isoform 1. Biochem. J. 2004, 379, 31–38. [Google Scholar] [CrossRef]
- Lee, B.L.; Li, X.; Liu, Y.; Sykes, B.D.; Fliegel, L. Structural and functional analysis of transmembrane XI of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 2009, 284, 11546–11556. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, J.; Lee, B.L.; Sykes, B.D.; Fliegel, L. Structural and functional analysis of transmembrane segment VI of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 2010, 285, 36656–36665. [Google Scholar] [CrossRef] [Green Version]
- Bullis, B.L.; Li, X.; Singh, D.N.; Berthiaume, L.G.; Fliegel, L. Properties of the Na+/H+ exchanger protein. Detergent-resistant aggregation and membrane microdistribution. Eur. J. Biochem. 2002, 269, 4887–4895. [Google Scholar] [CrossRef] [Green Version]
- Reddy, T.; Ding, J.; Li, X.; Sykes, B.D.; Rainey, J.K.; Fliegel, L. Structural and functional characterization of transmembrane segment IX of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 2008, 283, 22018–22030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Start Plasmid | Synthetic Oligonucleotide | Description | Calculated Molecular Wt |
|---|---|---|---|---|
| Wild type | A (Wt NHE HA tag) | N/A | 815-HA | 95220 |
| Wild type | B (WT NHE1 GFP tag) | N/A | 815-GFP | 121612 |
| NHE-700 | A | 5′-CCATGTCTCGGGCCCtCgagGGCTCAGACCCACTG-3′ | Stop @AA 700-HA | 81187 |
| NHE-675 | A | 5′-GGCAGAAGGCCCGGCtcgaGGAGCAGAAGATCAAC-3′ | Stop @AA 675-HA | 80229 |
| NHE-625 | A | 5′-CTGCCAGCACTGTCCctcGAgAAGGAGGAGGAGATC-3′ | Stop @AA 625-HA | 73953 |
| NHE-600 | A | 5′-GGCAAGATCCCCTCTctCGagTCCACCGTCTCCATG CAG-3′ | Stop @AA 600-HA | 71295 |
| NHE-582 | A | 5′-CCACAAGATGGAGATGctcgAGGCCATCGAGCTGGTG-3′ | Stop @AA 582-HA | 69569 |
| NHE-566 | A | 5′-GTGTCTGATAGCTGGCGAGCtCgagAAGGAGCCCCAGC TCATTG-3′ | Stop @AA 566-HA | 67579 |
| NHE-564 | A | 5′-GTGAAGAAGTGTCTGATAGCTctcGAGCGCTCCAAG GAGCCCCAGCTC-3′ | Stop @AA 564-HA | 67393 |
| NHE-562 | A | 5′-GAAATATGTGAAGAAGTGTCTcgagGCTGGCGAGC GCTCCAAG-3′ | Stop @AA 562-HA | 67096 |
| NHE-E566A | A | 5′-GTGTCTGATAGCTGGCgcGctggAGgatgacGGCCGCATCT TTTACCC-3′ | NHE-566 with last AA as A-HA | 67521 |
| NHE-E566K | A | 5′-GTGTCTGATAGCTGGCaAGctcgAGgatgacGGCCGCATC TTTTACCC-3′ | NHE-566 with last 4 AA as K-HA | 67578 |
| NHE-563-6A | A | 5′-ATATGTGAAGAAGTGTCTGgcAGCTgcCgcGctggAGgatgacGGCCGCATCTTTTACC-3′ | NHE-566 with last 4 AA as A-HA | 67493 |
| NHE-562-GFP | B | 5′-GAAATATGTGAAGAAGTGTCTcgagGCTGGCGAGC GCTCCAAG-3′ | Stop @AA 562-GFP | 90038 |
| NHE-564-GFP | B | 5′-GTGAAGAAGTGTCTGATAGCTctcGAGCGCTCCAAG GAGCCCCAGCTC-3′ | Stop @AA 564-GFP | 90335 |
| NHE-566-GFP | B | 5′-GTGTCTGATAGCTGGCGAGCtCgagAAGGAGCCCCAGCTCATTG-3′ | Stop @AA 566-GFP | 90521 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Dutta, D.; Jung, M.; Zimmermann, R.; Fliegel, L. Amino Acids 563–566 of the Na+/H+ Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity. Int. J. Mol. Sci. 2020, 21, 1737. https://doi.org/10.3390/ijms21051737
Li X, Dutta D, Jung M, Zimmermann R, Fliegel L. Amino Acids 563–566 of the Na+/H+ Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity. International Journal of Molecular Sciences. 2020; 21(5):1737. https://doi.org/10.3390/ijms21051737
Chicago/Turabian StyleLi, Xiuju, Debajyoti Dutta, Martin Jung, Richard Zimmermann, and Larry Fliegel. 2020. "Amino Acids 563–566 of the Na+/H+ Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity" International Journal of Molecular Sciences 21, no. 5: 1737. https://doi.org/10.3390/ijms21051737
APA StyleLi, X., Dutta, D., Jung, M., Zimmermann, R., & Fliegel, L. (2020). Amino Acids 563–566 of the Na+/H+ Exchanger Isoform 1 C-Terminal Cytosolic Tail Prevent Protein Degradation and Stabilize Protein Expression and Activity. International Journal of Molecular Sciences, 21(5), 1737. https://doi.org/10.3390/ijms21051737