The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy


Abstract
1. Introduction
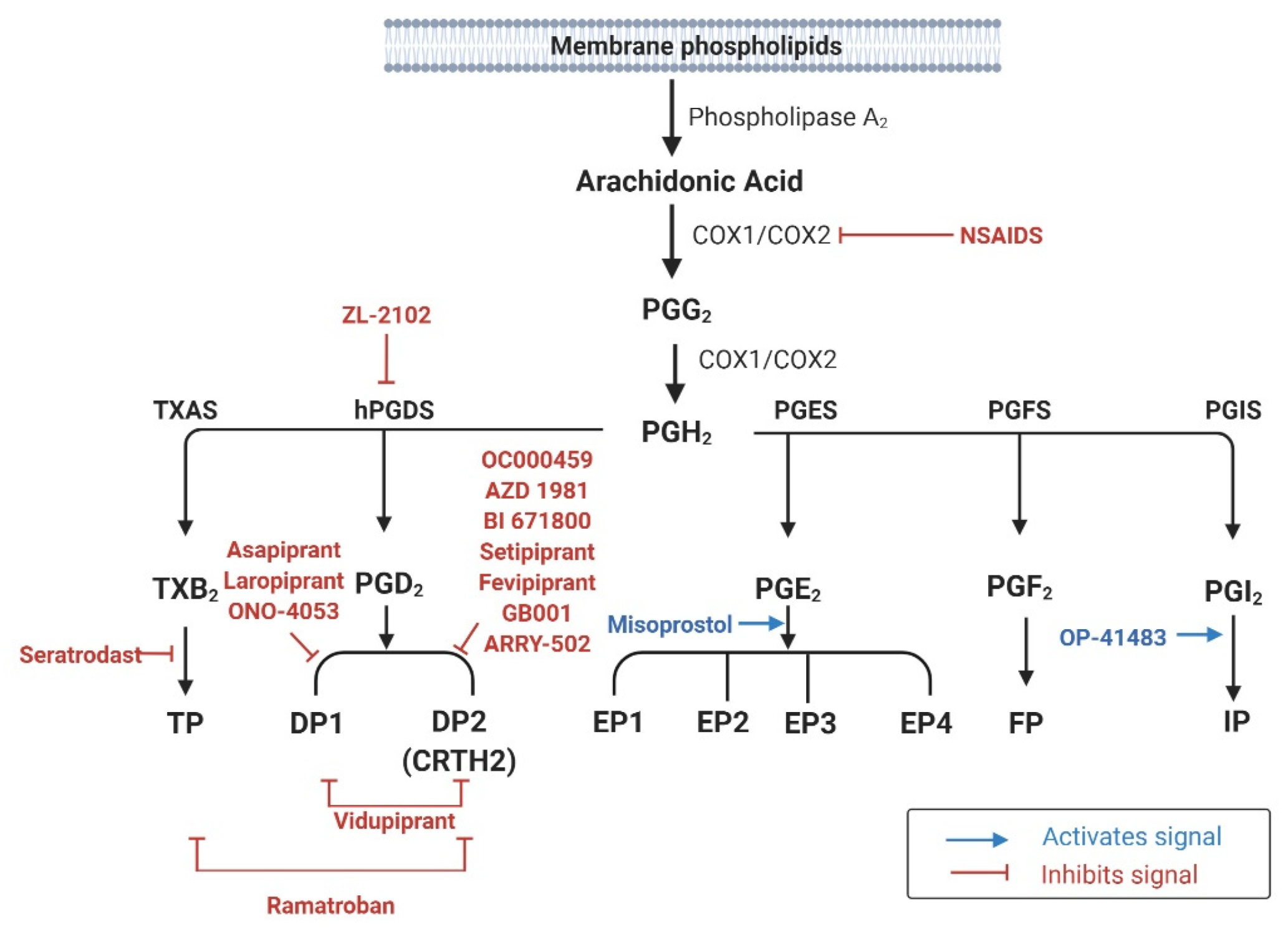
2. Biosynthesis of Prostaglandins
2.1. Phospholipase A2 and Arachidonic Acid
2.2. The Cyclooxygenase Pathway
2.3. Generation of Individual PGs and Their Receptors
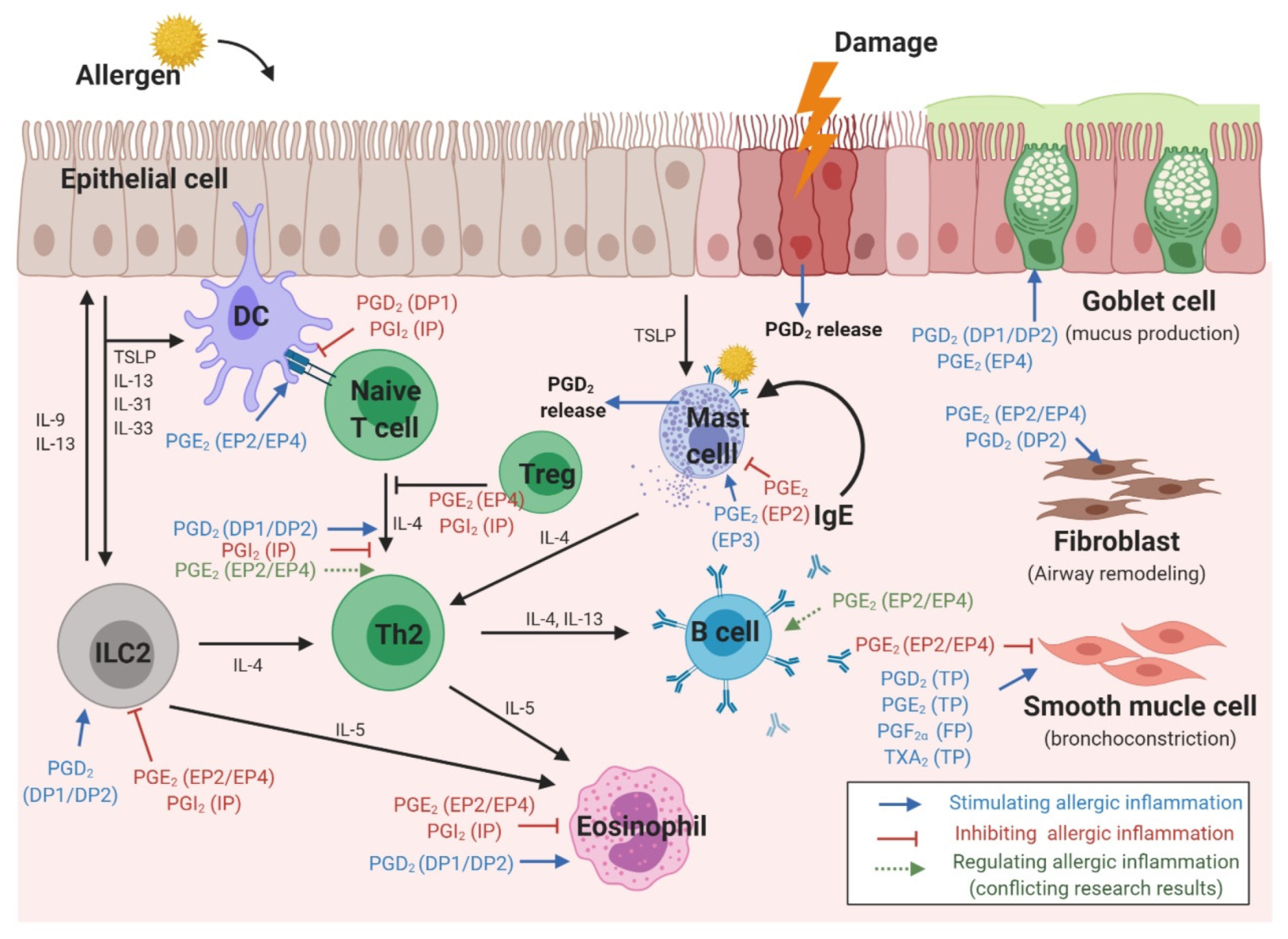
3. The Role of Prostaglandins in Various Cell Types Involved in Allergic Reactions
3.1. Epithelial Cells
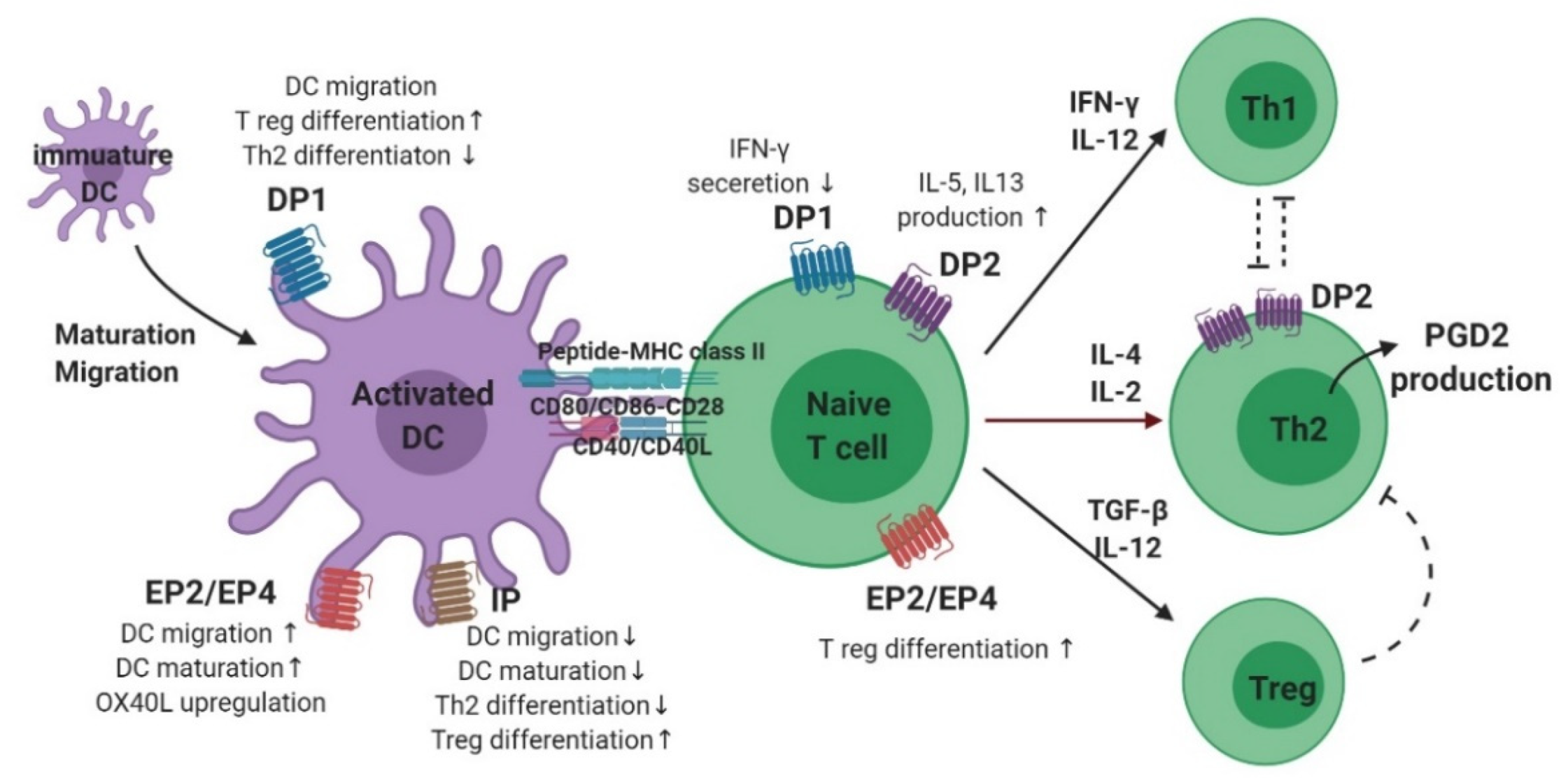
3.2. Dendritic Cells
3.3. T Cells
3.4. B Cells
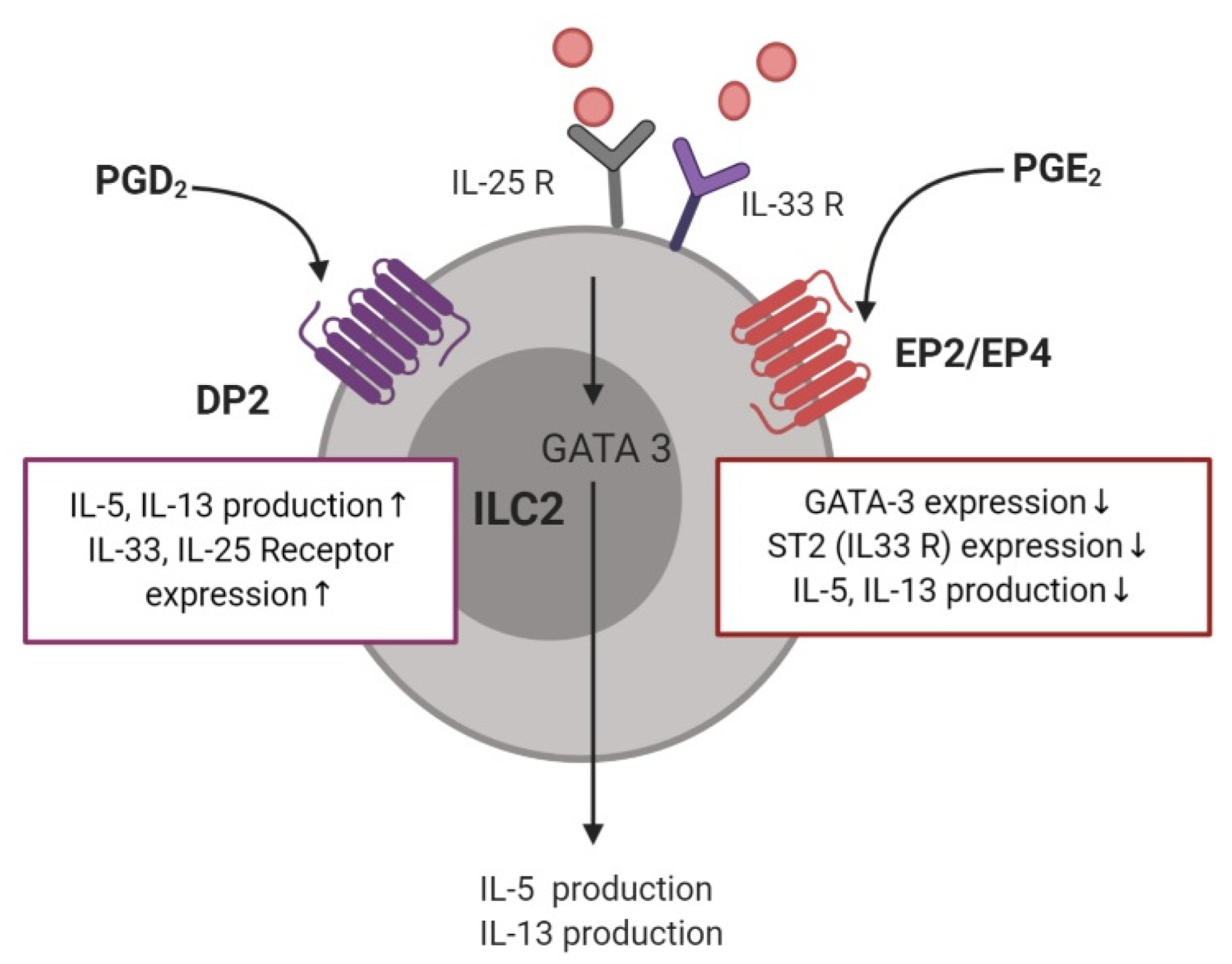
3.5. Type 2 Innate Lymphoid Cells
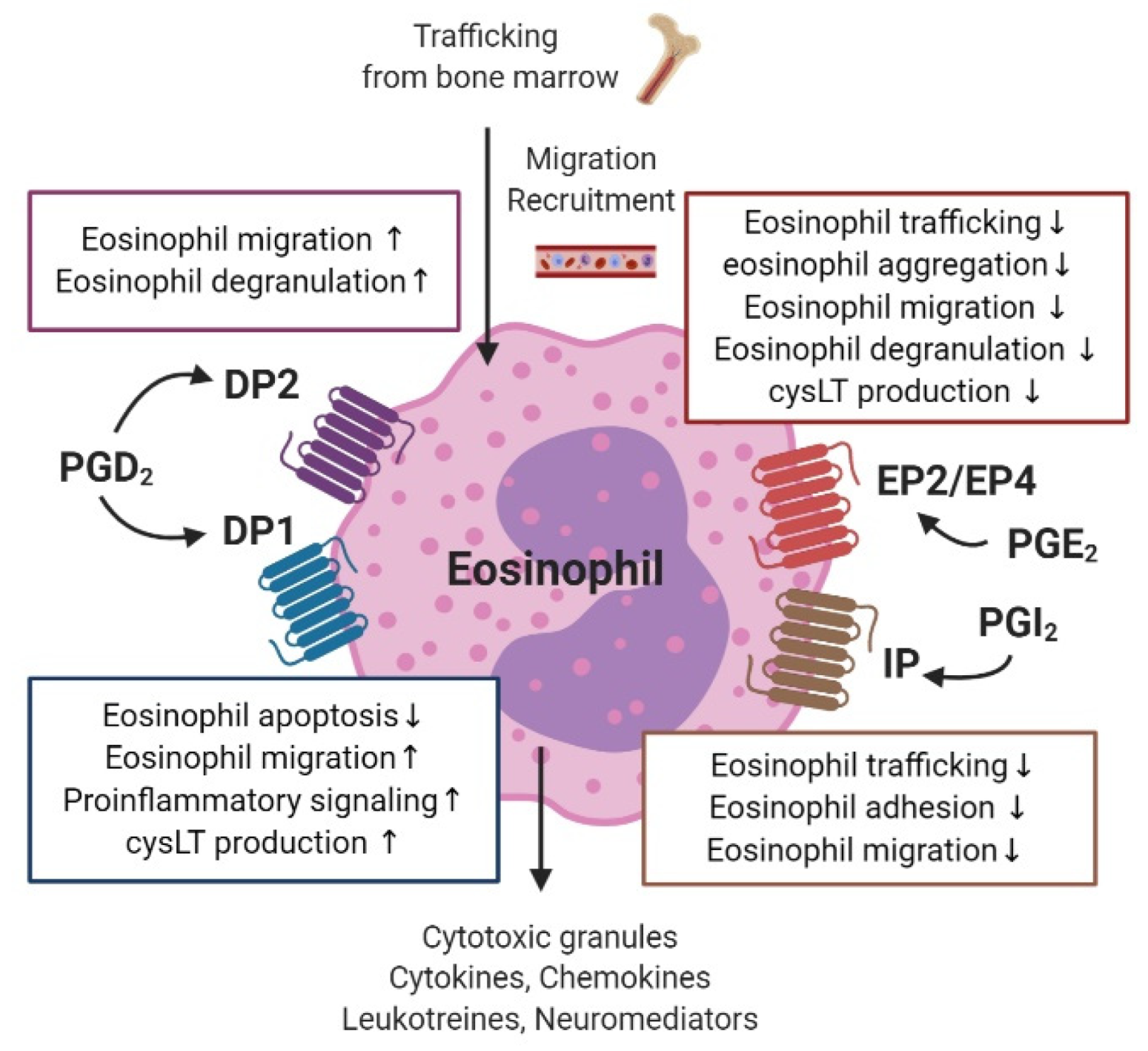
3.6. Eosinophils
3.7. Mast Cells
3.8. Smooth Muscle Cells
3.9. Fibroblasts (in Nasal Polyps)
4. Clinical Studies of Prostaglandins in Allergic Airway Diseases
4.1. Asthma
4.2. Allergic Rhinitis
4.3. Aspirin-Exacerbated Respiratory Disease
4.4. Nasal Polyps
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| AERD | aspirin exacerbated respiratory disease |
| cAMP | cyclic adenosine 3′,5′-monophosphate |
| BAL | bronchoalveolar lavage |
| COX | cyclooxygenase |
| cPGES | cytosolic prostaglandin E synthase |
| cysLT | Cysteinyl leukotriene |
| DC | dendritic cell |
| FEV1 | forced expiratory volume 1 |
| H-PGDS | hematopoietic-prostaglandin D synthase |
| IgE | immunoglobulin E |
| IL | interleukin |
| ILC | innate lymphoid cell |
| INF-γ | interferon γ |
| L-PGDS | lipocalin-prostaglandin D synthase |
| LPS | lipopolysaccharide |
| LT | leukotriene |
| LTC4 | leukotrieneC4 |
| LTD4 | leukotrieneD4 |
| LTE4 | leukotrieneE4 |
| mPGES | microsomal prostaglandin E synthase |
| NALF | nasal lavage fluid |
| NSAID | nonsteroidal anti-inflammatory drug |
| PG | prostaglandin |
| PGD2 | prostaglandin D2 |
| PGE2 | prostaglandin E2 |
| PGF2α | prostaglandin F2α |
| PGFS | prostaglandin F synthase |
| PGG2 | prostaglandin G2 |
| PGH2 | prostaglandin H2 |
| PGI2 | prostaglandin I2 |
| PLA2 | phospholipase A2 |
| TNF-α | tumor necrosis factor α |
| Treg | regulatory T cell |
| TSLP | thymic stromal lymphopoietin |
| TXA2 | thromboxane A2 |
| TXB2 | thromboxane B2 |
References
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Cyclooxygenase pathways. Acta Biochim. Pol. 2014, 61, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Debeuf, N.; Lambrecht, B.N. Eicosanoid Control Over Antigen Presenting Cells in Asthma. Front. Immunol. 2018, 9, 2006. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Piper, P.J.; Vane, J.R. Release of additional factors in anaphylaxis and its antagonism by anti-inflammatory drugs. Nature 1969, 223, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Claar, D.; Hartert, T.V.; Peebles, R.S., Jr. The role of prostaglandins in allergic lung inflammation and asthma. Expert Rev. Respir. Med. 2015, 9, 55–72. [Google Scholar] [CrossRef]
- Nakayama, T. Introduction to “allergic inflammation”. Immunol. Rev. 2017, 278, 5–7. [Google Scholar] [CrossRef]
- Elieh Ali Komi, D.; Bjermer, L. Mast Cell-Mediated Orchestration of the Immune Responses in Human Allergic Asthma: Current Insights. Clin. Rev. Allergy Immunol. 2019, 56, 234–247. [Google Scholar] [CrossRef]
- White, A.A.; Stevenson, D.D. Aspirin-Exacerbated Respiratory Disease. N. Engl. J. Med. 2018, 379, 1060–1070. [Google Scholar] [CrossRef]
- Sanak, M. Eicosanoid Mediators in the Airway Inflammation of Asthmatic Patients: What is New? Allergy Asthma Immunol. Res. 2016, 8, 481–490. [Google Scholar] [CrossRef]
- Laidlaw, T.M. Pathogenesis of NSAID-induced reactions in aspirin-exacerbated respiratory disease. World J. Otorhinolaryngol. Head Neck Surg. 2018, 4, 162–168. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Hallstrand, T.S.; Lai, Y.; Ni, Z.; Oslund, R.C.; Henderson, W.R., Jr.; Gelb, M.H.; Wenzel, S.E. Relationship between levels of secreted phospholipase A(2) groups IIA and X in the airways and asthma severity. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2011, 41, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef]
- Hata, A.N.; Breyer, R.M. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004, 103, 147–166. [Google Scholar] [CrossRef]
- Narumiya, S.; FitzGerald, G.A. Genetic and pharmacological analysis of prostanoid receptor function. J. Clin. Investig. 2001, 108, 25–30. [Google Scholar] [CrossRef]
- Seo, M.J.; Oh, D.K. Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Prog. Lipid Res. 2017, 66, 50–68. [Google Scholar] [CrossRef]
- Lewis, R.A.; Soter, N.A.; Diamond, P.T.; Austen, K.F.; Oates, J.A.; Roberts, L.J., 2nd. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J. Immunol. 1982, 129, 1627–1631. [Google Scholar]
- Luna-Gomes, T.; Magalhaes, K.G.; Mesquita-Santos, F.P.; Bakker-Abreu, I.; Samico, R.F.; Molinaro, R.; Calheiros, A.S.; Diaz, B.L.; Bozza, P.T.; Weller, P.F.; et al. Eosinophils as a novel cell source of prostaglandin D2: Autocrine role in allergic inflammation. J. Immunol. 2011, 187, 6518–6526. [Google Scholar] [CrossRef]
- Okano, M.; Fujiwara, T.; Sugata, Y.; Gotoh, D.; Masaoka, Y.; Sogo, M.; Tanimoto, W.; Yamamoto, M.; Matsumoto, R.; Eguchi, N.; et al. Presence and characterization of prostaglandin D2-related molecules in nasal mucosa of patients with allergic rhinitis. Am. J. Rhinol. 2006, 20, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Brightling, C.E.; Brusselle, G.; Altman, P. The impact of the prostaglandin D2 receptor 2 and its downstream effects on the pathophysiology of asthma. Allergy 2019. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L.; Freezer, N.J.; Ritter, W.; O’Toole, S.; Howarth, P.H. Prostaglandin D2-induced bronchoconstriction is mediated only in part by the thromboxane prostanoid receptor. Eur. Respir. J. 1995, 8, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakashima, K.; Kamei, D.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Ohmiya, Y.; Watanabe, K.; Kudo, I. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 2003, 278, 37937–37947. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar]
- Komoto, J.; Yamada, T.; Watanabe, K.; Woodward, D.F.; Takusagawa, F. Prostaglandin F2alpha formation from prostaglandin H2 by prostaglandin F synthase (PGFS): Crystal structure of PGFS containing bimatoprost. Biochemistry 2006, 45, 1987–1996. [Google Scholar] [CrossRef]
- Nakayama, T. Prostacyclin analogues: Prevention of cardiovascular diseases. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 351–359. [Google Scholar] [CrossRef]
- Miyata, A.; Yokoyama, C.; Ihara, H.; Bandoh, S.; Takeda, O.; Takahashi, E.; Tanabe, T. Characterization of the human gene (TBXAS1) encoding thromboxane synthase. Eur. J. Biochem. 1994, 224, 273–279. [Google Scholar] [CrossRef]
- Ruan, K.H. Advance in understanding the biosynthesis of prostacyclin and thromboxane A2 in the endoplasmic reticulum membrane via the cyclooxygenase pathway. Mini Rev. Med. Chem. 2004, 4, 639–647. [Google Scholar] [CrossRef]
- Hirata, T.; Ushikubi, F.; Kakizuka, A.; Okuma, M.; Narumiya, S. Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J. Clin. Investig. 1996, 97, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Emergence of Biomolecular Pathways to Define Novel Asthma Phenotypes. Type-2 Immunity and Beyond. Am. J. Respir. Cell Mol. Biol. 2016, 55, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Parulekar, A.D.; Kao, C.C.; Diamant, Z.; Hanania, N.A. Targeting the interleukin-4 and interleukin-13 pathways in severe asthma: Current knowledge and future needs. Curr. Opin. Pulm. Med. 2018, 24, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Karta, M.R.; Broide, D.H.; Doherty, T.A. Insights into Group 2 Innate Lymphoid Cells in Human Airway Disease. Curr. Allergy Asthma Rep. 2016, 16, 8. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Boyce, J.A. Role of lipid mediators and control of lymphocyte responses in type 2 immunopathology. J. Allergy Clin. Immunol. 2018, 141, 1182–1190. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lee, S.N.; Aoyagi, H.; Yamasaki, Y.; Yoo, J.Y.; Park, B.; Shin, D.M.; Yoon, H.G.; Yoon, J.H. The extracellular signal-regulated kinase mitogen-activated protein kinase/ribosomal S6 protein kinase 1 cascade phosphorylates cAMP response element-binding protein to induce MUC5B gene expression via D-prostanoid receptor signaling. J. Biol. Chem. 2011, 286, 34199–34214. [Google Scholar] [CrossRef]
- Wright, D.H.; Ford-Hutchinson, A.W.; Chadee, K.; Metters, K.M. The human prostanoid DP receptor stimulates mucin secretion in LS174T cells. Br. J. Pharm. 2000, 131, 1537–1545. [Google Scholar] [CrossRef][Green Version]
- Sally, E.S.; Yassine, A.; Christopher, E.B. D prostanoid receptor 2 (chemoattractant receptor-homologous molecule expressed on TH2 cells) protein expression in asthmatic patients and its effects on bronchial epithelial cells. J. Allergy Clin. Immunol. 2015, 135, 395–406. [Google Scholar]
- Akaba, T.; Komiya, K.; Suzaki, I.; Kozaki, Y.; Tamaoki, J.; Rubin, B.K. Activating prostaglandin E2 receptor subtype EP4 increases secreted mucin from airway goblet cells. Pulm. Pharmacol. Ther. 2018, 48, 117–123. [Google Scholar] [CrossRef]
- Kowalski, M.L.; Pawliczak, R.; Wozniak, J.; Siuda, K.; Poniatowska, M.; Iwaszkiewicz, J.; Kornatowski, T.; Kaliner, M.A. Differential metabolism of arachidonic acid in nasal polyp epithelial cells cultured from aspirin-sensitive and aspirin-tolerant patients. Am. J. Respir. Crit. Care Med. 2000, 161, 391–398. [Google Scholar] [CrossRef]
- Schmidt, L.M.; Belvisi, M.G.; Bode, K.A.; Bauer, J.; Schmidt, C.; Suchy, M.T.; Tsikas, D.; Scheuerer, J.; Lasitschka, F.; Grone, H.J.; et al. Bronchial epithelial cell-derived prostaglandin E2 dampens the reactivity of dendritic cells. J. Immunol. 2011, 186, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.; Jacoby, D.B.; Nadel, J.A.; Lazarus, S.C. The effects of epithelial cell supernatant on contractions of isolated canine tracheal smooth muscle. Am. Rev. Respir. Dis. 1988, 138, 780–783. [Google Scholar] [CrossRef]
- Scandella, E.; Men, Y.; Legler, D.F.; Gillessen, S.; Prikler, L.; Ludewig, B.; Groettrup, M. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood 2004, 103, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Harizi, H.; Grosset, C.; Gualde, N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J. Leukoc. Biol. 2003, 73, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Krause, P.; Scandella, E.; Singer, E.; Groettrup, M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J. Immunol. 2006, 176, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; de Heer, H.J.; Soullie, T.; Hoogsteden, H.C.; Trottein, F.; Lambrecht, B.N. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J. Immunol. 2003, 171, 3936–3940. [Google Scholar] [CrossRef]
- Hammad, H.; Kool, M.; Soullie, T.; Narumiya, S.; Trottein, F.; Hoogsteden, H.C.; Lambrecht, B.N. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J. Exp. Med. 2007, 204, 357–367. [Google Scholar] [CrossRef]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Vos, N.; Hoogsteden, H.C.; Lambrecht, B.N. Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. J. Clin. Investig. 2007, 117, 464–472. [Google Scholar] [CrossRef]
- Zhou, W.; Hashimoto, K.; Goleniewska, K.; O’Neal, J.F.; Ji, S.; Blackwell, T.S.; Fitzgerald, G.A.; Egan, K.M.; Geraci, M.W.; Peebles, R.S., Jr. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J. Immunol. 2007, 178, 702–710. [Google Scholar] [CrossRef]
- Kalinski, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. Dendritic cells, obtained from peripheral blood precursors in the presence of PGE2, promote Th2 responses. Adv. Exp. Med. Biol. 1997, 417, 363–367. [Google Scholar] [CrossRef]
- Kalinski, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J. Immunol. 1997, 159, 28–35. [Google Scholar] [PubMed]
- Kalinski, P.; Schuitemaker, J.H.; Hilkens, C.M.; Kapsenberg, M.L. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: The levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 1998, 161, 2804–2809. [Google Scholar] [PubMed]
- Walker, W.; Rotondo, D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology 2004, 111, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.M.M.; Ritter, M.; Del Fresno, C.; Jonasdottir, H.S.; van der Ham, A.J.; Pelgrom, L.R.; Schramm, G.; Layland, L.E.; Sancho, D.; Prazeres da Costa, C.; et al. Dectin-1/2-induced autocrine PGE2 signaling licenses dendritic cells to prime Th2 responses. PloS Biol. 2018, 16, e2005504. [Google Scholar] [CrossRef] [PubMed]
- Harizi, H.; Norbert, G. Inhibition of IL-6, TNF-alpha, and cyclooxygenase-2 protein expression by prostaglandin E2-induced IL-10 in bone marrow-derived dendritic cells. Cell Immunol. 2004, 228, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, E.; Jing, H.; Ganea, D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cell Immunol. 2003, 223, 120–132. [Google Scholar] [CrossRef]
- Harizi, H.; Juzan, M.; Pitard, V.; Moreau, J.F.; Gualde, N. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 2002, 168, 2255–2263. [Google Scholar] [CrossRef]
- Rieser, C.; Papesh, C.; Herold, M.; Bock, G.; Ramoner, R.; Klocker, H.; Bartsch, G.; Thurnher, M. Differential deactivation of human dendritic cells by endotoxin desensitization: Role of tumor necrosis factor-alpha and prostaglandin E2. Blood 1998, 91, 3112–3117. [Google Scholar] [CrossRef]
- Rieser, C.; Bock, G.; Klocker, H.; Bartsch, G.; Thurnher, M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: Synergistic activation of interleukin 12 production. J. Exp. Med. 1997, 186, 1603–1608. [Google Scholar] [CrossRef]
- Chizzolini, C.; Chicheportiche, R.; Alvarez, M.; de Rham, C.; Roux-Lombard, P.; Ferrari-Lacraz, S.; Dayer, J.M. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 2008, 112, 3696–3703. [Google Scholar] [CrossRef]
- Wong, T.H.; Gau, R.J.; Chen, Y.F.; Shen, H.H.; Lin, C.T.; Chen, S.L.; Suen, J.L. Dendritic cells treated with a prostaglandin I2 analog, iloprost, promote antigen-specific regulatory T cell differentiation in mice. Int. Immunopharmacol. 2019, 79, 106106. [Google Scholar] [CrossRef]
- Tanaka, K.; Hirai, H.; Takano, S.; Nakamura, M.; Nagata, K. Effects of prostaglandin D2 on helper T cell functions. Biochem. Biophys. Res. Commun. 2004, 316, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Mitson-Salazar, A.; Yin, Y.; Wansley, D.L.; Young, M.; Bolan, H.; Arceo, S.; Ho, N.; Koh, C.; Milner, J.D.; Stone, K.D.; et al. Hematopoietic prostaglandin D synthase defines a proeosinophilic pathogenic effector human T(H)2 cell subpopulation with enhanced function. J. Allergy Clin. Immunol. 2016, 137, 907–918. [Google Scholar] [CrossRef]
- Hilvering, B.; Hinks, T.S.C.; Stoger, L.; Marchi, E.; Salimi, M.; Shrimanker, R.; Liu, W.; Chen, W.; Luo, J.; Go, S.; et al. Synergistic activation of pro-inflammatory type-2 CD8(+) T lymphocytes by lipid mediators in severe eosinophilic asthma. Mucosal Immunol. 2018, 11, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Wambre, E.; Bajzik, V.; DeLong, J.H.; O’Brien, K.; Nguyen, Q.A.; Speake, C.; Gersuk, V.H.; DeBerg, H.A.; Whalen, E.; Ni, C.; et al. A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Palikhe, N.S.; Laratta, C.; Nahirney, D.; Vethanayagam, D.; Bhutani, M.; Vliagoftis, H.; Cameron, L. Elevated levels of circulating CD4(+) CRTh2(+) T cells characterize severe asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2016, 46, 825–836. [Google Scholar] [CrossRef]
- Shrestha Palikhe, N.; Bosonea, A.M.; Laratta, C.; Gandhi, V.D.; Nahirney, D.; Hillaby, A.; Bowen, M.; Bhutani, M.; Mayers, I.; Cameron, L.; et al. Stability of peripheral blood immune markers in patients with asthma. Allergy Asthma Clin. Immunol. 2019, 15, 30. [Google Scholar] [CrossRef]
- Snijdewint, F.G.; Kalinski, P.; Wierenga, E.A.; Bos, J.D.; Kapsenberg, M.L. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J. Immunol. 1993, 150, 5321–5329. [Google Scholar]
- Bao, Y.S.; Zhang, P.; Xie, R.J.; Wang, M.; Wang, Z.Y.; Zhou, Z.; Zhai, W.J.; Feng, S.Z.; Han, M.Z. The regulation of CD4+ T cell immune responses toward Th2 cell development by prostaglandin E2. Int. Immunopharmacol. 2011, 11, 1599–1605. [Google Scholar] [CrossRef]
- Yao, C.; Hirata, T.; Soontrapa, K.; Ma, X.; Takemori, H.; Narumiya, S. Prostaglandin E(2) promotes Th1 differentiation via synergistic amplification of IL-12 signalling by cAMP and PI3-kinase. Nat. Commun. 2013, 4, 1685. [Google Scholar] [CrossRef]
- Zaslona, Z.; Okunishi, K.; Bourdonnay, E.; Domingo-Gonzalez, R.; Moore, B.B.; Lukacs, N.W.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E(2) suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J. Allergy Clin. Immunol. 2014, 133, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Sakata, D.; Esaki, Y.; Li, Y.; Matsuoka, T.; Kuroiwa, K.; Sugimoto, Y.; Narumiya, S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 2009, 15, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Aoki, T.; Thumkeo, D.; Siriwach, R.; Yao, C.; Narumiya, S. T cell-intrinsic prostaglandin E2-EP2/EP4 signaling is critical in pathogenic TH17 cell-driven inflammation. J. Allergy Clin. Immunol. 2019, 143, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guan, K.; Zhou, Y.; Wu, J.; Wang, Y.; Wang, W. Prostaglandin E2 signal inhibits T regulatory cell differentiation during allergic rhinitis inflammation through EP4 receptor. World Allergy Organ. J. 2019, 12, 100090. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, C.; Wang, W.; Jin, R.; Li, Q.; Ge, Q.; Guan, Y.; Zhang, Y. Prostaglandins E2 signal mediated by receptor subtype EP2 promotes IgE production in vivo and contributes to asthma development. Sci. Rep. 2016, 6, 20505. [Google Scholar] [CrossRef]
- Roper, R.L.; Conrad, D.H.; Brown, D.M.; Warner, G.L.; Phipps, R.P. Prostaglandin E2 promotes IL-4-induced IgE and IgG1 synthesis. J. Immunol. 1990, 145, 2644–2651. [Google Scholar]
- Fedyk, E.R.; Phipps, R.P. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc. Natl. Acad. Sci. USA 1996, 93, 10978–10983. [Google Scholar] [CrossRef]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 2013, 5, 174ra126. [Google Scholar] [CrossRef]
- Tojima, I.; Matsumoto, K.; Kikuoka, H.; Hara, S.; Yamamoto, S.; Shimizu, S.; Kouzaki, H.; Shimizu, T. Evidence for the induction of Th2 inflammation by group 2 innate lymphoid cells in response to prostaglandin D2 and cysteinyl leukotrienes in allergic rhinitis. Allergy 2019, 74, 2417–2426. [Google Scholar] [CrossRef]
- Xue, L.; Salimi, M.; Panse, I.; Mjosberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Oyesola, O.O.; Duque, C.; Huang, L.C.; Larson, E.M.; Fruh, S.P.; Webb, L.M.; Peng, S.A.; Tait Wojno, E.D. The Prostaglandin D2 Receptor CRTH2 Promotes IL-33-Induced ILC2 Accumulation in the Lung. J. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Smith, S.G.; Salter, B.; El-Gammal, A.; Oliveria, J.P.; Obminski, C.; Watson, R.; O’Byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Allergen-induced Increases in Sputum Levels of Group 2 Innate Lymphoid Cells in Subjects with Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Hochdorfer, T.; Israelsson, E.; Hasselberg, A.; Cavallin, A.; Thorn, K.; Muthas, D.; Shojaee, S.; Luer, K.; Muller, M.; et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J. Allergy Clin. Immunol. 2019, 144, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Eastman, J.J.; Cavagnero, K.J.; Deconde, A.S.; Kim, A.S.; Karta, M.R.; Broide, D.H.; Zuraw, B.L.; White, A.A.; Christiansen, S.C.; Doherty, T.A. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2017, 140, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hardman, C.; Chen, W.; Luo, J.; Batty, P.; Chen, Y.L.; Nahler, J.; Wu, Y.; Pavord, I.D.; Erpenbeck, V.J.; Sandham, D.A.; et al. Fevipiprant, a selective prostaglandin D2 receptor 2 antagonist, inhibits human group 2 innate lymphoid cell aggregation and function. J. Allergy Clin. Immunol. 2019, 143, 2329–2333. [Google Scholar] [CrossRef]
- Maric, J.; Ravindran, A.; Mazzurana, L.; Van Acker, A.; Rao, A.; Kokkinou, E.; Ekoff, M.; Thomas, D.; Fauland, A.; Nilsson, G.; et al. Cytokine-induced endogenous production of prostaglandin D2 is essential for human group 2 innate lymphoid cell activation. J. Allergy Clin. Immunol. 2019, 143, 2202–2214. [Google Scholar] [CrossRef]
- Maric, J.; Ravindran, A.; Mazzurana, L.; Bjorklund, A.K.; Van Acker, A.; Rao, A.; Friberg, D.; Dahlen, S.E.; Heinemann, A.; Konya, V.; et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J. Allergy Clin. Immunol. 2018, 141, 1761–1773. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, W.; Zhao, C.; Wang, Y.; Wu, H.; Sun, X.; Guan, Y.; Zhang, Y. Prostaglandin E2 Inhibits Group 2 Innate Lymphoid Cell Activation and Allergic Airway Inflammation Through E-Prostanoid 4-Cyclic Adenosine Monophosphate Signaling. Front. Immunol. 2018, 9, 501. [Google Scholar] [CrossRef]
- Zhou, W.; Toki, S.; Zhang, J.; Goleniewksa, K.; Newcomb, D.C.; Cephus, J.Y.; Dulek, D.E.; Bloodworth, M.H.; Stier, M.T.; Polosuhkin, V.; et al. Prostaglandin I2 Signaling and Inhibition of Group 2 Innate Lymphoid Cell Responses. Am. J. Respir. Crit. Care Med. 2016, 193, 31–42. [Google Scholar] [CrossRef]
- Peinhaupt, M.; Sturm, E.M.; Heinemann, A. Prostaglandins and Their Receptors in Eosinophil Function and As Therapeutic Targets. Front. Med. (Lausanne) 2017, 4, 104. [Google Scholar] [CrossRef] [PubMed]
- Sedej, M.; Schroder, R.; Bell, K.; Platzer, W.; Vukoja, A.; Kostenis, E.; Heinemann, A.; Waldhoer, M. D-type prostanoid receptor enhances the signaling of chemoattractant receptor-homologous molecule expressed on T(H)2 cells. J. Allergy Clin. Immunol. 2012, 129, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ueki, S.; Adachi, T.; Bourdeaux, J.; Oyamada, H.; Yamada, Y.; Hamada, K.; Kanda, A.; Kayaba, H.; Chihara, J. Expression of PPARgamma in eosinophils and its functional role in survival and chemotaxis. Immunol. Lett. 2003, 86, 183–189. [Google Scholar] [CrossRef]
- Gervais, F.G.; Cruz, R.P.; Chateauneuf, A.; Gale, S.; Sawyer, N.; Nantel, F.; Metters, K.M.; O’Neill G, P. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J. Allergy Clin. Immunol. 2001, 108, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Monneret, G.; Gravel, S.; Diamond, M.; Rokach, J.; Powell, W.S. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 2001, 98, 1942–1948. [Google Scholar] [CrossRef]
- Shamri, R.; Dubois, G.; Erpenbeck, V.J.; Mankuta, D.; Sandham, D.A.; Levi-Schaffer, F. Fevipiprant, a DP2 receptor antagonist, inhibits eosinophil migration towards mast cells. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2019, 49, 255–257. [Google Scholar] [CrossRef]
- Heinemann, A.; Schuligoi, R.; Sabroe, I.; Hartnell, A.; Peskar, B.A. Delta 12-prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J. Immunol. 2003, 170, 4752–4758. [Google Scholar] [CrossRef]
- Schratl, P.; Sturm, E.M.; Royer, J.F.; Sturm, G.J.; Lippe, I.T.; Peskar, B.A.; Heinemann, A. Hierarchy of eosinophil chemoattractants: Role of p38 mitogen-activated protein kinase. Eur J. Immunol. 2006, 36, 2401–2409. [Google Scholar] [CrossRef]
- Schratl, P.; Royer, J.F.; Kostenis, E.; Ulven, T.; Sturm, E.M.; Waldhoer, M.; Hoefler, G.; Schuligoi, R.; Lippe, I.T.; Peskar, B.A.; et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J. Immunol. 2007, 179, 4792–4799. [Google Scholar] [CrossRef]
- Mesquita-Santos, F.P.; Bakker-Abreu, I.; Luna-Gomes, T.; Bozza, P.T.; Diaz, B.L.; Bandeira-Melo, C. Co-operative signalling through DP(1) and DP(2) prostanoid receptors is required to enhance leukotriene C(4) synthesis induced by prostaglandin D(2) in eosinophils. Br. J. Pharm. 2011, 162, 1674–1685. [Google Scholar] [CrossRef]
- Feng, X.; Ramsden, M.K.; Negri, J.; Baker, M.G.; Payne, S.C.; Borish, L.; Steinke, J.W. Eosinophil production of prostaglandin D2 in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2016, 138, 1089–1097. [Google Scholar] [CrossRef]
- Teixeira, M.M.; al-Rashed, S.; Rossi, A.G.; Hellewell, P.G. Characterization of the prostanoid receptors mediating inhibition of PAF-induced aggregation of guinea-pig eosinophils. Br. J. Pharm. 1997, 121, 77–82. [Google Scholar] [CrossRef]
- Butchers, P.R.; Vardey, C.J. The effect of prostanoids on the function of human eosinophils. Agents Actions Suppl. 1990, 31, 103–112. [Google Scholar] [CrossRef]
- Luschnig-Schratl, P.; Sturm, E.M.; Konya, V.; Philipose, S.; Marsche, G.; Frohlich, E.; Samberger, C.; Lang-Loidolt, D.; Gattenlohner, S.; Lippe, I.T.; et al. EP4 receptor stimulation down-regulates human eosinophil function. Cell Mol. Life Sci. 2011, 68, 3573–3587. [Google Scholar] [CrossRef]
- Sturm, E.M.; Schratl, P.; Schuligoi, R.; Konya, V.; Sturm, G.J.; Lippe, I.T.; Peskar, B.A.; Heinemann, A. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J. Immunol. 2008, 181, 7273–7283. [Google Scholar] [CrossRef]
- Peacock, C.D.; Misso, N.L.; Watkins, D.N.; Thompson, P.J. PGE 2 and dibutyryl cyclic adenosine monophosphate prolong eosinophil survival in vitro. J. Allergy Clin. Immunol. 1999, 104, 153–162. [Google Scholar] [CrossRef]
- Konya, V.; Philipose, S.; Balint, Z.; Olschewski, A.; Marsche, G.; Sturm, E.M.; Schicho, R.; Peskar, B.A.; Schuligoi, R.; Heinemann, A. Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors. Eur J. Immunol. 2011, 41, 2379–2389. [Google Scholar] [CrossRef]
- Pal, K.; Ramsden, M.; Shim, Y.M.; Borish, L.; Payne, S.C.; Steinke, J.W. Suppression of aspirin-mediated eosinophil activation by prostaglandin E2: Relevance to aspirin and nonsteroidal anti-inflammatory drug hypersensitivity. Ann. Allergy Asthma Immunol. 2019, 123, 503–506. [Google Scholar] [CrossRef]
- Sturm, E.M.; Schuligoi, R.; Konya, V.; Sturm, G.J.; Heinemann, A. Inhibitory effect of prostaglandin I2 on bone marrow kinetics of eosinophils in the guinea pig. J. Leukoc. Biol. 2011, 90, 285–291. [Google Scholar] [CrossRef]
- Konya, V.; Sturm, E.M.; Schratl, P.; Beubler, E.; Marsche, G.; Schuligoi, R.; Lippe, I.T.; Peskar, B.A.; Heinemann, A. Endothelium-derived prostaglandin I(2) controls the migration of eosinophils. J. Allergy Clin. Immunol. 2010, 125, 1105–1113. [Google Scholar] [CrossRef]
- Baothman, B.K.; Smith, J.; Kay, L.J.; Suvarna, S.K.; Peachell, P.T. Prostaglandin D2 generation from human lung mast cells is catalysed exclusively by cyclooxygenase-1. Eur. J. Pharm. 2018, 819, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, K.M.; Cahill, K.N.; Katz, H.R.; Murphy, K.C.; Feng, C.; Lee-Sarwar, K.; Lai, J.; Bhattacharyya, N.; Israel, E.; Boyce, J.A.; et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2016, 137, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Campos-Alberto, E.; Yoshimura, T.; Bredo, G.; Rieger, A.M.; Puttagunta, L.; Barreda, D.R.; Befus, A.D.; Cameron, L. Expression of DP2 (CRTh2), a prostaglandin D(2) receptor, in human mast cells. PLoS ONE 2014, 9, e108595. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Abdu, S.; Maguire, T.J.A.; Hopkins, C.; Till, S.J.; Woszczek, G. Prostaglandin D2 receptors in human mast cells. Allergy 2019. [Google Scholar] [CrossRef]
- Feng, C.; Beller, E.M.; Bagga, S.; Boyce, J.A. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood 2006, 107, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.J.; Yeo, W.W.; Peachell, P.T. Prostaglandin E2 activates EP2 receptors to inhibit human lung mast cell degranulation. Br. J. Pharm. 2006, 147, 707–713. [Google Scholar] [CrossRef]
- Torres, R.; Picado, C.; de Mora, F. The PGE2-EP2-mast cell axis: An antiasthma mechanism. Mol. Immunol. 2015, 63, 61–68. [Google Scholar] [CrossRef]
- Bradbury, D.; Clarke, D.; Seedhouse, C.; Corbett, L.; Stocks, J.; Knox, A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J. Biol. Chem. 2005, 280, 29993–30000. [Google Scholar] [CrossRef]
- Pavord, I.D.; Wong, C.S.; Williams, J.; Tattersfield, A.E. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am. Rev. Respir. Dis. 1993, 148, 87–90. [Google Scholar] [CrossRef]
- Benyahia, C.; Gomez, I.; Kanyinda, L.; Boukais, K.; Danel, C.; Leseche, G.; Longrois, D.; Norel, X. PGE(2) receptor (EP(4)) agonists: Potent dilators of human bronchi and future asthma therapy? Pulm. Pharmacol. Ther. 2012, 25, 115–118. [Google Scholar] [CrossRef]
- Buckley, J.; Birrell, M.A.; Maher, S.A.; Nials, A.T.; Clarke, D.L.; Belvisi, M.G. EP4 receptor as a new target for bronchodilator therapy. Thorax 2011, 66, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, N.; Belvisi, M.G.; Patel, H.J.; Yacoub, M.H.; Chung, K.F.; Mitchell, J.A. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells. Relevance to asthma therapy. Am. J. Respir. Cell Mol. Biol. 2001, 24, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Aso, H.; Ito, S.; Mori, A.; Suganuma, N.; Morioka, M.; Takahara, N.; Kondo, M.; Hasegawa, Y. Differential regulation of airway smooth muscle cell migration by E-prostanoid receptor subtypes. Am. J. Respir. Cell Mol. Biol. 2013, 48, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.; Kaul, H.; Berair, R.; Gonem, S.; Singapuri, A.; Sutcliffe, A.J.; Chachi, L.; Biddle, M.S.; Kaur, D.; Bourne, M.; et al. DP2 antagonism reduces airway smooth muscle mass in asthma by decreasing eosinophilia and myofibroblast recruitment. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Smith, A.P.; Cuthbert, M.F.; Dunlop, L.S. Effects of inhaled prostaglandins E1, E2, and F2alpha on the airway resistance of healthy and asthmatic man. Clin. Sci. Mol. Med. 1975, 48, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Basili, S.; Vieri, M.; Cipollone, F.; Santarone, S.; Alessandri, C.; Gazzaniga, P.; Cordova, C.; Violi, F. Enhanced thromboxane biosynthesis in patients with chronic obstructive pulmonary disease. The Chronic Obstructive Bronchitis and Haemostasis Study Group. Am. J. Respir. Crit. Care Med. 1997, 156, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Cao, Y.; Zhang, Y.; Edvinsson, L.; Xu, C.B. Enhanced airway smooth muscle cell thromboxane receptor signaling via activation of JNK MAPK and extracellular calcium influx. Eur J. Pharm. 2011, 650, 629–638. [Google Scholar] [CrossRef]
- Han, D.Y.; Cho, J.S.; Moon, Y.M.; Lee, H.R.; Lee, H.M.; Lee, B.D.; Baek, B.J. Effect of prostaglandin e2 on vascular endothelial growth factor production in nasal polyp fibroblasts. Allergy Asthma Immunol. Res. 2013, 5, 224–231. [Google Scholar] [CrossRef]
- Roca-Ferrer, J.; Garcia-Garcia, F.J.; Pereda, J.; Perez-Gonzalez, M.; Pujols, L.; Alobid, I.; Mullol, J.; Picado, C. Reduced expression of COXs and production of prostaglandin E(2) in patients with nasal polyps with or without aspirin-intolerant asthma. J. Allergy Clin. Immunol. 2011, 128, 66–72. [Google Scholar] [CrossRef]
- Cahill, K.N.; Raby, B.A.; Zhou, X.; Guo, F.; Thibault, D.; Baccarelli, A.; Byun, H.M.; Bhattacharyya, N.; Steinke, J.W.; Boyce, J.A.; et al. Impaired E Prostanoid2 Expression and Resistance to Prostaglandin E2 in Nasal Polyp Fibroblasts from Subjects with Aspirin-Exacerbated Respiratory Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 34–40. [Google Scholar] [CrossRef]
- Kanai, K.; Okano, M.; Fujiwara, T.; Kariya, S.; Haruna, T.; Omichi, R.; Makihara, S.I.; Hirata, Y.; Nishizaki, K. Effect of prostaglandin D2 on VEGF release by nasal polyp fibroblasts. Allergol. Int. 2016, 65, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, V.J.; Popov, T.A.; Miller, D.; Weinstein, S.F.; Spector, S.; Magnusson, B.; Osuntokun, W.; Goldsmith, P.; Weiss, M.; Beier, J. The oral CRTh2 antagonist QAW039 (fevipiprant): A phase II study in uncontrolled allergic asthma. Pulm. Pharmacol. Ther. 2016, 39, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bateman, E.D.; Guerreros, A.G.; Brockhaus, F.; Holzhauer, B.; Pethe, A.; Kay, R.A.; Townley, R.G. Fevipiprant, an oral prostaglandin DP2 receptor (CRTh2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Gonem, S.; Berair, R.; Singapuri, A.; Hartley, R.; Laurencin, M.F.M.; Bacher, G.; Holzhauer, B.; Bourne, M.; Mistry, V.; Pavord, I.D.; et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: A single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir. Med. 2016, 4, 699–707. [Google Scholar] [CrossRef]
- Wenzel, S.; Chantry, D.; Eberhardt, C.; Hopkins, R.; Saunders, M.; Anderson, L.; Aitchinson, R.; Bell, S.; Izuhara, K.; Ono, J.; et al. ARRY-502, a potent, selective, oral CRTh2 antagonist reduces Th2 mediators in patients with mild to moderate Th2-driven asthma. Eur. Respir. J. 2014, 44, 4836. [Google Scholar]
- Kuna, P.; Bjermer, L.; Tornling, G. Two Phase II randomized trials on the CRTh2 antagonist AZD1981 in adults with asthma. Drug Des. c. 2016, 10, 2759–2770. [Google Scholar] [CrossRef]
- Pettipher, R.; Hunter, M.G.; Perkins, C.M.; Collins, L.P.; Lewis, T.; Baillet, M.; Steiner, J.; Bell, J.; Payton, M.A. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy 2014, 69, 1223–1232. [Google Scholar] [CrossRef]
- Horak, F.; Zieglmayer, P.; Zieglmayer, R.; Lemell, P.; Collins, L.P.; Hunter, M.G.; Steiner, J.; Lewis, T.; Payton, M.A.; Perkins, C.M.; et al. The CRTH2 antagonist OC000459 reduces nasal and ocular symptoms in allergic subjects exposed to grass pollen, a randomised, placebo-controlled, double-blind trial. Allergy 2012, 67, 1572–1579. [Google Scholar] [CrossRef]
- Ortega, H.; Fitzgerald, M.; Raghupathi, K.; Tompkins, C.A.; Shen, J.; Dittrich, K.; Pattwell, C.; Singh, D. A phase 2 study to evaluate the safety, efficacy and pharmacokinetics of DP2 antagonist GB001 and to explore biomarkers of airway inflammation in mild-to-moderate asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef]
- Hall, I.P.; Fowler, A.V.; Gupta, A.; Tetzlaff, K.; Nivens, M.C.; Sarno, M.; Finnigan, H.A.; Bateman, E.D.; Rand Sutherland, E. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm. Pharmacol. Ther. 2015, 32, 37–44. [Google Scholar] [CrossRef]
- Diamant, Z.; Sidharta, P.N.; Singh, D.; O’Connor, B.J.; Zuiker, R.; Leaker, B.R.; Silkey, M.; Dingemanse, J. Setipiprant, a selective CRTH2 antagonist, reduces allergen-induced airway responses in allergic asthmatics. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2014, 44, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Ratner, P.; Andrews, C.P.; Hampel, F.C.; Martin, B.; Mohar, D.E.; Bourrelly, D.; Danaietash, P.; Mangialaio, S.; Dingemanse, J.; Hmissi, A.; et al. Efficacy and safety of setipiprant in seasonal allergic rhinitis: Results from Phase 2 and Phase 3 randomized, double-blind, placebo- and active-referenced studies. Allergy Asthma Clin. Immunol. 2017, 13, 18. [Google Scholar] [CrossRef]
- Terada, N.; Yamakoshi, T.; Hasegawa, M.; Tanikawa, H.; Maesako, K.; Ishikawa, K.; Konno, A. The effect of ramatroban (BAY u 3405), a thromboxane A2 receptor antagonist, on nasal cavity volume and minimum cross-sectional area and nasal mucosal hemodynamics after nasal mucosal allergen challenge in patients with perennial allergic rhinitis. Acta Otolaryngol. Suppl. 1998, 537, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Yamakoshi, T.; Hasegawa, M.; Tanikawa, H.; Nagata, H.; Maesako, K.-i.; Konno, A. Effect of a thromboxane A2 receptor antagonist ramatroban (BAY u 3405), on inflammatory cells, chemical mediators and non-specific nasal hyperreactivity after allergen challenge in patients with perennial allergic rhinitis. Allergol. Int. 1998, 47, 59–67. [Google Scholar] [CrossRef][Green Version]
- Aizawa, H.; Inoue, H.; Nakano, H.; Matsumoto, K.; Yoshida, M.; Fukuyama, S.; Koto, H.; Hara, N. Effects of thromboxane A2 antagonist on airway hyperresponsiveness, exhaled nitric oxide, and induced sputum eosinophils in asthmatics. Prostaglandins Leukot Essent Fat. Acids 1998, 59, 185–190. [Google Scholar] [CrossRef]
- Hoshino, M.; Sim, J.; Shimizu, K.; Nakayama, H.; Koya, A. Effect of AA-2414, a thromboxane A2 receptor antagonist, on airway inflammation in subjects with asthma. J. Allergy Clin. Immunol. 1999, 103, 1054–1061. [Google Scholar] [CrossRef]
- Okubo, K.; Hashiguchi, K.; Takeda, T.; Baba, K.; Kitagoh, H.; Miho, H.; Tomomatsu, H.; Yamaguchi, S.; Odani, M.; Yamamotoya, H. A randomized controlled phase II clinical trial comparing ONO-4053, a novel DP1 antagonist, with a leukotriene receptor antagonist pranlukast in patients with seasonal allergic rhinitis. Allergy 2017, 72, 1565–1575. [Google Scholar] [CrossRef]
- Bianco, S.; Robuschi, M.; Grugni, A.; Ceserani, R.; Gandolfi, C. Effect of prostacyclin on antigen induced immediate bronchoconstriction in asthmatic patients. Prostaglandins Med. 1979, 3, 39–45. [Google Scholar] [CrossRef]
- Liu, M.C.; Bleecker, E.R.; Lichtenstein, L.M.; Kagey-Sobotka, A.; Niv, Y.; McLemore, T.L.; Permutt, S.; Proud, D.; Hubbard, W.C. Evidence for elevated levels of histamine, prostaglandin D2, and other bronchoconstricting prostaglandins in the airways of subjects with mild asthma. Am. Rev. Respir. Dis. 1990, 142, 126–132. [Google Scholar] [CrossRef]
- Liu, M.C.; Hubbard, W.C.; Proud, D.; Stealey, B.A.; Galli, S.J.; Kagey-Sobotka, A.; Bleecker, E.R.; Lichtenstein, L.M. Immediate and late inflammatory responses to ragweed antigen challenge of the peripheral airways in allergic asthmatics. Cellular, mediator, and permeability changes. Am. Rev. Respir. Dis. 1991, 144, 51–58. [Google Scholar] [CrossRef]
- Fajt, M.L.; Gelhaus, S.L.; Freeman, B.; Uvalle, C.E.; Trudeau, J.B.; Holguin, F.; Wenzel, S.E. Prostaglandin D(2) pathway upregulation: Relation to asthma severity, control, and TH2 inflammation. J. Allergy Clin. Immunol. 2013, 131, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Pavord, I.D.; Ward, R.; Woltmann, G.; Wardlaw, A.J.; Sheller, J.R.; Dworski, R. Induced sputum eicosanoid concentrations in asthma. Am. J. Respir. Crit. Care Med. 1999, 160, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Galdiero, M.R.; Pecoraro, A.; Pucino, V.; Criscuolo, G.; Triassi, M.; Varricchi, G. Prostaglandin D2 receptor antagonists in allergic disorders: Safety, efficacy, and future perspectives. Expert Opin. Investig. Drugs 2019, 28, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wendell, S.G.; Fan, H.; Zhang, C. G Protein-Coupled Receptors in Asthma Therapy: Pharmacology and Drug Action. Pharmacol. Rev. 2020, 72, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Min, J.; Jiang, H.; Mao, B. Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) antagonists in asthma: A systematic review and meta-analysis protocol. Bmj Open 2018, 8, e020882. [Google Scholar] [CrossRef] [PubMed]
- Murillo, J.C.; Dimov, V.; Gonzalez-Estrada, A. An evaluation of fevipiprant for the treatment of asthma: A promising new therapy? Expert Opin. Pharmacother. 2018, 19, 2087–2093. [Google Scholar] [CrossRef]
- White, C.; Wright, A.; Brightling, C. Fevipiprant in the treatment of asthma. Expert Opin. Investig. Drugs 2018, 27, 199–207. [Google Scholar] [CrossRef]
- Erpenbeck, V.J.; Popov, T.A.; Miller, D.; Weinstein, S.F.; Spector, S.; Magnusson, B.; Osuntokun, W.; Goldsmith, P.; Weiss, M.; Beier, J. Data on the oral CRTh2 antagonist QAW039 (fevipiprant) in patients with uncontrolled allergic asthma. Data Brief. 2016, 9, 199–205. [Google Scholar] [CrossRef][Green Version]
- Kao, C.C.; Parulekar, A.D. Spotlight on fevipiprant and its potential in the treatment of asthma: Evidence to date. J. Asthma Allergy 2019, 12, 1–5. [Google Scholar] [CrossRef]
- Pelaia, C.; Crimi, C.; Vatrella, A.; Busceti, M.T.; Gaudio, A.; Garofalo, E.; Bruni, A.; Terracciano, R.; Pelaia, G. New treatments for asthma: From the pathogenic role of prostaglandin d2 to the therapeutic effects of fevipiprant. Pharmacol. Res. 2019, 155, 104490. [Google Scholar] [CrossRef]
- Study of Efficacy and Safety of QAW039 in Patients with Severe Asthma Inadequately Controlled with Standard of Care Asthma Treatment. Available online: https://clinicaltrials.gov/ct2/show/NCT02555683 (accessed on 8 January 2020).
- Study of Efficacy and Safety of QAW039 in Patients with Severe Asthma Inadequately Controlled with Standard of Care Asthma Treatment. Available online: https://clinicaltrials.gov/ct2/show/NCT02563067 (accessed on 8 January 2020).
- Study of Efficacy and Safety of QAW039 When Added to Standard-of-Care Asthma Therapy in Patients with Uncontrolled Asthma. Available online: https://clinicaltrials.gov/ct2/show/NCT03215758 (accessed on 8 January 2020).
- Study of Efficacy and Safety of QAW039 When Added to Standard-of-Care Asthma Therapy in Patients with Uncontrolled Asthma. Available online: https://clinicaltrials.gov/ct2/show/NCT03226392 (accessed on 8 January 2020).
- Asano, K.; Sagara, H.; Ichinose, M.; Hirata, M.; Nakajima, A.; Ortega, H.; Tohda, Y. A Phase 2a Study of DP2 antagonist GB001 for Asthma. J. Allergy Clin. Immunol. Pract. 2019. [Google Scholar] [CrossRef] [PubMed]
- Langfang Xinghe Industry Co., Ltd. Study of the Tolerability and Pharmacokinetic of ZL-2102 with an Investigation of Food Effect in Healthy Male Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT02397005 (accessed on 8 January 2020).
- Aggarwal, S.; Moodley, Y.P.; Thompson, P.J.; Misso, N.L. Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum: Association with asthma severity and eosinophilic inflammation. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2010, 40, 85–93. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Watson, R.M.; O’Byrne, P.M. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am. J. Respir. Crit. Care Med. 1999, 159, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, W.; Szmidt, M. A six week double blind, placebo controlled, crossover study of the effect of misoprostol in the treatment of aspirin sensitive asthma. Thorax 1999, 54, 900–904. [Google Scholar] [CrossRef][Green Version]
- Mathe, A.A.; Hedqvist, P.; Holmgren, A.; Svanborg, N. Bronchial hyperreactivity to prostaglandin F 2 and histamine in patients with asthma. Br. Med. J. 1973, 1, 193–196. [Google Scholar] [CrossRef]
- Kharitonov, S.A.; Sapienza, M.A.; Barnes, P.J.; Chung, K.F. Prostaglandins E2 and F2alpha reduce exhaled nitric oxide in normal and asthmatic subjects irrespective of airway caliber changes. Am. J. Respir. Crit. Care Med. 1998, 158, 1374–1378. [Google Scholar] [CrossRef]
- Fujimura, M.; Ozawa, S.; Matsuda, T. Effect of oral administration of a prostacyclin analog (OP-41483) on pulmonary function and bronchial responsiveness in stable asthmatic subjects. J. Asthma 1991, 28, 419–424. [Google Scholar] [CrossRef]
- Bianco, S.; Robuschi, M.; Ceserani, R.; Gandolfi, C. Effects of prostacyclin on aspecifically and specifically induced bronchoconstriction in asthmatic patients. Eur J. Respir. Dis. Suppl. 1980, 106, 81–87. [Google Scholar]
- Hardy, C.C.; Bradding, P.; Robinson, C.; Holgate, S.T. Bronchoconstrictor and antibronchoconstrictor properties of inhaled prostacyclin in asthma. J. Appl. Physiol. (1985) 1988, 64, 1567–1574. [Google Scholar] [CrossRef]
- Naclerio, R.M.; Creticos, P.S.; Norman, P.S.; Lichtenstein, L.M. Mediator release after nasal airway challenge with allergen. Am. Rev. Respir. Dis. 1986, 134, 1102. [Google Scholar] [CrossRef]
- Shirasaki, H.; Kikuchi, M.; Kanaizumi, E.; Himi, T. Accumulation of CRTH2-positive leukocytes in human allergic nasal mucosa. Ann. Allergy Asthma Immunol. 2009, 102, 110–115. [Google Scholar] [CrossRef]
- Ciebiada, M.; Gorski, P.; Antczak, A. Evaluation of eicosanoids in nasal lavage as biomarkers of inflammation in patients with allergic rhinitis. Arch. Med. Sci. 2014, 10, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L.; Smith, S.; Harrison, J.; Ritter, W.; Howarth, P.H. The effect of BAY u 3405, a thromboxane receptor antagonist, on prostaglandin D2-induced nasal blockage. J. Allergy Clin. Immunol. 1993, 91, 903–909. [Google Scholar] [CrossRef]
- Kajiwara, D.; Aoyagi, H.; Shigeno, K.; Togawa, M.; Tanaka, K.; Inagaki, N.; Miyoshi, K. Role of hematopoietic prostaglandin D synthase in biphasic nasal obstruction in guinea pig model of experimental allergic rhinitis. Eur. J. Pharm. 2011, 667, 389–395. [Google Scholar] [CrossRef]
- Nabe, T.; Kuriyama, Y.; Mizutani, N.; Shibayama, S.; Hiromoto, A.; Fujii, M.; Tanaka, K.; Kohno, S. Inhibition of hematopoietic prostaglandin D synthase improves allergic nasal blockage in guinea pigs. Prostaglandins Other Lipid Mediat. 2011, 95, 27–34. [Google Scholar] [CrossRef]
- Steinke, J.W.; Wilson, J.M. Aspirin-exacerbated respiratory disease: Pathophysiological insights and clinical advances. J. Asthma Allergy 2016, 9, 37–43. [Google Scholar] [CrossRef][Green Version]
- Li, K.L.; Lee, A.Y.; Abuzeid, W.M. Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management. Med. Sci. 2019, 7, 45. [Google Scholar] [CrossRef]
- Steinke, J.W.; Borish, L. Factors driving the aspirin exacerbated respiratory disease phenotype. Am. J. Rhinol. Allergy 2015, 29, 35–40. [Google Scholar] [CrossRef]
- Parker, A.R.; Ayars, A.G.; Altman, M.C.; Henderson, W.R., Jr. Lipid Mediators in Aspirin-Exacerbated Respiratory Disease. Immunol. Allergy Clin. North. Am. 2016, 36, 749–763. [Google Scholar] [CrossRef]
- Mastalerz, L.; Tyrak, K.E.; Ignacak, M.; Konduracka, E.; Mejza, F.; Cmiel, A.; Buczek, M.; Kot, A.; Oles, K.; Sanak, M. Prostaglandin E2 decrease in induced sputum of hypersensitive asthmatics during oral challenge with aspirin. Allergy 2019, 74, 922–932. [Google Scholar] [CrossRef]
- Adamusiak, A.M.; Stasikowska-Kanicka, O.; Lewandowska-Polak, A.; Danilewicz, M.; Wagrowska-Danilewicz, M.; Jankowski, A.; Kowalski, M.L.; Pawliczak, R. Expression of arachidonate metabolism enzymes and receptors in nasal polyps of aspirin-hypersensitive asthmatics. Int. Arch. Allergy Immunol. 2012, 157, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Machado-Carvalho, L.; Torres, R.; Perez-Gonzalez, M.; Alobid, I.; Mullol, J.; Pujols, L.; Roca-Ferrer, J.; Picado, C. Altered expression and signalling of EP2 receptor in nasal polyps of AERD patients: Role in inflammation and remodelling. Rhinology 2016, 54, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.M.; Simon, R.A.; Woessner, K.M.; Wineinger, N.E.; White, A.A. Effect of misoprostol on patients with aspirin-exacerbated respiratory disease undergoing aspirin challenge and desensitization. Ann. Allergy Asthma Immunol. 2017, 119, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.N.; Bensko, J.C.; Boyce, J.A.; Laidlaw, T.M. Prostaglandin D(2): A dominant mediator of aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2015, 135, 245–252. [Google Scholar] [CrossRef]
- Szczeklik, A.; Sladek, K.; Dworski, R.; Nizankowska, E.; Soja, J.; Sheller, J.; Oates, J. Bronchial aspirin challenge causes specific eicosanoid response in aspirin-sensitive asthmatics. Am. J. Respir. Crit. Care Med. 1996, 154, 1608–1614. [Google Scholar] [CrossRef]
- Rusznak, M.; Peebles, R.S., Jr. Prostaglandin E2 in NSAID-exacerbated respiratory disease: Protection against cysteinyl leukotrienes and group 2 innate lymphoid cells. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 38–45. [Google Scholar] [CrossRef]
- Bochenek, G.; Nagraba, K.; Nizankowska, E.; Szczeklik, A. A controlled study of 9alpha,11beta-PGF2 (a prostaglandin D2 metabolite) in plasma and urine of patients with bronchial asthma and healthy controls after aspirin challenge. J. Allergy Clin. Immunol. 2003, 111, 743–749. [Google Scholar] [CrossRef]
- Nizankowska, E.; Czerniawska-Mysik, G.; Szczeklik, A. Lack of effect of i.v. prostacyclin on aspirin-induced asthma. Eur. J. Respir. Dis. 1986, 69, 363–368. [Google Scholar]
- Xie, L.; Liu, A.G.; Cui, Y.H.; Zhang, Y.P.; Liao, B.; Li, N.N.; Wang, X.S. Expression profiles of prostaglandin E2 receptor subtypes in aspirin tolerant adult Chinese with chronic rhinosinusitis. Am. J. Rhinol. Allergy 2015, 29, 322–328. [Google Scholar] [CrossRef]
- Perez-Novo, C.A.; Watelet, J.B.; Claeys, C.; Van Cauwenberge, P.; Bachert, C. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J. Allergy Clin. Immunol. 2005, 115, 1189–1196. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, G.E.; Lee, B.J.; Kwon, S.W.; Lee, S.H.; Kim, H.S.; Jang, Y.J. Natural killer cells regulate eosinophilic inflammation in chronic rhinosinusitis. Sci. Rep. 2016, 6, 27615. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Fujiwara, T.; Yamamoto, M.; Sugata, Y.; Matsumoto, R.; Fukushima, K.; Yoshino, T.; Shimizu, K.; Eguchi, N.; Kiniwa, M.; et al. Role of prostaglandin D2 and E2 terminal synthases in chronic rhinosinusitis. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2006, 36, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Meng, Q.; Scadding, G.; Parikh, A.; Corrigan, C.J.; Lee, T.H. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J. Allergy Clin. Immunol. 2006, 117, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Machado-Carvalho, L.; Roca-Ferrer, J.; Picado, C. Prostaglandin E2 receptors in asthma and in chronic rhinosinusitis/nasal polyps with and without aspirin hypersensitivity. Respir. Res. 2014, 15, 100. [Google Scholar] [CrossRef]
- Xie, L.; Liu, A.G.; Peng, L.Y.; Wang, S.J.; Zhang, Y.P.; Wang, X.S. Expression of E-prostanoid receptors in nasal polyp tissues of smoking and nonsmoking patients with chronic rhinosinusitis. PLoS ONE 2018, 13, e0200989. [Google Scholar] [CrossRef]
- Yamamoto, M.; Okano, M.; Fujiwara, T.; Kariya, S.; Higaki, T.; Nagatsuka, H.; Tsujigiwa, H.; Yamada, M.; Yoshino, T.; Urade, Y.; et al. Expression and characterization of PGD2 receptors in chronic rhinosinusitis: Modulation of DP and CRTH2 by PGD2. Int. Arch. Allergy Immunol. 2009, 148, 127–136. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Production | Receptor | Downstream Signaling | Receptor Expression |
|---|---|---|---|---|
| PGD2 | mast cells, eosinophils, T cells, dendritic cells, macrophages, endothelial cells, platelets, lung parenchyma | DP1 | ↑cAMP | mucus-secreting goblet cells, nasal serous glands, vascular endothelial cells, T cells, dendritic cells, eosinophils |
| DP2 (CRTH2) | ↓cAMP, ↑Ca2+ | T cells, basophils, eosinophils, ILC2 | ||
| PGE2 | epithelial cells, fibroblasts, macrophage, smooth muscle cells, platelets | EP1 | ↑Ca2+ | T cells, dendritic cells, B cells, smooth muscle cells |
| EP2 | ↑cAMP | T cells, dendritic cells, B cells, ILC2, mast cells, basophils, smooth muscle cells | ||
| EP3 | ↓cAMP | T cells, B cells, dendritic cells, smooth muscle cells | ||
| EP4 | ↑cAMP | T cells, B cells, dendritic cells, smooth muscle cells | ||
| PGF2α | lung parenchyma, vascular smooth muscle cells, peripheral blood lymphocytes | FP | ↑IP3/DAG/Ca2+ | none |
| PGI2 | endothelial cells, vascular smooth muscle cells, lung parenchyma | IP | ↑cAMP | T cells, dendritic cells, B cells, ILC2, endothelial cells, platelets |
| TXA2 | platelets, vascular smooth muscle cells, macrophages | TP | ↑IP3/DAG/Ca2+, ↑↓cAMP | megakaryocytes, monocytes |
| Drug and Dose | Indication (Sample Size) | Key Results | Ref. |
|---|---|---|---|
| DP2 antagonist | |||
| Fevipiprant (QAW039), oral administration, 500 mg daily for 28 days | Mild to moderate uncontrolled allergic asthma (n = 170) | Improvement of lung function in patients with FEV1 <70% | [132] |
| Fevipiprant (QAW039), oral administration, 1–450 mg daily or 2–150 mg twice daily, with inhaled budesonide 200 µg twice a day, for 12 weeks | Allergic asthma uncontrolled by a low-dose inhaled corticosteroid (n = 2598) | Total daily dose of 150 mg (150 mg once or 75 mg twice per a day) showed an improvement in forced expiratory volume | [133] |
| Fevipiprant (QAW039), oral administration, 225 mg twice daily for 12 weeks | Moderate to severe asthma with serum eosinophil ≥ 2% (n = 61) | Decreased sputum eosinophil count | [134] |
| ARRY-502, oral administration, 200 mg twice daily for four weeks | Mild allergic asthma (n = 184) | Reduction of FeNO level and decreased serum markers of Th2 inflammation | [135] |
| AZD1981, oral administration, 100 mg twice daily | Stable asthma withdrawn from inhaled corticosteroid (n = 209) | No efficacy on morning peak expiratory flow | [136] |
| AZD1981, oral administration, 50–1000 mg twice daily, for four weeks, with an inhaled corticosteroid | Asthma uncontrolled by inhaled corticosteroid (n = 510) | 400 mg group showed improved FEV1, significant improvement in questionnaire score and FEV1 in atopic subgroup | [136] |
| OC000549, oral administration, 25 mg daily/200 mg daily/100 mg twice daily, for 12 weeks, with use of short-acting β2 agonist | Mild to moderate asthma (n = 460) | Improved FEV1 (prominent in eosinophilic subjects), lower incidence of symptom exacerbation and respiratory infection | [137] |
| OC000549, oral administration, 200 mg twice daily for eight days | Seasonal allergic rhinitis (n = 35) | Reduced grass-pollen induced nasal and ocular symptoms | [138] |
| GB001, oral administration, 30 mg daily for 28 days, with use of low dose inhaled fluticasone propionate | Mild to moderate atopic asthma (n = 36) | Improved FEV1 (prominent in patients with high FeNO or high blood eosinophil) | [139] |
| BI671800, oral administration, 50/200/400 mg twice daily for six weeks | Mild to moderate asthma (n = 389) | Greater improvement in FEV1 compared to moderate doses of fluticasone | [140] |
| BI671800, oral administration, 400 mg twice daily with inhaled fluticasone (88 µg) | Mild to moderate asthma with inhaled corticosteroid (n = 243) | Improvement in FEV1 compared to placebo; however, not significantly improved over montelukast | [140] |
| Setipiprant (ACT-129968), oral administration, 1000 mg twice daily for five days, washout period of three weeks | Stable allergic asthma (n = 15) | Reduction in both allergen-induced late asthmatic responses and airway hyper-responsiveness | [141] |
| Setipiprant (ACT-129968), oral administration, 100/500/1000 mg twice daily or 1000 mg daily for two weeks | Seasonal allergic rhinitis (n = 557) | Dose-related improvements in both nasal and ocular symptom scores | [142] |
| Setipiprant (ACT-129968), oral administration, 1000 mg twice daily for two weeks | Seasonal allergic rhinitis (n = 604) | No significant effect on either nasal or ocular symptom scores | [142] |
| Dual antagonist for DP2 and TP | |||
| Ramatroban (BYAu3405), oral administration, 150 mg twice daily for four weeks | Perianal allergic rhinitis (n = 10) | Inhibitory effect on allergen challenge-induced nasal mucosal swelling | [143] |
| Ramatroban (BYAu3405), oral administration, 150 mg twice daily for four weeks | Perianal allergic rhinitis (n = 11) | Inhibitory effect on histamine-induced nasal reactivity, decreased eosinophil counts in nasal lavage fluid | [144] |
| TP antagonist | |||
| Seratrodast, oral administration, 80 mg daily for four weeks | Asthma (n = 14) | Decreased airway hyper-responsiveness, no definite effect on exhaled nitric oxide and sputum eosinophils | [145] |
| AA02414, oral administration, 80 mg daily for four months | Asthma (n = 31) | Improved symptom score, peak expiratory flow, and bronchial responsiveness to metacholine, decreased activated eosinophil infiltration | [146] |
| DP1 antagonist | |||
| ONO-4053, oral administration, 300 mg daily for two weeks | Seasonal allergic rhinitis (Japanese cedar pollen) (n = 200) | Greater improvement in nasal symptoms compared to either placebo or pranlukast | [147] |
| PGI2 analogue | |||
| OP-41483, oral administration, 200 µg 4 times daily for four weeks | Stable asthma (n = 8) | No direct effect on bronchial responsiveness | [148] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Lee, S.H.; Kim, T.H. The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy. Int. J. Mol. Sci. 2020, 21, 1851. https://doi.org/10.3390/ijms21051851
Lee K, Lee SH, Kim TH. The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy. International Journal of Molecular Sciences. 2020; 21(5):1851. https://doi.org/10.3390/ijms21051851
Chicago/Turabian StyleLee, Kijeong, Sang Hag Lee, and Tae Hoon Kim. 2020. "The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy" International Journal of Molecular Sciences 21, no. 5: 1851. https://doi.org/10.3390/ijms21051851
APA StyleLee, K., Lee, S. H., & Kim, T. H. (2020). The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy. International Journal of Molecular Sciences, 21(5), 1851. https://doi.org/10.3390/ijms21051851