Biology and Therapeutic Targets of Colorectal Serrated Adenocarcinoma; Clues for a Histologically Based Treatment against an Aggressive Tumor

 ,
,
Abstract
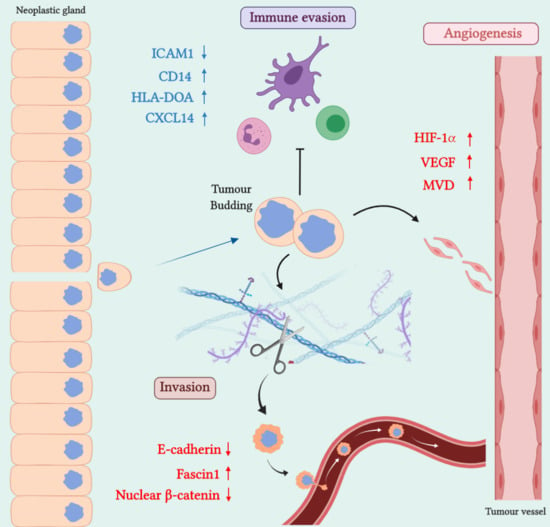
:
1. Introduction
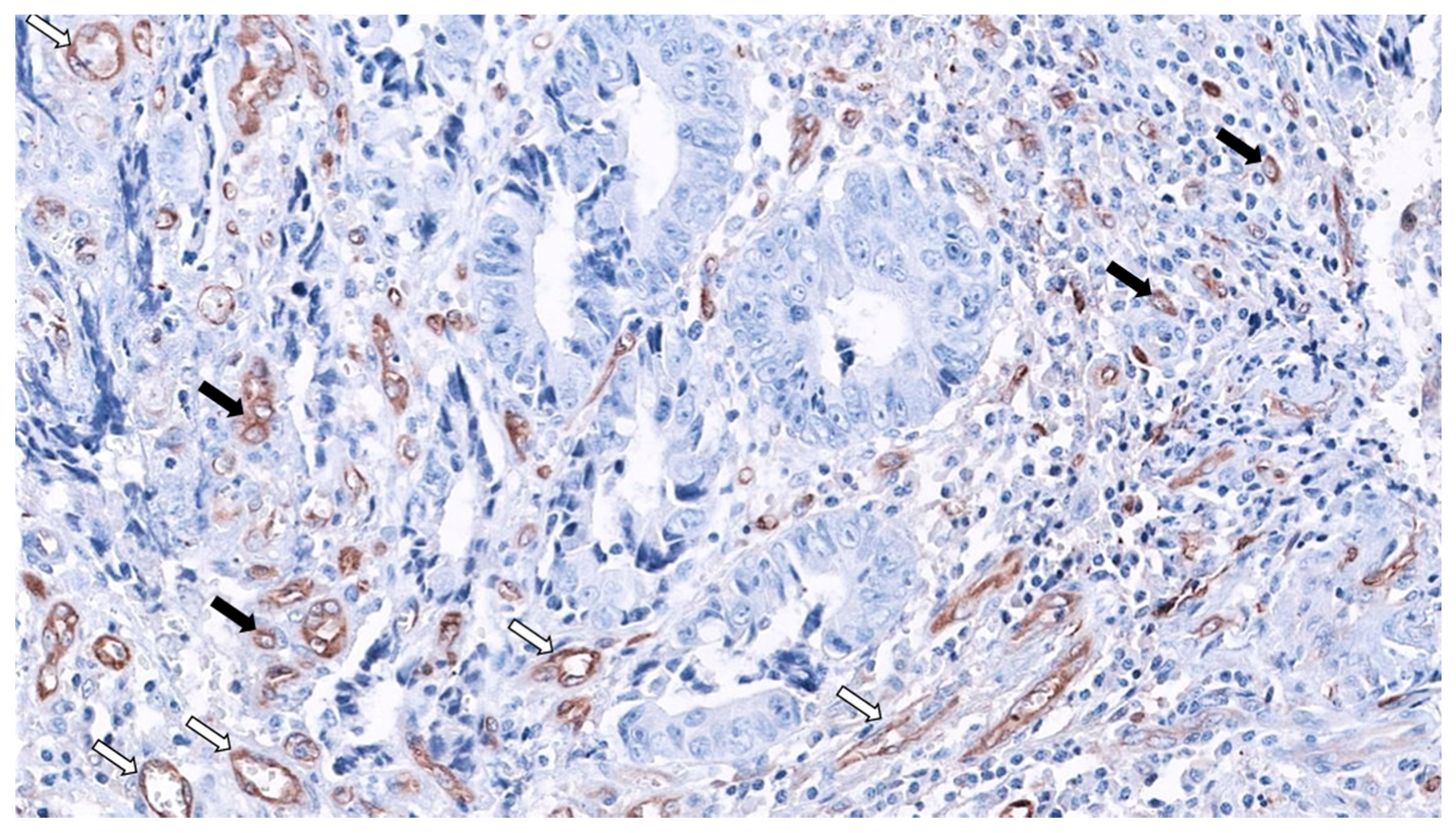
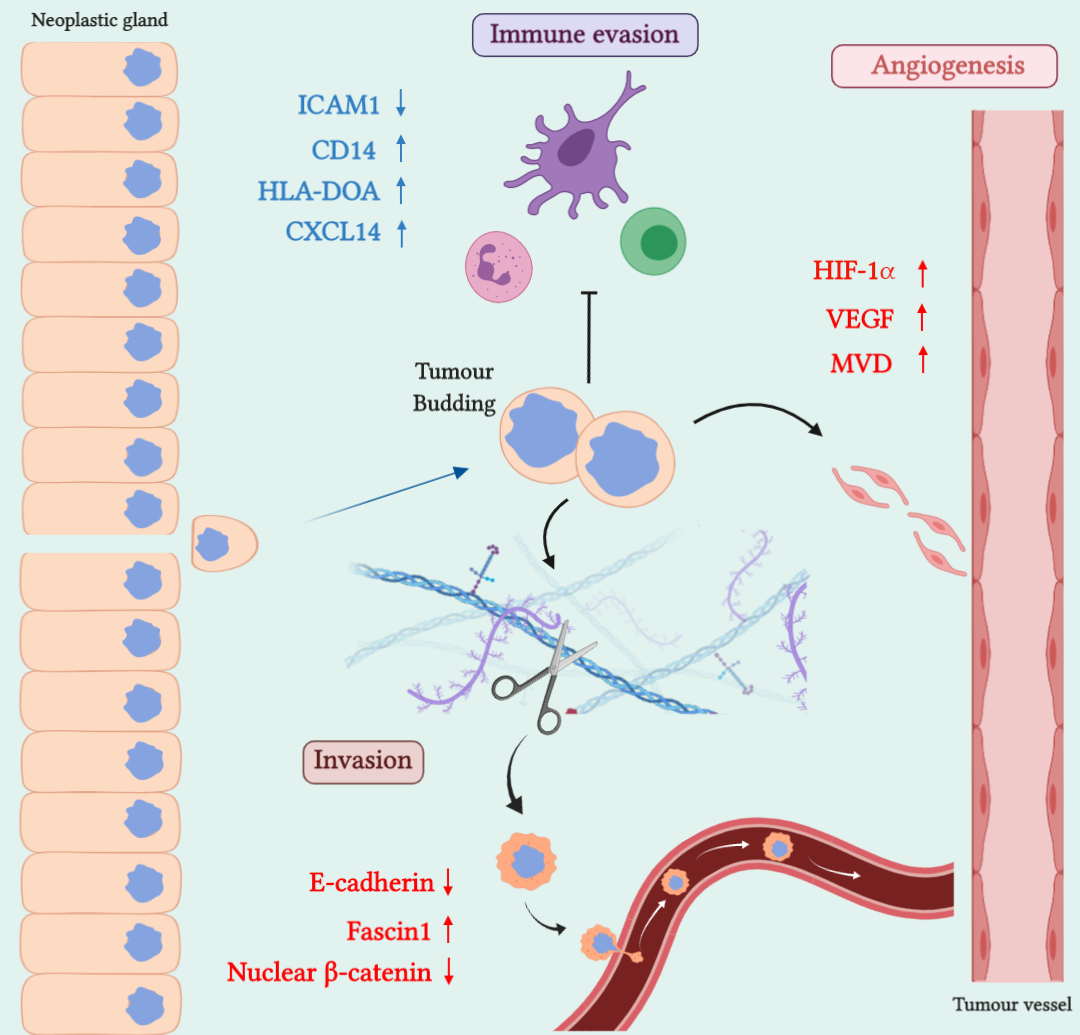
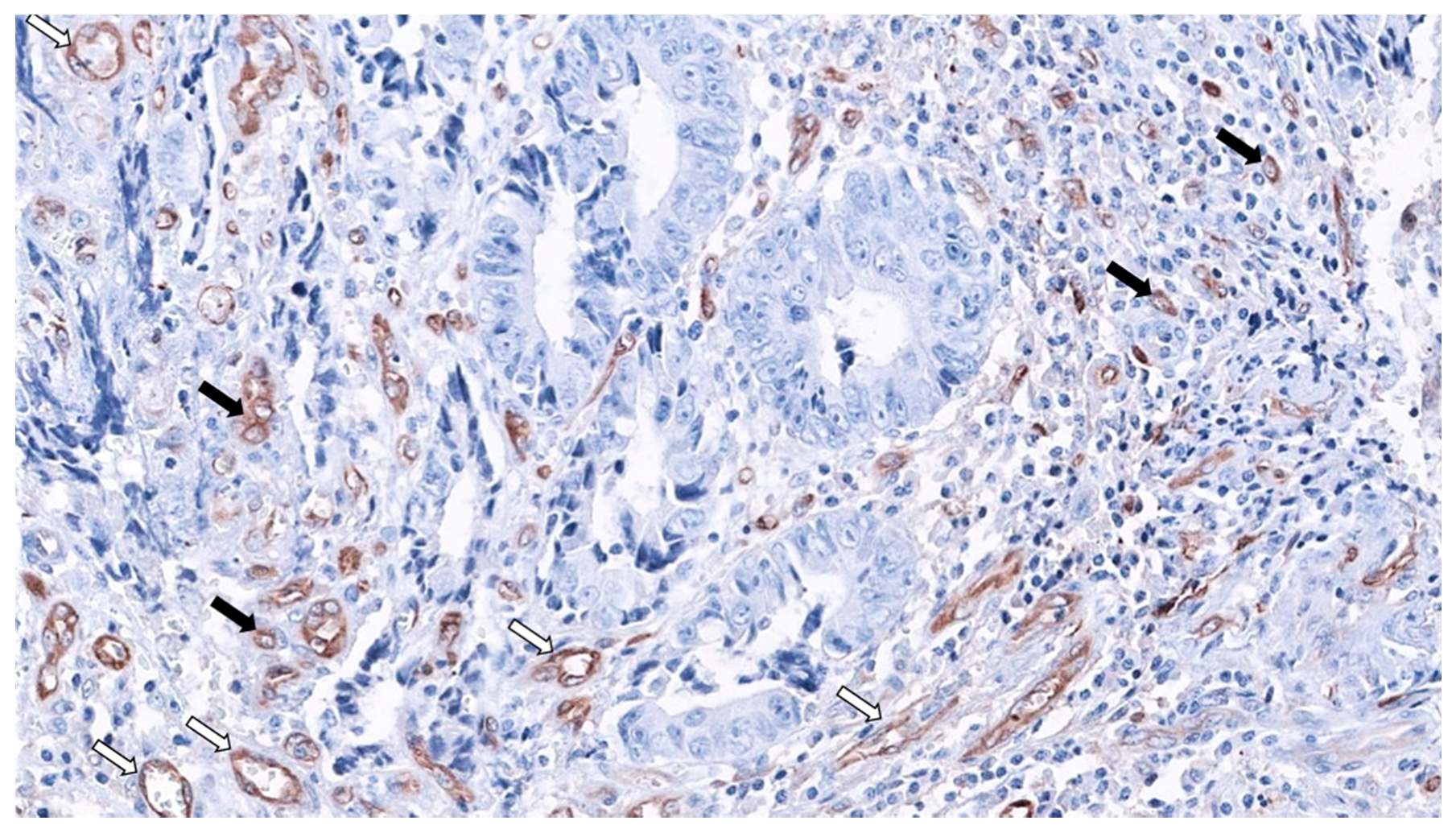
2. SAC Shows an Upregulation of Angiogenesis Markers
Therapeutic Opportunities Against Angiogenesis in SAC
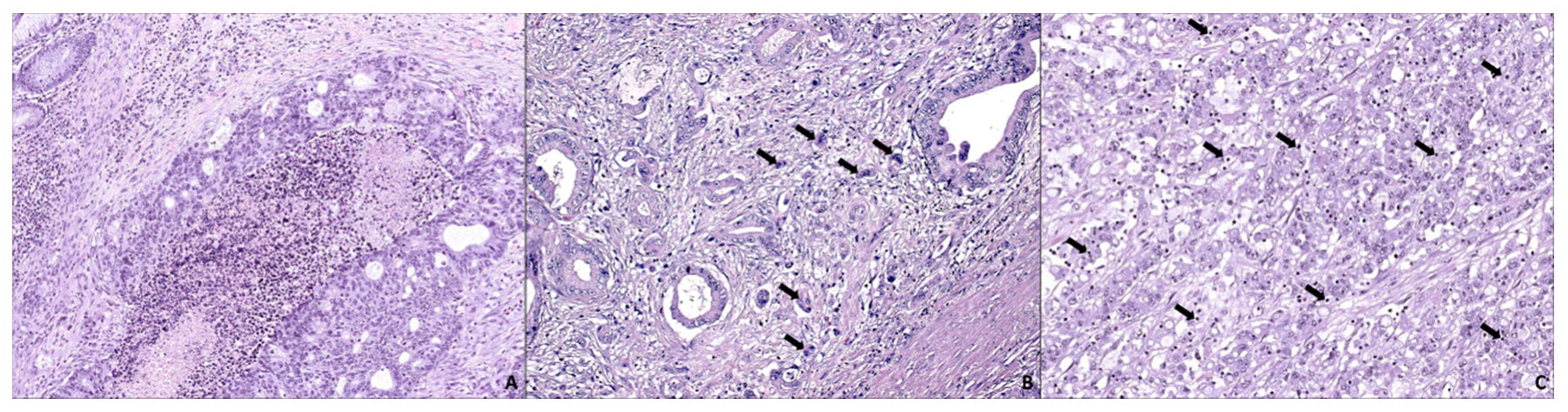
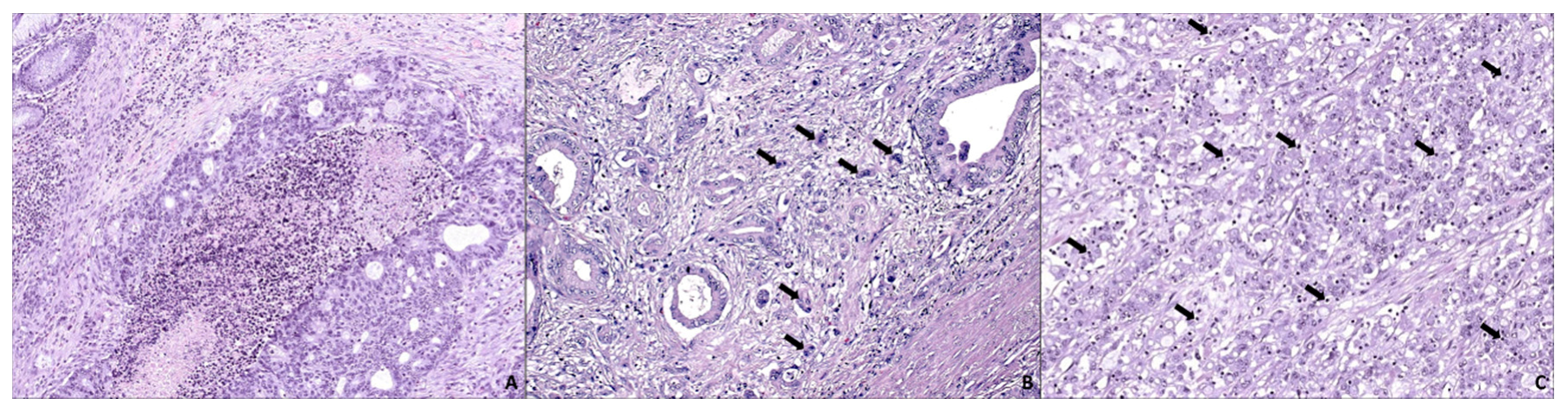
3. SAC is Especially Capable of Avoiding the Immune Response
Therapeutic Approaches for Avoiding Immune Evasion in SAC
4. SAC Displays an Active Invasive Front
Possible Therapies Targeting SAC Invasive Front
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Stefanius, K.; Ylitalo, L.; Tuomisto, A.; Kuivila, R.; Kantola, T.; Sirniö, P.; Karttunen, T.J.; Mäkinen, M.J. Frequent mutations of KRAS in addition to BRAF in colorectal serrated adenocarcinoma. Histopathology 2011, 58, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Solano, J.; Conesa-Zamora, P.; Carbonell, P.; Trujillo-Santos, J.; Torres-Moreno, D.D.; Pagán-Gómez, I.; Rodríguez-Braun, E.; Pérez-Guillermo, M. Colorectal serrated adenocarcinoma shows a different profile of oncogene mutations, MSI status and DNA repair protein expression compared to conventional and sporadic MSI-H carcinomas. Int. J. Cancer 2012, 131, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Snover, D.C.; Jass, J.R.; Fenoglio-Preiser, C.; Batts, K.P. Serrated polyps of the large intestine: A morphologic and molecular review of an evolving concept. Am. J. Clin. Pathol. 2005, 124, 380–391. [Google Scholar] [CrossRef]
- García-Solano, J.; Turpin-Sevilla, M.; García-García, F.; Carbonell-Muñoz, R.; Torres-Moreno, D.; Conesa, A.; Conesa-Zamora, P. Differences in gene expression profiling and biomarkers between histological colorectal carcinoma subsets from the serrated pathway. Histopathology 2019, 75, 496–507. [Google Scholar] [CrossRef]
- Bellizzi, A.M.; Frankel, W.L. Colorectal cancer due to deficiency in DNA mismatch repair function: A review. Adv. Anat. Pathol. 2009, 16, 405–417. [Google Scholar] [CrossRef]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. Carcinoma of the colon and rectum. In WHO Classification of Tumors of the Digestive System, 4th ed.; IARC: Lyon, France, 2010; pp. 134–146. [Google Scholar]
- Nagtegaal, I.; Odze, R.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Makinen, M.J. Colorectal serrated adenocarcinoma. Histopathology 2007, 50, 131–150. [Google Scholar] [CrossRef]
- García-Solano, J.; Pérez-Guillermo, M.; Conesa-Zamora, P.; Acosta-Ortega, J.; Trujillo-Santos, J.; Cerezuela-Fuentes, P.; Mäkinen, M.J. Clinicopathologic study of 85 colorectal serrated adenocarcinomas: Further insights into the full recognition of a new subset of colorectal carcinoma. Hum. Pathol. 2010, 41, 1359–1368. [Google Scholar] [CrossRef]
- Conesa-Zamora, P.; García-Solano, J.; García-García, F.; Turpin, M.; Trujillo-Santos, J.; Torres-Moreno, D.; Pérez-Guillermo, M. Expression profiling shows differential molecular pathways and provides potential new diagnostic biomarkers for colorectal serrated adenocarcinoma. Int. J. Cancer 2013, 132, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Kahi, C.J. Screening Relevance of Sessile Serrated Polyps. Clin. Endosc. 2019, 52, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailie, L.; Loughrey, M.B.; Coleman, H.G. Lifestyle Risk Factors for Serrated Colorectal Polyps: A Systematic Review and Meta-analysis. Gastroenterology. Gastroenterology 2017, 152, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crockett, S.D.; Nagtegaal, I.D. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 2019, 157, 949–966. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.C.; Alpern, Z.A. Smoking and the Increased Risk for Serrated Polyps: Implications for Screening and Surveillance. J. Clin. Gastroenterol. 2019, 53, 319–321. [Google Scholar] [CrossRef]
- Yu, J.; Chen, Y.; Fu, X.; Zhou, X.; Peng, Y.; Shi, L.; Wu, Y. Invasive Fusobacterium Nucleatum May Play a Role in the Carcinogenesis of Proximal Colon Cancer Through the Serrated Neoplasia Pathway. Int. J. cancer 2016, 139, 1318–1326. [Google Scholar] [CrossRef] [Green Version]
- García-Solano, J.; Conesa-Zamora, P.; Trujillo-Santos, J.; Mäkinen, M.J.; Pérez-Guillermo, M. Tumour budding and other prognostic pathological features at invasive margins in serrated colorectal adenocarcinoma: A comparative study with conventional carcinoma. Histopathology 2011, 59, 1046–1056. [Google Scholar] [CrossRef]
- García-Solano, J.; Conesa-Zamora, P.; Trujillo-Santos, J.; Torres-Moreno, D.; Mäkinen, M.J.; Pérez-Guillermo, M. Immunohistochemical expression profile of β-catenin, E-cadherin, P-cadherin, laminin-5γ2 chain, and SMAD4 in colorectal serrated adenocarcinoma. Hum. Pathol. 2012, 43, 1094–1102. [Google Scholar] [CrossRef]
- Conesa-zamora, P.; García-solano, J.; Turpin, C.; Sebastián-león, P.; Torres-moreno, D.; Estrada, E.; Tuomisto, A.; Wilce, J.; Mäkinen, M.J.; Pérez-Guillermo, M.; et al. Methylome profiling reveals functions and genes which are differentially methylated in serrated compared to conventional colorectal carcinoma. Clin. Epigenet. 2015, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.J.; Marsh Durban, V.; Meniel, V.; Williams, G.T.; Clarke, A.R. PTEN loss and KRAS activation leads to the formation of serrated adenomas and metastatic carcinoma in the mouse intestine. J. Pathol. 2014, 233, 27–38. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Duran, A.; L’Hermitte, A.; Shelton, P.M.; Nakanishi, N.; Reina-Campos, M.; Moscat, J. Simultaneous Loss of Both Atypical Protein Kinase C Genes in the Intestinal Epithelium Drives Serrated Intestinal Cancer by Impairing Immunosurveillance. Immunity 2018, 49, 1132–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, Y.; Diaz-meco, M.T.; Moscat, J. Serrated Colorectal Cancer: The Road Less Travelled? Trends Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Laiho, P.; Kokko, A.; Vanharanta, S.; Salovaara, R.; Sammalkorpi, H.; Tuppurainen, K.; Davalos, V.; Schwartz, S., Jr.; Arango, D. Serrated carcinomas form a subclass of colorectal cancer with distinct molecular basis. Oncogene 2007, 26, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomisto, A.; García-solano, J.; Sirniö, P.; Väyrynen, J. HIF-1 α expression and high microvessel density are characteristic features in serrated colorectal cancer. Virchows Arch. Int. J. Pathol. 2016, 469, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Gil, A.; Pérez-Sanz, F.; García-Solano, J.; Alburquerque-González, B.; Parreño-González, M.A.; Legaz-García, M.C.; Fernández-Breis, J.T.; Rodriguez-Braun, E.; Pimentel, P.; Tuomisto, A.; et al. ColPortal, an integrative multiomic platform for analysing epigenetic interactions in colorectal cancer. Sci. Data 2019, 6, 255. [Google Scholar] [CrossRef] [Green Version]
- García-Solano, J.; Turpin, M.; Torres-Moreno, D.; Huertas-López, F.; Tuomisto, A.; Mäkinen, M.J.; Conesa, A.; Conesa-Zamora, P. Two histologically colorectal carcinomas subsets from the serrated pathway show different methylome signatures and diagnostic biomarkers. Clin. Epigenet. 2018, 10, 141. [Google Scholar] [CrossRef]
- Kondelova, A.; Alburquerque-González, B.; Vychytilova-Faltejskova, P.; García-Solano, J.; Prochazka, V.; Kala, Z.; Pérez, F.; Slaby, O.; Conesa-Zamora, P. miR-181a-2* expression is different amongst carcinomas from the colorectal serrated route. Mutagenesis 2019, 30. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Battaglin, F.; Puccini, A.; Intini, R.; Schirripa, M.; Ferro, A.; Bergamo, F.; Lonardi, S.; Zagonel, V.; Lenz, H.; Loupakis, F.; et al. The role of tumor angiogenesis as a therapeutic target in colorectal cancer. Expert Rev. Anticancer Ther. 2018, 18, 251–266. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Salvatore, L.; Masi, G.; Sensi, E.; Schirripa, M.; Michelucci, A.; Pfanner, E.; Brunetti, I.; Lupi, C.; et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur. J. Cancer 2014, 50, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Battaglin, F.; Wang, J.; Lo, J.H.; Soni, S.; Zhang, W. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 2019, 81, 101912. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 242–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Hamid, O. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Housseau, F. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Jakoncic, J.; Zhang, J.J.; Huang, X. Migrastatin analogues target fascin to block tumour metastasis. Nature 2010, 464, 1062–1066. [Google Scholar] [CrossRef] [Green Version]
- Montoro-García, S.; Alburquerque-González, B.; Bernabé-García, A.; Rodrigues, P.C.; den-Haan, H.; Luque, I.; Nicolás, F.J.; Pérez-Sánchez, H.; Cayuela, M.L.; Salo, T.; et al. Novel anti-invasive properties of a fascin inhibitor on colorectal cancer cells. J. Mol. Med. 2020, in press. [Google Scholar]
- Alburquerque-González, B.; Bernabé-García, M.; Montoro-García, S.; Bernabé-García, Á.; Rodrigues, P.C.; Ruiz Sanz, J.; López-Calderón, F.F.; Luque, I.; Nicolas, F.J.; Cayuela, M.L.; et al. New role of antidepressant imipramine as a fascin1 inhibitor in colorectal cancer cells. Exp. Mol. Med. 2020, 52, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Sauer, T.; Lang, U.E. Is Imipramine Helpful in the Treatment of Cancer? A Case Presentation and Discussion of Possible Clinical Implications. Biomed. J. Sci. Tech. Res. 2018, 4, 4194–4195. [Google Scholar] [CrossRef]
- Walker, A.J.; Card, T.; Bates, T.E.; Muir, K. Tricyclic antidepressants and the incidence of certain cancers: A study using the GPRD. Br. J. Cancer 2011, 104, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jiang, J.; Yu, X.; Xia, W.; Yu, P.; Wang, K.; Zhao, Z.; Chen, Z.; Chen, O.W.; Jiang, J.; et al. Endothelial cells in colorectal cancer. World J. Gastrointest Oncol. 2019, 11, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrive, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Moreno-sánchez, R. HIF-1 modulates energy metabolism in cancer cells by inducing over- expression of specific glycolytic isoforms HIF-1α modulates energy metabolism in cancer cells by modifying the status of glycolytic enzymes. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1-α and HIF2-α: Sibling rivalry in hypoxic tumor growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.; Paraskeva, E.; Baxevanidou, K.; Simos, G.; Papacharalambous, C.; Samara, M.; Koukoulis, G. HIF-1α in colorectal carcinoma: Review of the literature. J. Balk. Union Oncol. 2015, 20, 680–689. [Google Scholar]
- Morimoto, T.; Mitomi, H.; Saito, T. Distinct profile of HIF1α, PTCH, EphB2, or DNA Repair Protein Expression and BRAF Mutation in Colorectal Serrated Adenoma. J. Gastroenterol. Hepatol. 2014, 29, 1192–1199. [Google Scholar] [CrossRef]
- Jiang, Y.; Fan, L.; Jiang, C.; Zhang, Y.; Luo, H.; Tang, Z.; Xia, D.; Wang, M. Expression and significance of PTEN, hypoxia-inducible factor-1 alpha in colorectal adenoma and adenocarcinoma. World J. Gastroenterol. 2003, 9, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Simiantonaki, N.; Taxeidis, M.; Jayasinghe, C.; Kurzik-dumke, U.; Kirkpatrick, C.J. Hypoxia-inducible factor 1 alpha expression increases during colorectal carcinogenesis and tumor progression. BMC Cancer 2008, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigopoulos, D.N.; Tsiambas, E.; Lazaris, A.C. Deregulation of EGFR/VEGF/HIF-1a Signaling Pathway in Colon Adenocarcinoma Based on Tissue Microarrays Analysis. J. Balk. Union Oncol. 2010, 15, 107–115. [Google Scholar]
- Chen, Z.; He, X.; Xia, W.; Huang, Q.; Zhang, Z.; Ye, J.; Ni, C. Prognostic value and clinicopathological differences of HIFs in colorectal cancer: Evidence from meta-analysis. PLoS ONE 2013, 8, e80337. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, M.; Comolli, G.; Torchio, M.; Mazzini, G.; Danova, M. Circulating Endothelial Cells and Their Subpopulations: Role as Predictive Biomarkers in Antiangiogenic Therapy for Colorectal Cancer. Clin. Colorectal Cancer 2015, 14, 11–17. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Hohmann, N.; Niethammer, A.G.; Friedrich, T.; Lubenau, H.; Ulrich, A.; Buechler, M.W.; Pianka, F.; Springer, M.; Breiner, K.M.; et al. Anti-angiogenic activity of VXM01, an oral T-cell vaccine against VEGF receptor 2, in patients with advanced pancreatic cancer: A randomized, placebo-controlled, phase 1 trial. Oncoimmunology 2015, 4, e1001217. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Yu, H.; Shi, X.; Xu, K.; Tang, Q.; Liang, B.; Hu, S.; Bao, Y. microRNA-497 inhibits invasion and metastasis of colorectal cancer cells by targeting vascular endothelial growth factor-A. Cell Prolif. 2016, 49, 69–78. [Google Scholar] [CrossRef]
- Seeber, A.; Gunsilius, E.; Gastl, G.; Pircher, A. Anti-angiogenics: Their value in colorectal cancer therapy. Oncol. Res. Treat. 2018, 41, 188–193. [Google Scholar] [CrossRef]
- Coxon, A.; Bready, J.; Min, H.; Kaufman, S.; Leal, J.; Yu, D.; Lee, T.A.; Sun, J.; Estrada, J.; Bolon, B. Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: Implications for AMG 386, an angiopoietin-1/2-neutralizing peptibody. Mol. Cancer Ther. 2010, 9, 2641–2651. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, C.; Dai, Y.; Huang, C.; Han, Q.; Jing, L.; Ma, Y.; Xu, Y.; Liu, Y.; Zhao, L.; et al. Cinobufagin suppresses colorectal cancer angiogenesis by disrupting the endothelial mammalian target of rapamycin/inducible factor 1-α axis. Cancer Sci. 2019, 110, 1724–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataria, N.; Martinez, C.A.; Kerr, B.; Zaiter, S.S.; Morgan, M.; McAlpine, S.R.; Cook, K.M. C-terminal HSP90 inhibitors block the HIF-1 hypoxic response by degrading HIF-1α through the oxygen-dependent degradation pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. 2019, 53, 480–495. [Google Scholar] [CrossRef]
- Chen, D.; Huang, J.; Liu, K.; Zhang, L.; Yang, Z.; Chuai, Z.; Wang, Y.; Shi, D.; Huang, Q.; Fu, W. BRAF V600E mutation and its association with clinicopathological features of colorectal cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e90607. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Rashid, A.; Lee, J.; Kim, S.G.; Hamilton, S.R. Frequent CpG island methylation in serrated adenomas of the colorectum. Am. J. Pathol. 2003, 162, 815–822. [Google Scholar] [CrossRef]
- Morris, V.K. Systemic therapy in BRAF V600E-mutant metastatic colorectal cancer: Recent advances and future strategies. Curr. Colorectal Cancer Rep. 2019, 15, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Ou, F.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D. Impact of Consensus Molecular Subtype on survival in patients with metastatic colorectal cancer: Results from CALGB/SWOG 80405. J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.; Innocenti, F. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or re. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Gutting, T.; Weber, C.A.; Weidner, P.; Herweck, F.; Henn, S.; Friedrich, T.; Yin, S.; Kzhyshkowska, J.; Gaiser, T.; Janssen, K.P.; et al. PPARγ-activation increases intestinal M1 macrophages and mitigates formation of serrated adenomas in mutant KRAS mice. Oncoimmunology 2018, 7, e1423168. [Google Scholar] [CrossRef] [Green Version]
- Deschoolmeester, V.; Baay, M.; Lardon, F.; Pauwels, P.; Peeters, M. Immune Cells in Colorectal Cancer: Prognostic Relevance and Role of MSI. Cancer Microenviron. J. Int. Cancer Microenviron. Soc. 2011, 4, 377–392. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Andersen, M.H. The role of dendritic cells in cancer. Semin. Immunopathol. 2017, 39, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, V.G.; Rubio, C.; Martínez-Fernández, M.; Segovia, C.; López-Calderón, F.; Garín, M.I.; Dueñas, M. BMP4 Induces M2 Macrophage Polarization and Favors Tumor Progression in Bladder Cancer. Clin. Cancer Res. 2017, 23, 7388–7399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Shunyakov, L.; Ryan, C.K.; Sahasrabudhe, D.M.; Khorana, A.A. The influence of host response on colorectal cancer prognosis. Clin. Colorectal Cancer 2004, 4, 38–45. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Pagès, F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Kocián, P.; Šedivcová, M.; Drgáč, J.; Cerná, K.; Hoch, J.; Kodet, R.; Fialová, A. Tumor-infiltrating lymphocytes and dendritic cells in human colorectal cancer: Their relationship to KRAS mutational status and disease recurrence. Hum. Immunol. 2011, 72, 1022–1028. [Google Scholar] [CrossRef]
- Roxburgh, C.S.; McMillan, D.C. The role of the in situ local inflammatory response in predicting recurrence and survival in patients with primary operable colorectal cancer. Cancer Treat. Rev. 2012, 38, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, J.P.; Kantola, T.; Väyrynen, S.A.; Klintrup, K.; Bloigu, R.; Karhu, T.; Mäkinen, M.J. The relationships between serum cytokine levels and tumor infiltrating immune cells and their clinical significance in colorectal cancer. Int. J. Cancer 2016, 139, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [Green Version]
- Engström, A.; Erlandsson, A.; Delbro, D.; Wijkander, J. Conditioned media from macrophages of M1, but not M2 phenotype, inhibit the proliferation of the colon cancer cell lines HT-29 and CACO-2. Int. J. Oncol. 2014, 44, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Stratton, M.R. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Shmulevich, I. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [Green Version]
- Catalano, I.; Grassi, E.; Bertotti, A.; Trusolino, L. Immunogenomics of Colorectal Tumors: Facts and Hypotheses on an Evolving Saga. Trends Cancer 2019, 5, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Galon, J. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Galon, J. Integrative analyses of colorectal cancer show Immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- García-Solano, J.; Conesa-Zamora, P.; Carbonell, P.; Trujillo-Santos, J.; Torres-Moreno, D.; Rodriguez-Braun, E.; Vicente-Ortega, V.; Pérez-Guillermo, M. Microsatellite pathologic score does not efficiently identify high microsatellite instability in colorectal serrated adenocarcinoma. Hum. Pathol. 2013, 44, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Smyth, M.J.; Teng, M. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a028530. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Hernández-Jiménez, E.; Avendaño-Ortiz, J.; Toledano, V.; Varela-Serrano, A.; Fernández-Navarro, I.; López-Collazo, E. Obstructive Sleep Apnea Monocytes Exhibit High Levels of Vascular Endothelial Growth Factor Secretion, Augmenting Tumor Progression. Mediat. Inflamm. 2018, 7373921. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, T.; Hida, S.; Kitazawa, M.; Fujii, C.; Kobayashi, A.; Takeoka, M.; Taniguchi, S.; Miyagawa, S. Fascin1 suppresses RIG-I–like receptor signaling and interferon-β production by associating with IκB kinase ϵ (IKKϵ) in colon cancer. J. Biol. Chem. 2018, 293, 6326–6336. [Google Scholar] [CrossRef] [Green Version]
- Blake, S.J.; Teng, M.W. Role of IL-17 and IL-22 in autoimmunity and cancer. Actas Dermosifiliogr. 2014, 105, 41–50. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantero-Cid, R.; Casas-Martín, J.; Hernández-Jiménez, E.; Cubillos-zapata, C.; Varela-serrano, A.; Avendaño-ortiz, J.; Casarrubios, M.; Montalbán-Hernández, K.; Villacañas-Gil, I.; Guerra-Ppastrián, L.; et al. PD-L1/PD-1 crosstalk in colorectal cancer: Are we targeting the right cells? BMC Cancer 2018, 18, 945. [Google Scholar] [CrossRef] [PubMed]
- Natalwala, A.; Spychal, R.; Tselepis, C. Epithelial-mesenchymal transition mediated tumourigenesis in the gastrointestinal tract. World J. Gastroenterol. 2008, 14, 3792–3797. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kondo, S.; Itakura, T.; Tokunaga, M.; Hatayama, S.; Katayama, K.; Sugimoto, Y. SNAIL- and SLUG-induced side population phenotype of HCT116 human colorectal cancer cells and its regulation by BET inhibitors. Biochem. Biophys. Res. Commun. 2020, 521, 152–157. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A.; Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition Find the latest version: Review series The basics of epithelial-mesenchymal transition. Clin. Investig. 2010, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prall, F. Tumour budding in colorectal carcinoma. Histopathology 2007, 50, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Schmalhofer, O.; Brabletz, T. Gastrointestinal stem cells in development and cancer. J. Pathol. 2009, 217, 307–317. [Google Scholar] [CrossRef]
- Conacci-sorrell, M.; Simcha, I.; Ben-yedidia, T.; Blechman, J.; Savagner, P.; Ben-ze, A. Autoregulation of E-cadherin expression by cadherin–Cadherin interactions: The roles. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Spedding, M.; Peters, J.A.; Harmar, A.J. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br. J. Pharmacol. 2013, 170, 1459–1581. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Tao, Y.S.; Edwards, R.A.; Tubb, B.; Wang, S.; Bryan, J.; Mccrea, P.D. b-Catenin Associates with the Actin-bundling Protein Fascin in a Noncadherin Complex. Cell Biol. 1996, 134, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Kureishy, N.; Sapountzi, V.; Prag, S.; Anilkumar, N.; Adams, J.C. Fascins, and their roles in cell structure and function. Bioessays 2002, 24, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Skacel, M.; Lavery, I.C.; Mukherjee, A.L.; Casey, G.; Adams, J.C. Prognostic significance of fascin expression in advanced colorectal cancer: An immunohistochemical study of colorectal adenomas and adenocarcinomas. BMC Cancer 2006, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tan, V.Y.; Lewis, S.J.; Adams, J.C.; Martin, R.M. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: A systematic review and meta-analysis. BMC Med. 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignjevic, D.; Schoumacher, M.; Gavert, N.; Janssen, K.; Jih, G.; Lae, M.; Louvard, D.; Ben-ze, A.; Robine, S. Fascin, a novel target of b-Catenin-TCF signaling, is expressed at the invasive front of human colon cancer. Cancer Res. 2007, 6844–6854. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.K.; Han, S.; Xing, B.; Huang, J.; Liu, B.; Bordeleau, F.; Reinhart-King, C.A.; Zhang, J.J.; Huang, X.Y. Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization. Nat. Commun. 2015, 6, 7465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahchan, N.S.; Dudley, J.T.; Mazur, P.K.; Flores, N.; Yang, D.; Palmerton, A.; Sage, J. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013, 3, 1364–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Treatment | Target(s) | FDA Approved | Treatment Current Indication | Rationale for Indication in Serrated Cancers | Reference(s) | |
|---|---|---|---|---|---|---|
| Anti-angiogenic therapy | Bevacizumab + chemotherapy | VEGF-A | Yes | mCRC | SAC presents higher expression HIF-1, VEGF and MVD compared to CC | [31] |
| Ramucirumab + FOLFIRI | VEGFR-2 | Yes | mCRC | [31] | ||
| Aflibercept in combination with FOLFIRI | VEGF-A, VEGF-B and PIGF | Yes | mCRC | [31] | ||
| Bevacizumab + FOLFOXIRI | VEGF-A | Yes | mCRC* | [32] | ||
| Regorafenib | VEGFR-1, VEGFR-2, VEGFR-3, Tie-2,cKIT, RET, BRAF PDGFR, and FGFR | Yes | mCRC | [31,33] | ||
| Therapy for avoiding immune response | Anti-PD-L1 and Galunisertib | PD-L1 and TGFβR | Yes | MSS-SAC | High PD-L1 expression, lack of CD-8+ response | [22] |
| Atezolizumab and MEK inhibitors | PD-L1 and MEK | Yes | MSS-SAC | [22] | ||
| Ipilimumab and tremelimumab | CTLA-4 | Yes | MSIH classical SACs | Possible utility in classical SAC based on MSI-H status | [34] | |
| MDX1105, durvalumab, avelumab and atezolizumab | PD-L1(CD274) | Yes, MDX1105 in phase I study | MSIH classical SACs | [35] | ||
| Nivolumab and pembrolizumab | PD-1(CD279) | Yes | MSIH classical SACs | [36,37] | ||
| Possible therapies targeting SAC invasive front | Migrastatin | Fascin1 | No | None | Fascin1 overexpression in SAC Synthetic drug FDA approved drug | [38] |
| G2 | Fascin1 | No | None | [39] | ||
| Imipramine | Fascin1and GPCRs (G protein coupled receptors) | Yes | Antidepressant | [40,41,42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alburquerque-González, B.; López-Calderón, F.F.; López-Abellán, M.D.; Esteban-Gil, Á.; García-Solano, J.; Conesa-Zamora, P. Biology and Therapeutic Targets of Colorectal Serrated Adenocarcinoma; Clues for a Histologically Based Treatment against an Aggressive Tumor. Int. J. Mol. Sci. 2020, 21, 1991. https://doi.org/10.3390/ijms21061991
Alburquerque-González B, López-Calderón FF, López-Abellán MD, Esteban-Gil Á, García-Solano J, Conesa-Zamora P. Biology and Therapeutic Targets of Colorectal Serrated Adenocarcinoma; Clues for a Histologically Based Treatment against an Aggressive Tumor. International Journal of Molecular Sciences. 2020; 21(6):1991. https://doi.org/10.3390/ijms21061991
Chicago/Turabian StyleAlburquerque-González, Begoña, Fernando F. López-Calderón, María Dolores López-Abellán, Ángel Esteban-Gil, José García-Solano, and Pablo Conesa-Zamora. 2020. "Biology and Therapeutic Targets of Colorectal Serrated Adenocarcinoma; Clues for a Histologically Based Treatment against an Aggressive Tumor" International Journal of Molecular Sciences 21, no. 6: 1991. https://doi.org/10.3390/ijms21061991
APA StyleAlburquerque-González, B., López-Calderón, F. F., López-Abellán, M. D., Esteban-Gil, Á., García-Solano, J., & Conesa-Zamora, P. (2020). Biology and Therapeutic Targets of Colorectal Serrated Adenocarcinoma; Clues for a Histologically Based Treatment against an Aggressive Tumor. International Journal of Molecular Sciences, 21(6), 1991. https://doi.org/10.3390/ijms21061991