Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection of Cucurbit Leaf Crumple Virus



Abstract
:1. Introduction
2. Results
2.1. Optimization of LAMP Conditions for CuLCrV Detection
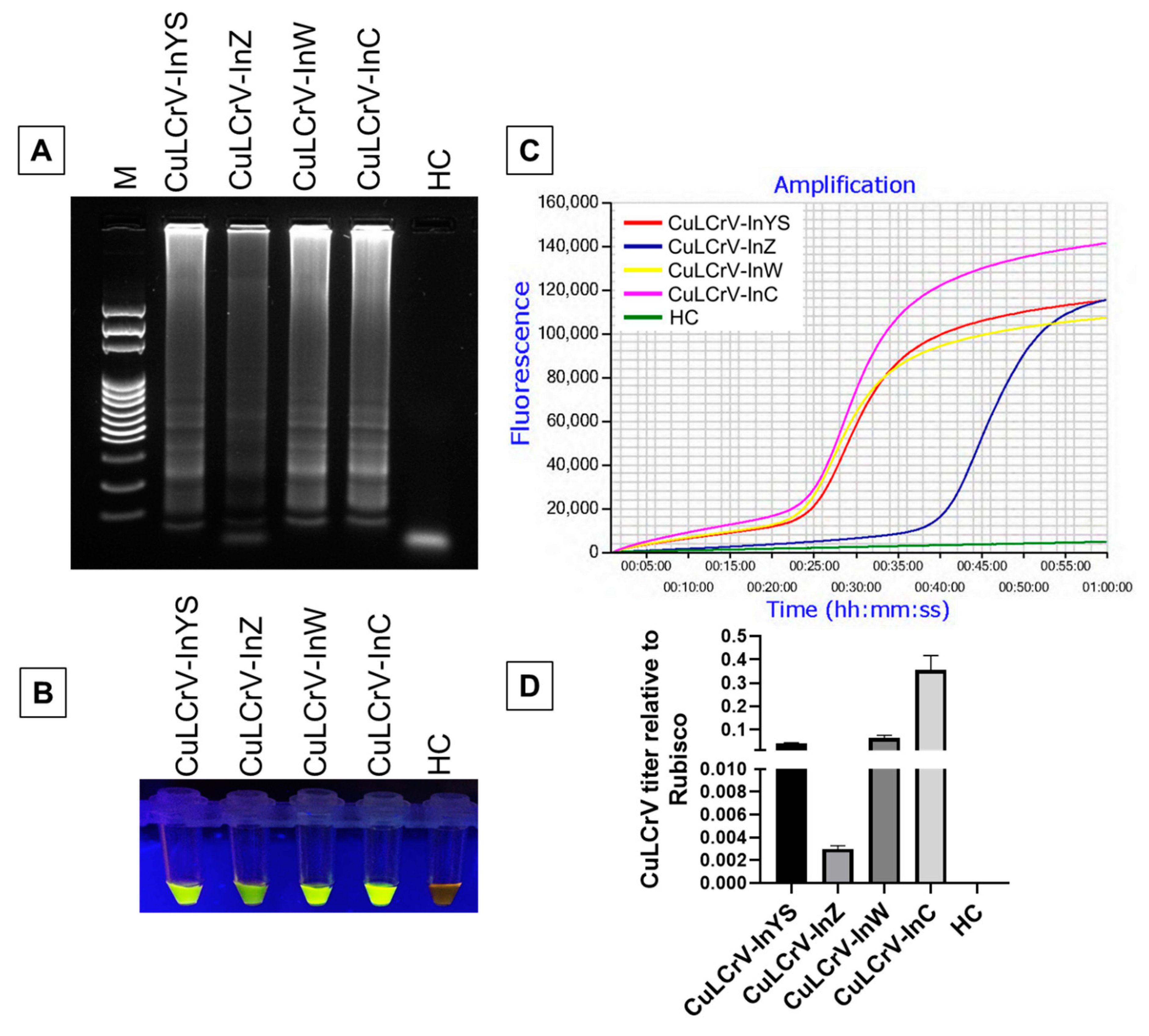
2.2. Evaluation of LAMP Primers with CuLCrV-Infected Samples
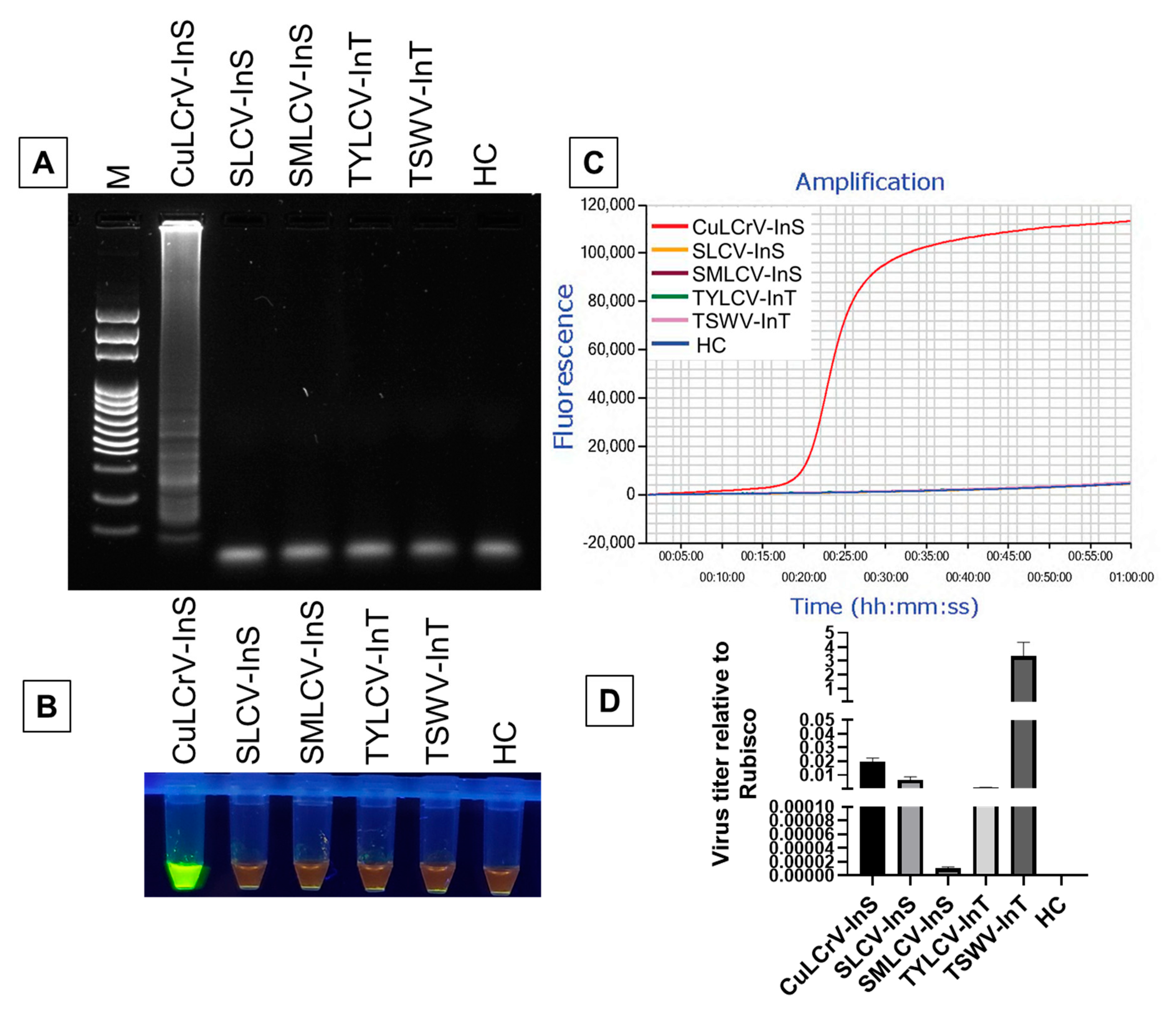
2.3. Specificity of LAMP Detection of CuLCrV
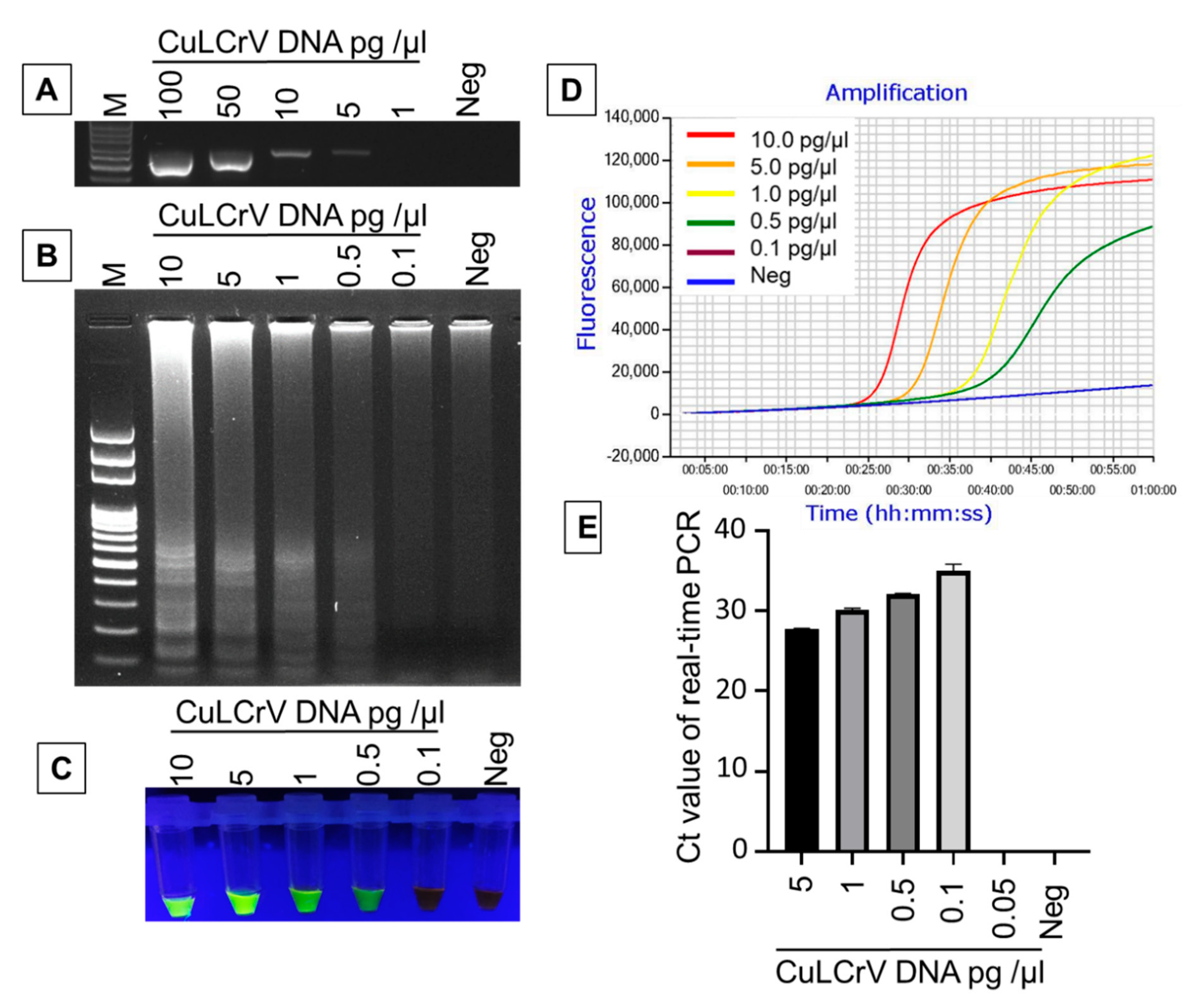
2.4. Comparative Sensitivity Analysis of LAMP Compared to PCR and qPCR
2.5. Diagnosis of CuLCrV Infected Field Samples
2.6. LAMP Amplification of Mixed Virus Infected Field Samples
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA and RNA Extraction
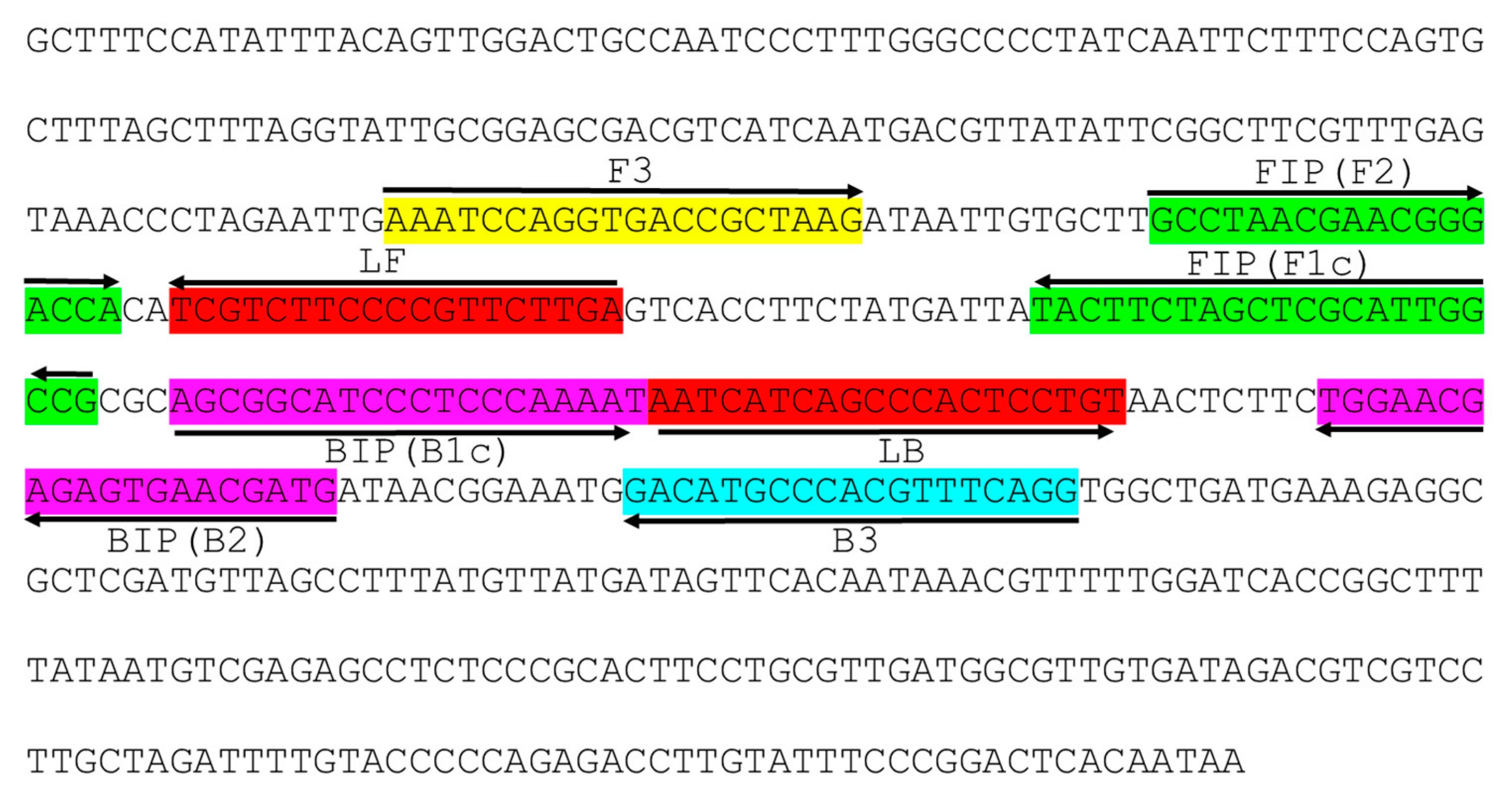
4.3. LAMP Primer Design and Comparison of CuLCrV DNA A Sequence with Other Begomovirus DNA Sequences
4.4. Optimization of LAMP Conditions to Detect CuLCrV Infection
4.5. Reaction Conditions of LAMP, PCR, and qPCR
4.6. Specificity Analysis of LAMP
4.7. Evaluation of LAMP with CuLCrV-Infected Field Samples
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BCaMV | Bean calico mosaic virus |
| CCYV | Cucurbit chlorotic yellows virus |
| CuLCrV | Cucurbit leaf crumple virus |
| CYSDV | Cucurbit yellow stunting disorder virus |
| LAMP | loop-mediated isothermal amplification |
| MCLCV | Melon chlorotic leaf curl virus |
| RFLP | Restriction fragment length polymorphism |
| SLCV | Squash leaf curl virus |
| SMLCV | Squash mild leaf curl virus |
| SqVYV | Squash Vein Yellowing Virus |
| SYMMV | Squash yellow mild mottle virus |
| ToCV | Tomato chlorosis virus |
| TSWV | Tomato spotted wilt virus |
| TYLCV | Tomato yellow leaf curl virus |
References
- Webb, S.; Akad, F.; Nyoike, T.; Liburd, O.; Polston, J. Whitefly-transmitted Cucurbit leaf crumple virus in Florida. Entomol. Nematol. 2008, 91, 460–465. [Google Scholar]
- Brown, J.K.; Idris, A.; Alteri, C.; Stenger, D.C. Emergence of a new cucurbit-infecting begomovirus species capable of forming viable reassortants with related viruses in the Squash leaf curl virus cluster. Phytopathology 2002, 92, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adkins, S.; Polston, J.; Turechek, W. Cucurbit leaf crumple virus Identified in Common Bean in Florida. Plant Dis. 2009, 93, 320. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Idris, A.; Olsen, M.; Miller, M.; Isakeit, T.; Anciso, J. Cucurbit leaf curl virus, a new whitefly transmitted geminivirus in Arizona, Texas, and Mexico. Plant Dis. 2000, 84, 809. [Google Scholar] [CrossRef]
- Akad, F.; Webb, S.; Nyoike, T.; Liburd, O.; Turechek, W.; Adkins, S.; Polston, J. Detection of Cucurbit leaf crumple virus in Florida cucurbits. Plant Dis. 2008, 92, 648. [Google Scholar] [CrossRef]
- Keinath, A.P.; Ling, K.-S.; Adkins, S.; Hasegawa, D.K.; Simmons, A.M.; Hoak, S.; Mellinger, H.C.; Kousik, C.S. First Report of Cucurbit Leaf Crumple Virus Infecting Three Cucurbit Crops in South Carolina. Plant Health Prog. 2018, 19, 322–323. [Google Scholar] [CrossRef]
- Adkins, S.; Webster, C.G.; Kousik, C.S.; Webb, S.E.; Roberts, P.D.; Stansly, P.A.; Turechek, W.W. Ecology and management of whitefly-transmitted viruses of vegetable crops in Florida. Virus Res. 2011, 159, 110–114. [Google Scholar] [CrossRef]
- Maia, G.S. Isolation, Identification and Characterization of Cucurbit Powdery Mildew in North Central Florida. Doctoral Dissertation, University of Florida, Gainesville, FL, USA, 2012. [Google Scholar]
- Turechek, W.; Adkins, S.; Kousik, C.; Webster, C.; Stansly, P.; Roberts, P. Epidemiological analysis of multi-virus infections of watermelon in experimental fields in southwest Florida. Phytopathology 2009, 99, S131. [Google Scholar]
- Dutta, B.; Myers, B.; Coolong, T.; Srinivasan, B.; Sparks, A. Whitefly-Transmitted Plant Viruses in Southern Georgia. University of Georgia Cooperative Extension Bulletin B1507. 2018. Available online: https://secure.caes.uga.edu/extension/publications/files/pdf/B%201507_2.PDF (accessed on 25 January 2020).
- Hagen, C.; Rojas, M.; Sudarshana, M.; Xoconostle-Cazares, B.; Natwick, E.; Turini, T.; Gilbertson, R. Biology and molecular characterization of Cucurbit leaf crumple virus, an emergent cucurbit-infecting begomovirus in the Imperial Valley of California. Plant Dis. 2008, 92, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Adkins, S.; Webster, C.G.; Baker, C.A.; Weaver, R.; Rosskopf, E.N.; Turechek, W.W. Detection of three whitefly-transmitted viruses infecting the cucurbit weed Cucumis melo var. dudaim in Florida. Plant Health Prog. 2009, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- McCreight, J.D.; Liu, H.-Y.; Turini, T.A. Genetic resistance to Cucurbit leaf crumple virus in melon. HortSci. 2008, 43, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Rojas, M. Use of degenerate primers in the polymerase chain reaction to detect whitefly-trans-mitted geminiviruses. Plant Dis. 1993, 77, 340–347. [Google Scholar] [CrossRef]
- Idris, A.; Mills-Lujan, K.; Martin, K.; Brown, J.K. Melon chlorotic leaf curl virus: Characterization and differential reassortment with closest relatives reveal adaptive virulence in the Squash leaf curl virus clade and host shifting by the host-restricted Bean calico mosaic virus. J. Virol. 2008, 82, 1959–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turechek, W.; Adkins, S.; Kousik, C.S.; Stansly, P.A.; Roberts, P.D. Patterns of multi-virus infections of watermelon at the plant and field levels in Florida. Hortsci. 2008, 43, 623–624. [Google Scholar]
- Hosseini, S.; Vázquez-Villegas, P.; Rito-Palomares, M.; Martinez-Chapa, S.O. Advantages, Disadvantages and Modifications of Conventional ELISA. In Enzyme-Linked Immunosorbent Assay (ELISA); Springer: Singapore, 2018; pp. 67–115. [Google Scholar]
- Satija, J.; Punjabi, N.; Mishra, D.; Mukherji, S. Plasmonic-ELISA: Expanding horizons. RSC Adv. 2016, 6, 85440–85456. [Google Scholar] [CrossRef]
- Buzdin, A.A. Nucleic acids hybridization: Potentials and limitations. In Nucleic Acids Hybridization Modern Applications; Springer: Dordrecht, The Netherlands, 2007; pp. 1–28. [Google Scholar]
- Holben, W.E.; Tiedje, J.M. Applications of nucleic acid hybridization in microbial ecology. Ecology 1988, 69, 561–568. [Google Scholar] [CrossRef]
- Wisler, G.; Duffus, J.; Liu, H.-Y.; Li, R. Ecology and epidemiology of whitefly-transmitted closteroviruses. Plant Dis. 1998, 82, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Ge, C.; Zhang, X.; Wang, J.; Zhou, M. A rapid detection method for the plant pathogen Sclerotinia sclerotiorum based on loop-mediated isothermal amplification (LAMP). Australas. Plant Pathol. 2014, 43, 61–66. [Google Scholar] [CrossRef]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.-J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 2011, 62, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.-S.; Wechter, W.; Somai, B.; Walcott, R.; Keinath, A. An improved real-time PCR system for broad-spectrum detection of Didymella bryoniae, the causal agent of gummy stem blight of cucurbits. Seed Sci. Technol. 2010, 38, 692–703. [Google Scholar] [CrossRef]
- Mori, Y.; Kanda, H.; Notomi, T. Loop-mediated isothermal amplification (LAMP): Recent progress in research and development. J. Infect. Chemother. 2013, 19, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Niessen, L. Current state and future perspectives of loop-mediated isothermal amplification (LAMP)-based diagnosis of filamentous fungi and yeasts. Appl. Microbiol. Biotechnol. 2015, 99, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakruddin, M.; Mannan, K.S.B.; Chowdhury, A.; Mazumdar, R.M.; Hossain, M.N.; Islam, S.; Chowdhury, M.A. Nucleic acid amplification: Alternative methods of polymerase chain reaction. J. Pharm. Bioallied Sci. 2013, 5, 245. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J. Advances in isothermal amplification: Novel strategies inspired by biological processes. Biosens. Bioelectron. 2015, 64, 196–211. [Google Scholar] [CrossRef]
- Parida, M.; Sannarangaiah, S.; Dash, P.K.; Rao, P.; Morita, K. Loop mediated isothermal amplification (LAMP): A new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol. 2008, 18, 407–421. [Google Scholar] [CrossRef]
- Tomita, N.; Mori, Y.; Kanda, H.; Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 2008, 3, 877. [Google Scholar] [CrossRef]
- Kalvatchev, Z.; Tsekov, I.; Kalvatchev, N. Loop-mediated amplification for sensitive and specific detection of viruses. Biotechnol. Biotechnol. Equip. 2010, 24, 1559–1561. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Qiu, Y.; Zheng, L.; Chen, J.; Yang, J. Development of a visual loop-mediated isothermal amplification method for rapid detection of the bacterial pathogen Pseudomonas putida of the large yellow croaker (Pseudosciaena crocea). J. Microbiol. Methods 2012, 89, 179–184. [Google Scholar] [CrossRef]
- Mori, Y.; Kitao, M.; Tomita, N.; Notomi, T. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J. Biochem. Biophys. Methods 2004, 59, 145–157. [Google Scholar] [CrossRef]
- Kuan, C.-P.; Wu, M.-T.; Lu, Y.-L.; Huang, H.-C. Rapid detection of Squash leaf curl virus by loop-mediated isothermal amplification. J. Virol. Methods 2010, 169, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-M.; Li, G.; Gao, Y.; Zhu, Y.-R.; Liu, J.; Zhu, X.-P. Reverse transcription loop-mediated isothermal amplification assay for detecting Tomato chlorosis virus. J. Virol. Methods 2015, 213, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Okuda, S.; Iwai, H. Detection of Cucurbit chlorotic yellows virus from Bemisia tabaci captured on sticky traps using reverse transcription loop-mediated isothermal amplification (RT-LAMP) and simple template preparation. J. Virol. Methods 2015, 221, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gu, Q.; Sun, H.; Li, H.; Sun, B.; Liang, X.; Yuan, Y.; Liu, R.; Shi, Y. One-step reverse transcription loop mediated isothermal amplification assay for sensitive and rapid detection of Cucurbit chlorotic yellows virus. J. Virol. Methods 2014, 195, 63–66. [Google Scholar] [CrossRef]
- Fukuta, S.; Kato, S.; Yoshida, K.; Mizukami, Y.; Ishida, A.; Ueda, J.; Kanbe, M.; Ishimoto, Y. Detection of Tomato yellow leaf curl virus by loop-mediated isothermal amplification reaction. J. Virol. Methods 2003, 112, 35–40. [Google Scholar] [CrossRef]
- Fukuta, S.; Ohishi, K.; Yoshida, K.; Mizukami, Y.; Ishida, A.; Kanbe, M. Development of immunocapture reverse transcription loop-mediated isothermal amplification for the detection of Tomato spotted wilt virus from chrysanthemum. J. Virol. Methods 2004, 121, 49–55. [Google Scholar] [CrossRef]
- Bhat, A.; Siljo, A.; Deeshma, K. Rapid detection of Piper yellow mottle virus and Cucumber mosaic virus infecting black pepper (Piper nigrum) by loop-mediated isothermal amplification (LAMP). J. Virol. Methods 2013, 193, 190–196. [Google Scholar] [CrossRef]
- Nie, X. Reverse transcription loop-mediated isothermal amplification of DNA for detection of Potato virus Y. Plant Dis. 2005, 89, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Almasi, M.A.; Manesh, M.E.; Jafary, H.; Dehabadi, S.M.H. Visual detection of Potato leafroll virus by loop-mediated isothermal amplification of DNA with the GeneFinder™ dye. J. Virol. Methods 2013, 192, 51–54. [Google Scholar] [CrossRef]
- Li, J.-Y.; Wei, Q.-W.; Liu, Y.; Tan, X.-Q.; Zhang, W.-N.; Wu, J.-Y.; Charimbu, M.K.; Hu, B.-S.; Cheng, Z.-B.; Yu, C. One-step reverse transcription loop-mediated isothermal amplification for the rapid detection of Cucumber green mottle mosaic virus. J. Virol. Methods 2013, 193, 583–588. [Google Scholar] [CrossRef]
- Fukuta, S.; Tamura, M.; Maejima, H.; Takahashi, R.; Kuwayama, S.; Tsuji, T.; Yoshida, T.; Itoh, K.; Hashizume, H.; Nakajima, Y. Differential detection of Wheat yellow mosaic virus, Japanese soil-borne wheat mosaic virus and Chinese wheat mosaic virus by reverse transcription loop-mediated isothermal amplification reaction. J. Virol. Methods 2013, 189, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Liu, Y.; Wang, X. Reverse transcription loop-mediated isothermal amplification of DNA for detection of Barley yellow dwarf viruses in China. J. Virol. Methods 2010, 169, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, S.; Iida, T.; Mizukami, Y.; Ishida, A.; Ueda, J.; Kanbe, M.; Ishimoto, Y. Detection of Japanese yam mosaic virus by RT-LAMP. Arch. Virol. 2003, 148, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; James, D. Use of reverse transcription loop-mediated isothermal amplification for the detection of Plum pox virus. J. Virol. Methods 2006, 138, 184–190. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Qian, Y.; Mu, J.; Shen, L.; Wang, F.; Yang, J. Rapid detection of Tobacco mosaic virus using the reverse transcription loop-mediated isothermal amplification method. Arch. Virol. 2010, 155, 1681–1685. [Google Scholar] [CrossRef]
- Le, D.T.; Netsu, O.; Uehara-Ichiki, T.; Shimizu, T.; Choi, I.-R.; Omura, T.; Sasaya, T. Molecular detection of nine rice viruses by a reverse-transcription loop-mediated isothermal amplification assay. J. Virol. Methods 2010, 170, 90–93. [Google Scholar] [CrossRef]
- Herrera-Vásquez, J.; Puchades, A.; Elvira-González, L.; Jaén-Sanjur, J.; Carpino, C.; Rubio, L.; Galipienso, L. Fast detection by loop-mediated isothermal amplification (LAMP) of the three begomovirus species infecting tomato in Panama. Eur. J. Plant Pathol. 2018, 151, 243–250. [Google Scholar] [CrossRef]
- Harper, S.; Ward, L.; Clover, G. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef]
- Waliullah, S.; Hudson, O.; Oliver, J.E.; Brannen, P.M.; Ji, P.; Ali, M.E. Comparative analysis of different molecular and serological methods for detection of Xylella fastidiosa in blueberry. PLoS ONE 2019, 14, e0221903. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Dai, T.; Zhang, H.; Wang, Y.; Zheng, X. Development of a Loop-Mediated Isothermal Amplification Assay to Detect Fusarium oxysporum. J. Phytopathol. 2015, 163, 63–66. [Google Scholar] [CrossRef]
- Tomlinson, J.; Dickinson, M.; Boonham, N. Detection of Botrytis cinerea by loop-mediated isothermal amplification. Lett. Appl. Microbiol. 2010, 51, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Gunimaladevi, I.; Kono, T.; Venugopal, M.; Sakai, M. Detection of Koi herpesvirus in common carp, Cyprinus carpio L., by loop-mediated isothermal amplification. J. Fish Dis. 2004, 27, 583–589. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Geographic Location | Symptomatic/Asymptomatic | No. of Samples Tested | Detected by qPCR | Detected by LAMP | Detected by PCR |
|---|---|---|---|---|---|---|
| Cucumber | Tift County | Symptomatic | 5 | 5 | 5 | 3 |
| Asymptomatic | 5 | 4 | 3 | 1 | ||
| Lowndes County | Symptomatic | 5 | 5 | 5 | 4 | |
| Asymptomatic | 5 | 3 | 2 | 1 | ||
| Winter Squash | Tift County | Symptomatic | 10 | 10 | 10 | 8 |
| Asymptomatic | 10 | 6 | 6 | 3 | ||
| Lowndes County | Symptomatic | 10 | 10 | 9 | 7 | |
| Asymptomatic | 10 | 5 | 5 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waliullah, S.; Ling, K.-S.; Cieniewicz, E.J.; Oliver, J.E.; Ji, P.; Ali, M.E. Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection of Cucurbit Leaf Crumple Virus. Int. J. Mol. Sci. 2020, 21, 1756. https://doi.org/10.3390/ijms21051756
Waliullah S, Ling K-S, Cieniewicz EJ, Oliver JE, Ji P, Ali ME. Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection of Cucurbit Leaf Crumple Virus. International Journal of Molecular Sciences. 2020; 21(5):1756. https://doi.org/10.3390/ijms21051756
Chicago/Turabian StyleWaliullah, Sumyya, Kai-Shu Ling, Elizabeth J. Cieniewicz, Jonathan E. Oliver, Pingsheng Ji, and Md Emran Ali. 2020. "Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection of Cucurbit Leaf Crumple Virus" International Journal of Molecular Sciences 21, no. 5: 1756. https://doi.org/10.3390/ijms21051756