Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy


{kind=link}
Abstract
:1. Introduction
2. How Do Cells Fuse with Each Other?
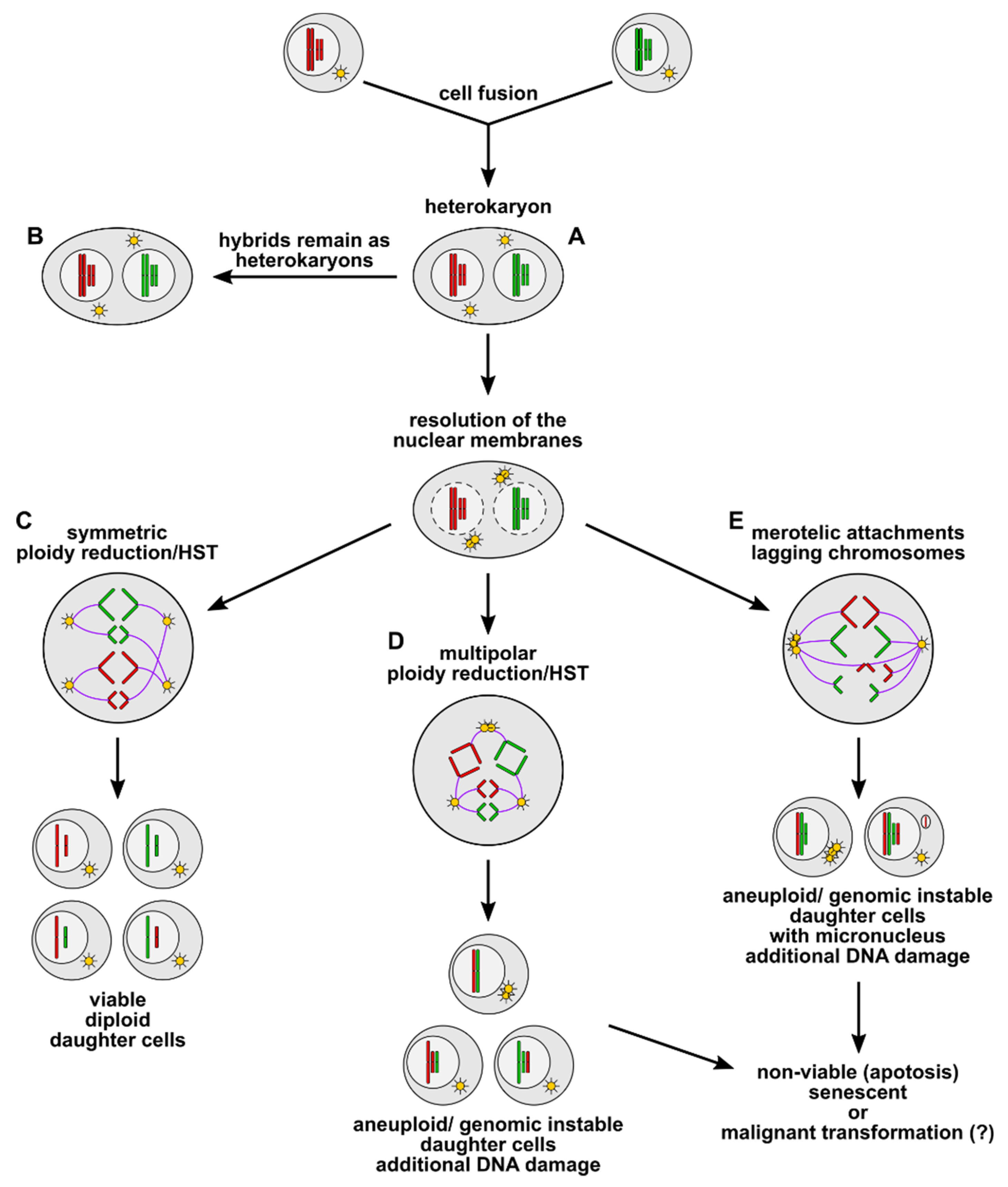
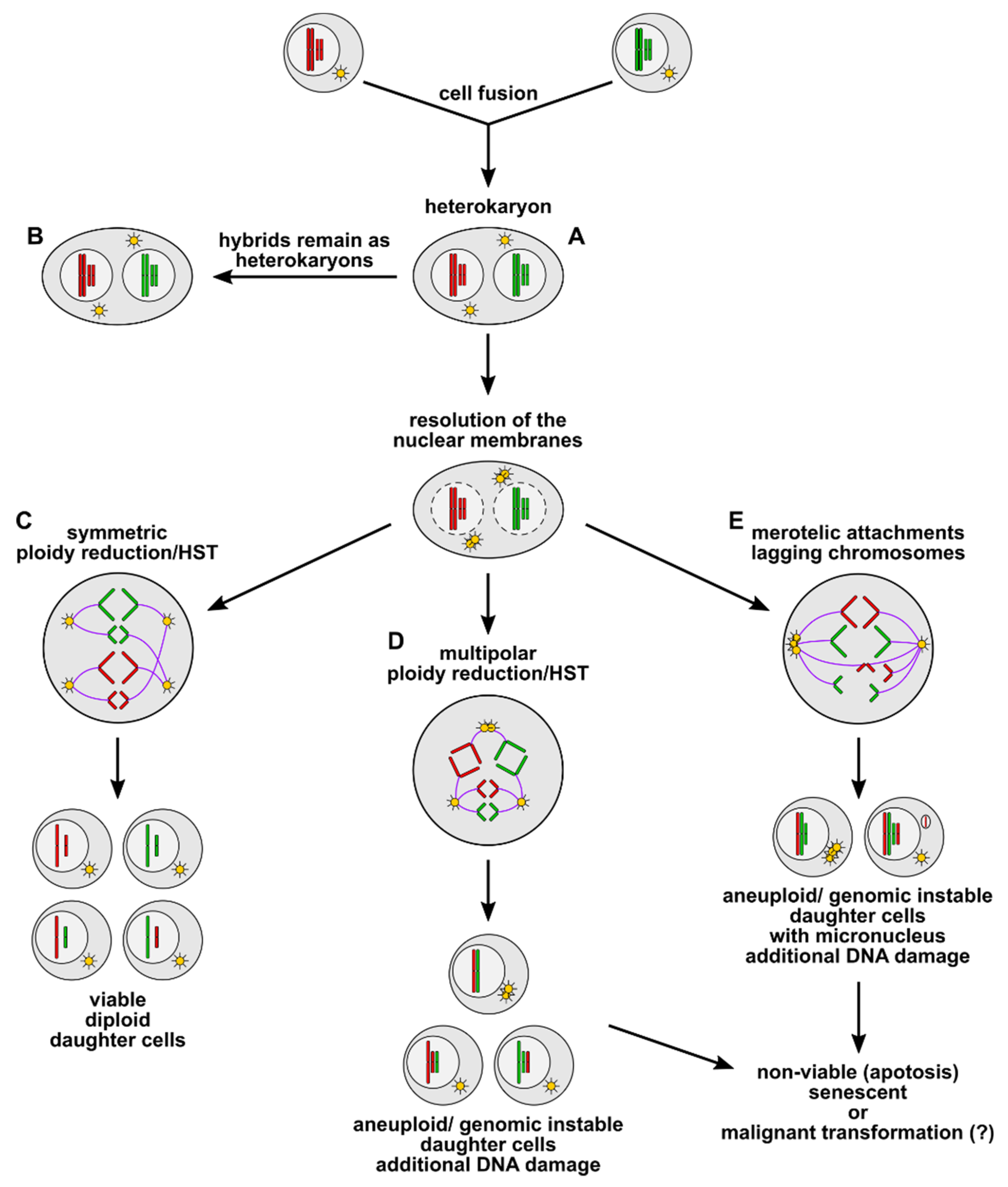
3. Cell Fusion as an Inducer of Polyploidy, Aneuploidy, and Genomic Instability
4. What Is the Fate of Cell Fusion-Derived Aneuploid and Genomic Instable Hybrid Cells?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMSC | bone marrow-derived stem cells |
| HST | heterokaryon-to-synkaryon transition |
| MSCs | mesenchymal stem/stromal cells |
| HSCs | hematopoietic stem cells |
| GFP | green fluorescent protein |
| Fah | fumarylacetoacetate hydrolase |
| NTBC | 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione |
| IL-4 | interleukin-4 |
| RANKL | receptor activator of NF-κB ligand |
| MMP-9 | matrix metallopeptidase 9 |
| DC-STAMP | dendrocyte expressed seven transmembrane protein |
| OC-STAMP | osteoclast stimulatory transmembrane protein |
| TNF-α | tumor necrosis factor-α |
References
- Grompe, M. The role of bone marrow stem cells in liver regeneration. Semin. Liver Dis. 2003, 23, 363–372. [Google Scholar]
- Jang, Y.Y.; Collector, M.I.; Baylin, S.B.; Diehl, A.M.; Sharkis, S.J. Hematopoietic stem cells convert into liver cells within days without fusion. Nat. Cell Biol. 2004, 6, 532–539. [Google Scholar] [CrossRef]
- Alison, M.R.; Poulsom, R.; Jeffery, R.; Dhillon, A.P.; Quaglia, A.; Jacob, J.; Novelli, M.; Prentice, G.; Williamson, J.; Wright, N.A. Hepatocytes from non-hepatic adult stem cells. Nature 2000, 406, 257. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Finegold, M.; Goodell, M.A. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J. Clin. Investig. 2004, 113, 1266–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M.; Pardal, R.; Garcia-Verdugo, J.M.; Fike, J.R.; Lee, H.O.; Pfeffer, K.; Lois, C.; Morrison, S.J.; Alvarez-Buylla, A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003, 425, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.M.; Blau, H.M. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezey, E.; Key, S.; Vogelsang, G.; Szalayova, I.; Lange, G.D.; Crain, B. Transplanted bone marrow generates new neurons in human brains. Proc. Natl. Acad. Sci. USA 2003, 100, 1364–1369. [Google Scholar] [CrossRef] [Green Version]
- Weimann, J.M.; Johansson, C.B.; Trejo, A.; Blau, H.M. Stable reprogrammed heterokaryons form spontaneously in Purkinje neurons after bone marrow transplant. Nat. Cell Biol. 2003, 5, 959–966. [Google Scholar] [CrossRef]
- Lagasse, E.; Connors, H.; Al-Dhalimy, M.; Reitsma, M.; Dohse, M.; Osborne, L.; Wang, X.; Finegold, M.; Weissman, I.L.; Grompe, M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 2000, 6, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Willenbring, H.; Bailey, A.S.; Foster, M.; Akkari, Y.; Dorrell, C.; Olson, S.; Finegold, M.; Fleming, W.H.; Grompe, M. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat. Med. 2004, 10, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, L.M.; Burns, L.; Eisenberg, C.A. Hematopoietic cells from bone marrow have the potential to differentiate into cardiomyocytes in vitro. Anat. Rec. 2003, 274A, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Green, R.; Capetenaki, Y.; Jackson, K.A.; Goodell, M.A. Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat. Med. 2003, 9, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Kawada, H.; Ogawa, M. Bone marrow origin of hematopoietic progenitors and stem cells in murine muscle. Blood 2001, 98, 2008–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBarge, M.A.; Blau, H.M. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell 2002, 111, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530. [Google Scholar] [CrossRef]
- Ferrand, J.; Noel, D.; Lehours, P.; Prochazkova-Carlotti, M.; Chambonnier, L.; Menard, A.; Megraud, F.; Varon, C. Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS ONE 2011, 6, e19569. [Google Scholar] [CrossRef] [Green Version]
- Silk, A.D.; Gast, C.E.; Davies, P.S.; Fakhari, F.D.; Vanderbeek, G.E.; Mori, M.; Wong, M.H. Fusion between hematopoietic and epithelial cells in adult human intestine. PLoS ONE 2013, 8, e55572. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.H.; Muzzonigro, T.M.; Bae, S.H.; LaPlante, J.M.; Hatch, H.M.; Petersen, B.E. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab. Investig. 2004, 84, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Power, C.; Rasko, J.E. Promises and challenges of stem cell research for regenerative medicine. Ann. Intern. Med. 2011, 155, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satija, N.K.; Singh, V.K.; Verma, Y.K.; Gupta, P.; Sharma, S.; Afrin, F.; Sharma, M.; Sharma, P.; Tripathi, R.P.; Gurudutta, G.U. Mesenchymal stem cell-based therapy: A new paradigm in regenerative medicine. J. Cell. Mol. Med. 2009, 13, 4385–4402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmar, T.; Seidel, J.; Zänker, K.S.; Niggemann, B. Carcinogenesis driven by bone marrow-derived stem cells. Contrib. Microbiol. 2006, 13, 156–169. [Google Scholar] [PubMed]
- Eisenberg, L.M.; Eisenberg, C.A. Stem cell plasticity, cell fusion, and transdifferentiation. Birth Defects Res. Part C Embryo Today 2003, 69, 209–218. [Google Scholar] [CrossRef]
- Newsome, P.N.; Johannessen, I.; Boyle, S.; Dalakas, E.; McAulay, K.A.; Samuel, K.; Rae, F.; Forrester, L.; Turner, M.L.; Hayes, P.C.; et al. Human cord blood-derived cells can differentiate into hepatocytes in the mouse liver with no evidence of cellular fusion. Gastroenterology 2003, 124, 1891–1900. [Google Scholar] [CrossRef]
- Wurmser, A.E.; Nakashima, K.; Summers, R.G.; Toni, N.; D’Amour, K.A.; Lie, D.C.; Gage, F.H. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature 2004, 430, 350–356. [Google Scholar] [CrossRef]
- Shi, D.; Reinecke, H.; Murry, C.E.; Torok-Storb, B. Myogenic fusion of human bone marrow stromal cells, but not hematopoietic cells. Blood 2004, 104, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Ylostalo, J.; Lynch, P.J.; Smith, J.; Perry, A.; Peister, A.; Wang, M.Y.; Prockop, D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc. Natl. Acad. Sci. USA 2003, 100, 2397–2402. [Google Scholar] [CrossRef] [Green Version]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef]
- Ying, Q.L.; Nichols, J.; Evans, E.P.; Smith, A.G. Changing potency by spontaneous fusion. Nature 2002, 416, 545–548. [Google Scholar] [CrossRef]
- Nygren, J.M.; Liuba, K.; Breitbach, M.; Stott, S.; Thoren, L.; Roell, W.; Geisen, C.; Sasse, P.; Kirik, D.; Bjorklund, A.; et al. Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nat. Cell Biol. 2008, 10, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Bustamante, O.; Alvarez-Barrientos, A.; Kofman, A.V.; Fabregat, I.; Bueren, J.A.; Theise, N.D.; Segovia, J.C. Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology 2006, 43, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.; Nakagawa, S.; Cortes, P.; di Vicino, U.; Romo, N.; Lluis, F.; Cosma, M.P. Controlled ploidy reduction of pluripotent 4n cells generates 2n cells during mouse embryo development. Sci. Adv. 2019, 5, eaax4199. [Google Scholar] [CrossRef] [Green Version]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wakefield, L.; Tarlow, B.D.; Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem Cell 2020, 26, 34–47.e3. [Google Scholar] [CrossRef]
- Sottile, F.; Aulicino, F.; Theka, I.; Cosma, M.P. Mesenchymal stem cells generate distinct functional hybrids in vitro via cell fusion or entosis. Sci. Rep. 2016, 6, 36863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, A.M.; Grompe, M.; Kurre, P. Intra-hematopoietic cell fusion as a source of somatic variation in the hematopoietic system. J. Cell Sci. 2012, 125, 2837–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrbaum, M.; Storchova, Z. Effects of aneuploidy on gene expression: Implications for cancer. FEBS J. 2016, 283, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Chunduri, N.K.; Storchova, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef]
- Pellman, D. Cell biology: Aneuploidy and cancer. Nature 2007, 446, 38–39. [Google Scholar] [CrossRef]
- Duncan, A.W.; Hanlon Newell, A.E.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012, 142, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W.; Hanlon Newell, A.E.; Bi, W.; Finegold, M.J.; Olson, S.B.; Beaudet, A.L.; Grompe, M. Aneuploidy as a mechanism for stress-induced liver adaptation. J. Clin. Investig. 2012, 122, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. Am. J. Pathol. 2019, 189, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Delespaul, L.; Merle, C.; Lesluyes, T.; Lagarde, P.; Le Guellec, S.; Perot, G.; Baud, J.; Carlotti, M.; Danet, C.; Fevre, M.; et al. Fusion-mediated chromosomal instability promotes aneuploidy patterns that resemble human tumors. Oncogene 2019, 38, 6083–6094. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.M.; Hearn, S.; Myers, M.P.; Lazebnik, Y. A primate virus generates transformed human cells by fusion. J. Cell Biol. 2005, 171, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell Fusion Connects Oncogenesis with Tumor Evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Zänker, K.S. Cell Fusion in Health and Disease: Volume I; Springer: Dordrecht, The Netherlands, 2011; Volume 1. [Google Scholar]
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [Green Version]
- Willkomm, L.; Bloch, W. State of the art in cell-cell fusion. Methods Mol. Biol. 2015, 1313, 1–19. [Google Scholar] [CrossRef]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of Mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64. [Google Scholar] [CrossRef]
- Huppertz, B.; Gauster, M. Trophoblast fusion. Adv. Exp. Med. Biol. 2011, 713, 81–95. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Soe, K.; Andersen, T.L.; Hobolt-Pedersen, A.S.; Bjerregaard, B.; Larsson, L.I.; Delaisse, J.M. Involvement of human endogenous retroviral syncytin-1 in human osteoclast fusion. Bone 2011, 48, 837–846. [Google Scholar] [CrossRef]
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 2006, 63, 1906–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.L.; Wang, M.; Xu, Z.; Huang, C.M.; Zhou, X.C.; Jiang, E.H.; Zhao, X.P.; Song, Y.; Song, K.; Shao, Z.; et al. Up-regulation of syncytin-1 contributes to TNF-alpha-enhanced fusion between OSCC and HUVECs partly via Wnt/beta-catenin-dependent pathway. Sci. Rep. 2017, 7, 40983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. In vitro fusion of normal and neoplastic breast epithelial cells with human mesenchymal stroma/stem cells (MSC) partially involves TNF receptor signaling. Stem Cells 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, A.; Pavlath, G.K. Molecular mechanisms of myoblast fusion across species. Adv. Exp. Med. Biol. 2011, 713, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Sondag, G.R.; Moussa, F.M.; Belcher, J.Y.; Yu, B.; Stinnett, H.; Novak, K.; Mbimba, T.; Khol, M.; Hankenson, K.D.; et al. Mutation in Osteoactivin Promotes Receptor Activator of NFkappaB Ligand (RANKL)-mediated Osteoclast Differentiation and Survival but Inhibits Osteoclast Function. J. Biol. Chem. 2015, 290, 20128–20146. [Google Scholar] [CrossRef] [Green Version]
- Mensah, K.A.; Ritchlin, C.T.; Schwarz, E.M. RANKL induces heterogeneous DC-STAMP(lo) and DC-STAMP(hi) osteoclast precursors of which the DC-STAMP(lo) precursors are the master fusogens. J. Cell. Physiol. 2010, 223, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.L.; Mikhailenko, I.; Tondravi, M.M.; Keegan, A.D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: Contribution of E-cadherin. J. Leukoc. Biol. 2007, 82, 1542–1553. [Google Scholar] [CrossRef]
- Papadaki, M.; Rinotas, V.; Violitzi, F.; Thireou, T.; Panayotou, G.; Samiotaki, M.; Douni, E. New Insights for RANKL as a Proinflammatory Modulator in Modeled Inflammatory Arthritis. Front. Immunol. 2019, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Qi, X.; Moreno, J.L.; Farber, D.L.; Keegan, A.D. NF-kappaB signaling participates in both RANKL- and IL-4-induced macrophage fusion: Receptor cross-talk leads to alterations in NF-kappaB pathways. J. Immunol. 2011, 187, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiler, J.; Mohr, M.; Zanker, K.S.; Dittmar, T. Matrix metalloproteinase-9 (MMP9) is involved in the TNF-alpha-induced fusion of human M13SV1-Cre breast epithelial cells and human MDA-MB-435-pFDR1 cancer cells. Cell Commun. Signal. 2018, 16, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-alpha, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Skokos, E.A.; Charokopos, A.; Khan, K.; Wanjala, J.; Kyriakides, T.R. Lack of TNF-alpha-induced MMP-9 production and abnormal E-cadherin redistribution associated with compromised fusion in MCP-1-null macrophages. Am. J. Pathol. 2011, 178, 2311–2321. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Zhu, F.; Zhang, H.Z.; Shang, Z.J. Tumor necrosis factor-alpha enhanced fusions between oral squamous cell carcinoma cells and endothelial cells via VCAM-1/VLA-4 pathway. Exp. Cell Res. 2012, 318, 1707–1715. [Google Scholar] [CrossRef]
- Mohr, M.; Tosun, S.; Arnold, W.H.; Edenhofer, F.; Zanker, K.S.; Dittmar, T. Quantification of cell fusion events human breast cancer cells and breast epithelial cells using a Cre-LoxP-based double fluorescence reporter system. Cell. Mol. Life Sci. 2015, 72, 3769–3782. [Google Scholar] [CrossRef]
- MacLauchlan, S.; Skokos, E.A.; Meznarich, N.; Zhu, D.H.; Raoof, S.; Shipley, J.M.; Senior, R.M.; Bornstein, P.; Kyriakides, T.R. Macrophage fusion, giant cell formation, and the foreign body response require matrix metalloproteinase 9. J. Leukoc. Biol. 2009, 85, 617–626. [Google Scholar] [CrossRef]
- Okada, Y. Sendai virus-induced cell fusion. Methods Enzymol. 1993, 221, 18–41. [Google Scholar]
- Record, M. Intercellular communication by exosomes in placenta: A possible role in cell fusion? Placenta 2014, 35, 297–302. [Google Scholar] [CrossRef]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [Green Version]
- Duelli, D.; Lazebnik, Y. Cell-to-cell fusion as a link between viruses and cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Rawling, J.; Cano, O.; Garcin, D.; Kolakofsky, D.; Melero, J.A. Recombinant Sendai viruses expressing fusion proteins with two furin cleavage sites mimic the syncytial and receptor-independent infection properties of respiratory syncytial virus. J. Virol. 2011, 85, 2771–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmulevitz, M.; Duncan, R. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 2000, 19, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Plafker, K.; Vorozhko, V.; Zuna, R.E.; Hanigan, M.H.; Gorbsky, G.J.; Plafker, S.M.; Angeletti, P.C.; Ceresa, B.P. Human papillomavirus 16 E5 induces bi-nucleated cell formation by cell-cell fusion. Virology 2009, 384, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Miyado, K.; Yoshida, K.; Yamagata, K.; Sakakibara, K.; Okabe, M.; Wang, X.; Miyamoto, K.; Akutsu, H.; Kondo, T.; Takahashi, Y.; et al. The fusing ability of sperm is bestowed by CD9-containing vesicles released from eggs in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12921–12926. [Google Scholar] [CrossRef] [Green Version]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Normand, G.; King, R.W. Understanding cytokinesis failure. Adv. Exp. Med. Biol. 2010, 676, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gnawali, N.; Hinman, A.W.; Mattingly, A.J.; Osimani, A.; Cimini, D. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 2019, 10, 2660–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef] [Green Version]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef]
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009, 28, 85–98. [Google Scholar] [CrossRef]
- Cimini, D.; Fioravanti, D.; Salmon, E.D.; Degrassi, F. Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J. Cell Sci. 2002, 115, 507–515. [Google Scholar]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Hetzer, M.W. Chromothripsis. Curr. Biol. 2015, 25, R397–R399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Akaike, T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc.) 1998, 63, 854–865. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Gerstein, A.C.; Fu, M.S.; Mukaremera, L.; Li, Z.; Ormerod, K.L.; Fraser, J.A.; Berman, J.; Nielsen, K. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio 2015, 6, e01340-15. [Google Scholar] [CrossRef] [Green Version]
- Millet, C.; Ausiannikava, D.; Le Bihan, T.; Granneman, S.; Makovets, S. Cell populations can use aneuploidy to survive telomerase insufficiency. Nat. Commun. 2015, 6, 8664. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.Y.; Wilson, N.R.; Mehta, S.; Hwang, S.S.; Hochstrasser, M. Loss of the SUMO protease Ulp2 triggers a specific multichromosome aneuploidy. Genes Dev. 2016, 30, 1881–1894. [Google Scholar] [CrossRef] [Green Version]
- Beaupere, C.; Dinatto, L.; Wasko, B.M.; Chen, R.B.; VanValkenburg, L.; Kiflezghi, M.G.; Lee, M.B.; Promislow, D.E.L.; Dang, W.; Kaeberlein, M.; et al. Genetic screen identifies adaptive aneuploidy as a key mediator of ER stress resistance in yeast. Proc. Natl. Acad. Sci. USA 2018, 115, 9586–9591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Teoh, F.; Tan, A.S.M.; Cao, Y.; Pavelka, N.; Berman, J. Aneuploidy Enables Cross-Adaptation to Unrelated Drugs. Mol. Biol. Evol. 2019, 36, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Carrell, D.T.; Wilcox, A.L.; Lowy, L.; Peterson, C.M.; Jones, K.P.; Erickson, L.; Campbell, B.; Branch, D.W.; Hatasaka, H.H. Elevated sperm chromosome aneuploidy and apoptosis in patients with unexplained recurrent pregnancy loss. Obstet. Gynecol. 2003, 101, 1229–1235. [Google Scholar] [CrossRef]
- Caneus, J.; Granic, A.; Rademakers, R.; Dickson, D.W.; Coughlan, C.M.; Chial, H.J.; Potter, H. Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol. Biol. Cell 2018, 29, 575–586. [Google Scholar] [CrossRef]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nakayama, Y.; Nambu, T.; Morishita, D.; Kawamoto, T.; Miyamoto, M.; Hirayama, T.; Okaniwa, M.; et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 2015, 6, 7668. [Google Scholar] [CrossRef] [Green Version]
- Estrada, J.C.; Torres, Y.; Benguria, A.; Dopazo, A.; Roche, E.; Carrera-Quintanar, L.; Perez, R.A.; Enriquez, J.A.; Torres, R.; Ramirez, J.C.; et al. Human mesenchymal stem cell-replicative senescence and oxidative stress are closely linked to aneuploidy. Cell Death Dis. 2013, 4, e691. [Google Scholar] [CrossRef] [Green Version]
- Biron-Shental, T.; Liberman, M.; Sharvit, M.; Sukenik-Halevy, R.; Amiel, A. Amniocytes from aneuploidy embryos have enhanced random aneuploidy and signs of senescence—Can these findings be related to medical problems? Gene 2015, 562, 232–235. [Google Scholar] [CrossRef]
- Meena, J.K.; Cerutti, A.; Beichler, C.; Morita, Y.; Bruhn, C.; Kumar, M.; Kraus, J.M.; Speicher, M.R.; Wang, Z.Q.; Kestler, H.A.; et al. Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J. 2015, 34, 1371–1384. [Google Scholar] [CrossRef] [Green Version]
- Miller, F.R.; McInerney, D.; Rogers, C.; Miller, B.E. Spontaneous fusion between metastatic mammary tumor subpopulations. J. Cell. Biochem. 1988, 36, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, W.F.; Greetham, J.; Bennett, D.C. Efficient spontaneous fusion between some co-cultured cells, especially murine melanoma cells. Cell Biol. Int. 1994, 18, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, X.; Wang, C.Y.; Hu, P.; Chu, C.Y.; Liu, S.; Zhau, H.E.; Chung, L.W. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörnen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811. https://doi.org/10.3390/ijms21051811
Dörnen J, Sieler M, Weiler J, Keil S, Dittmar T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. International Journal of Molecular Sciences. 2020; 21(5):1811. https://doi.org/10.3390/ijms21051811
Chicago/Turabian StyleDörnen, Jessica, Mareike Sieler, Julian Weiler, Silvia Keil, and Thomas Dittmar. 2020. "Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy" International Journal of Molecular Sciences 21, no. 5: 1811. https://doi.org/10.3390/ijms21051811
APA StyleDörnen, J., Sieler, M., Weiler, J., Keil, S., & Dittmar, T. (2020). Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. International Journal of Molecular Sciences, 21(5), 1811. https://doi.org/10.3390/ijms21051811