Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology

 , ,
, ,  and
and 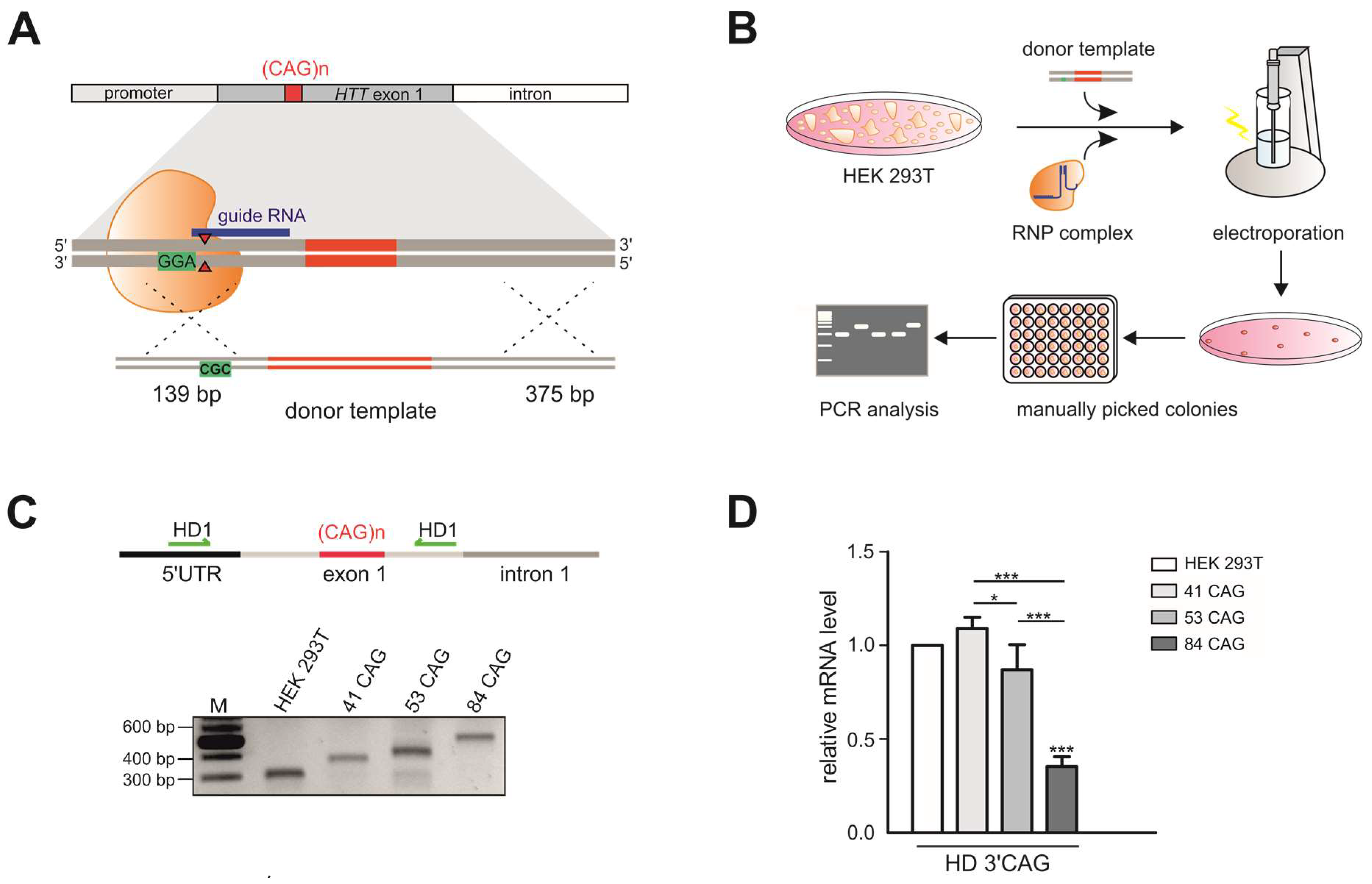{kind=link}
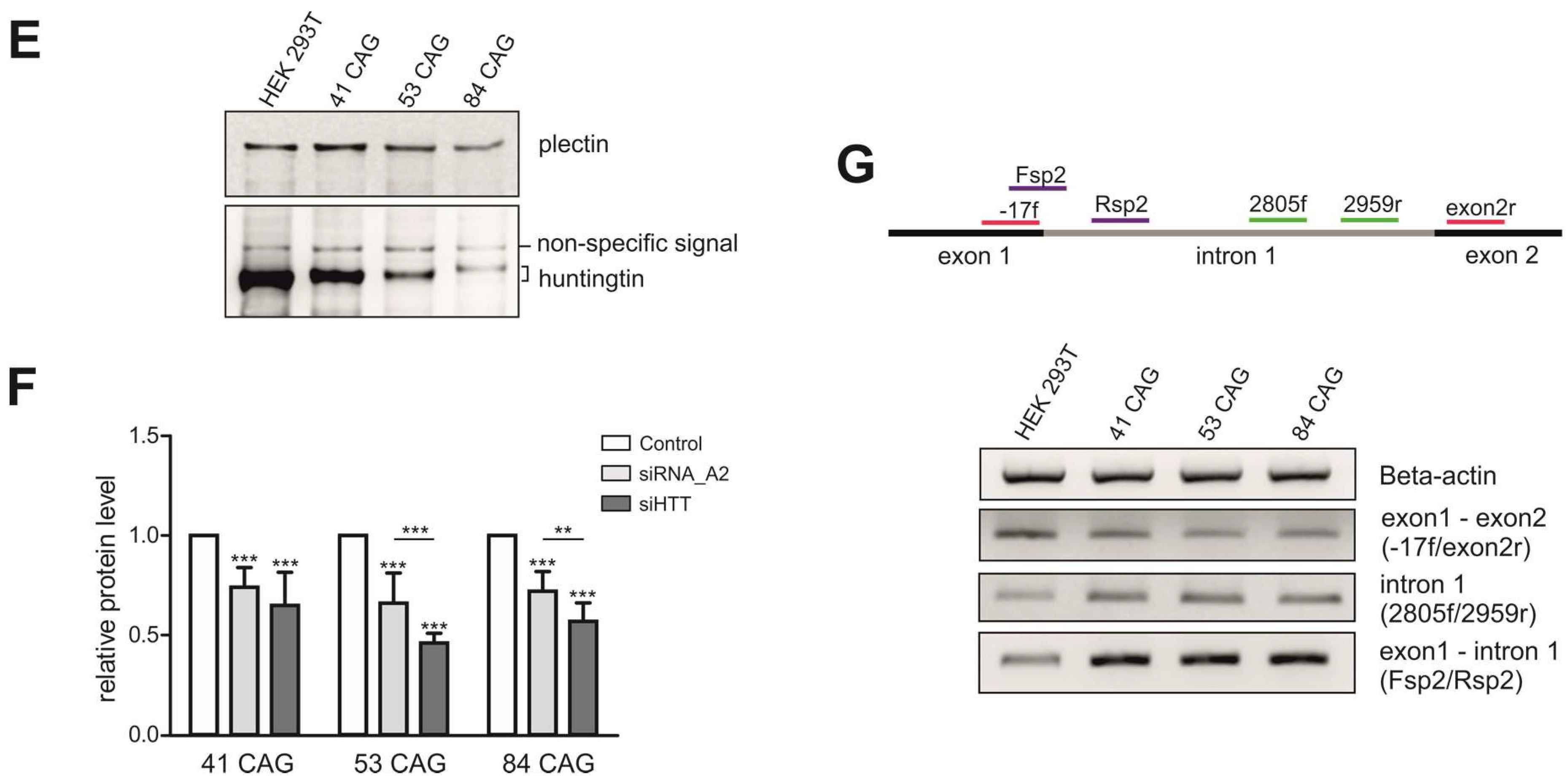{kind=link}
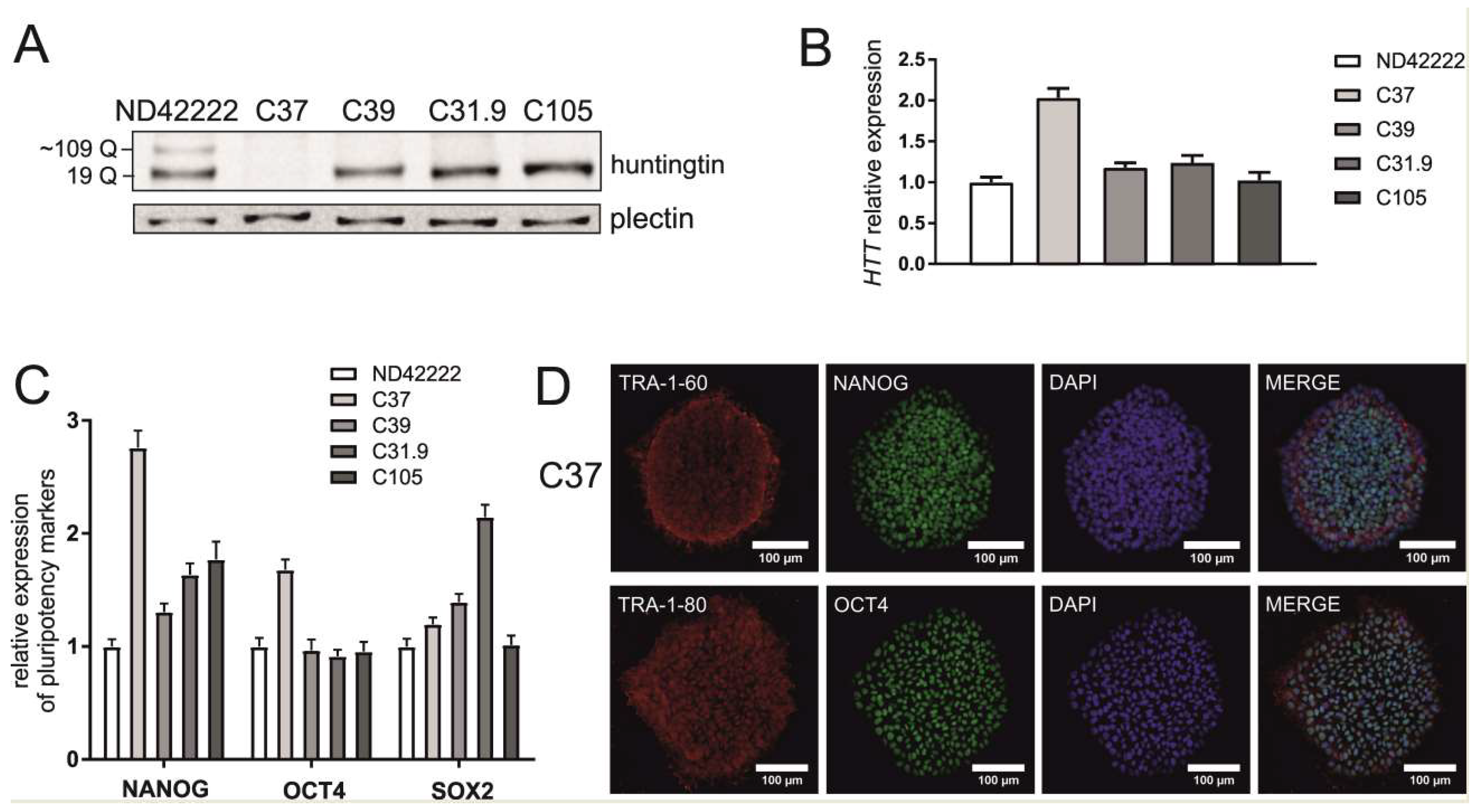{kind=link}
Abstract
Share and Cite
Dabrowska, M.; Ciolak, A.; Kozlowska, E.; Fiszer, A.; Olejniczak, M. Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology. Int. J. Mol. Sci. 2020, 21, 1854. https://doi.org/10.3390/ijms21051854
Dabrowska M, Ciolak A, Kozlowska E, Fiszer A, Olejniczak M. Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology. International Journal of Molecular Sciences. 2020; 21(5):1854. https://doi.org/10.3390/ijms21051854
Chicago/Turabian StyleDabrowska, Magdalena, Agata Ciolak, Emilia Kozlowska, Agnieszka Fiszer, and Marta Olejniczak. 2020. "Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology" International Journal of Molecular Sciences 21, no. 5: 1854. https://doi.org/10.3390/ijms21051854
APA StyleDabrowska, M., Ciolak, A., Kozlowska, E., Fiszer, A., & Olejniczak, M. (2020). Generation of New Isogenic Models of Huntington’s Disease Using CRISPR-Cas9 Technology. International Journal of Molecular Sciences, 21(5), 1854. https://doi.org/10.3390/ijms21051854