PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
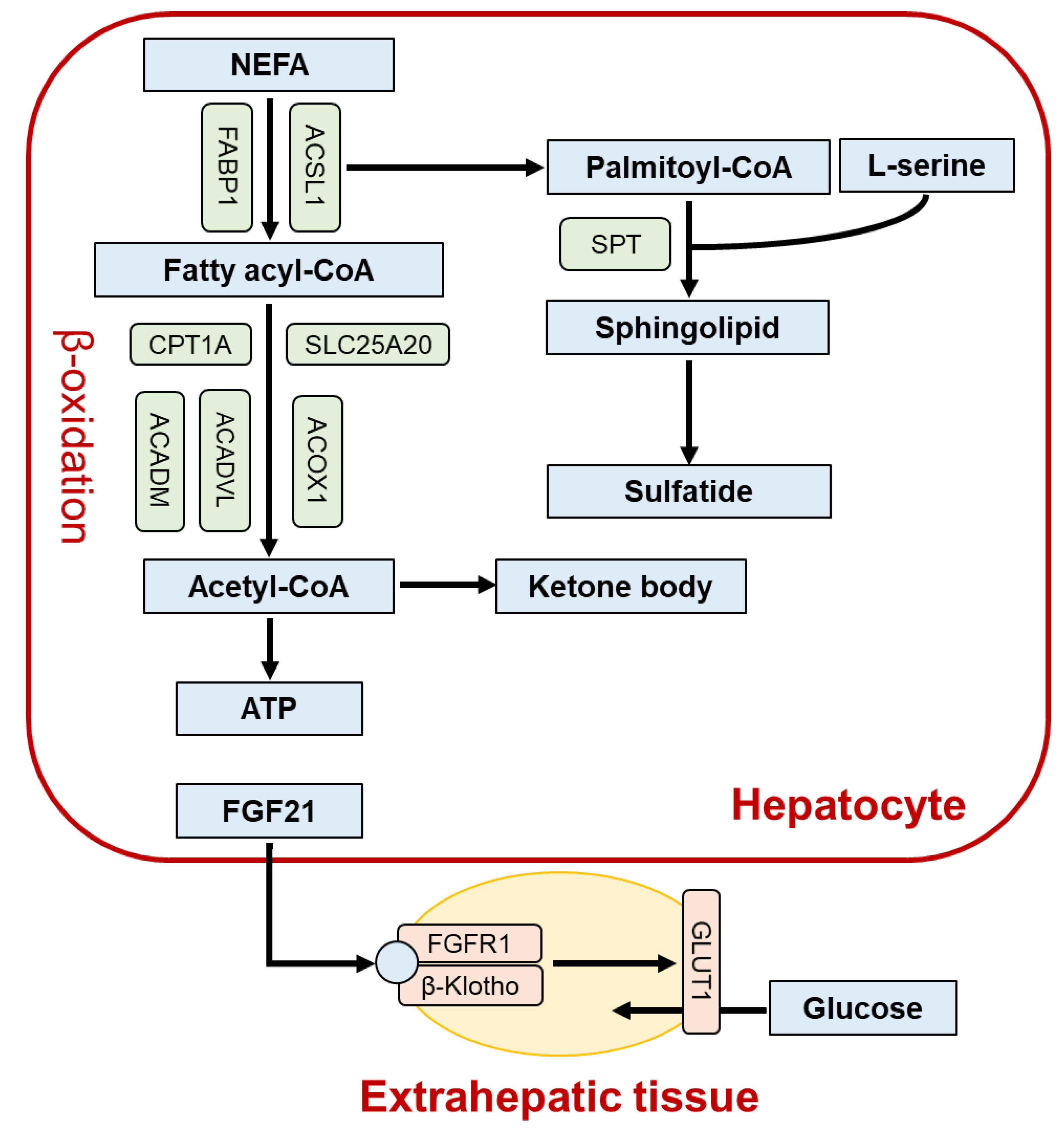
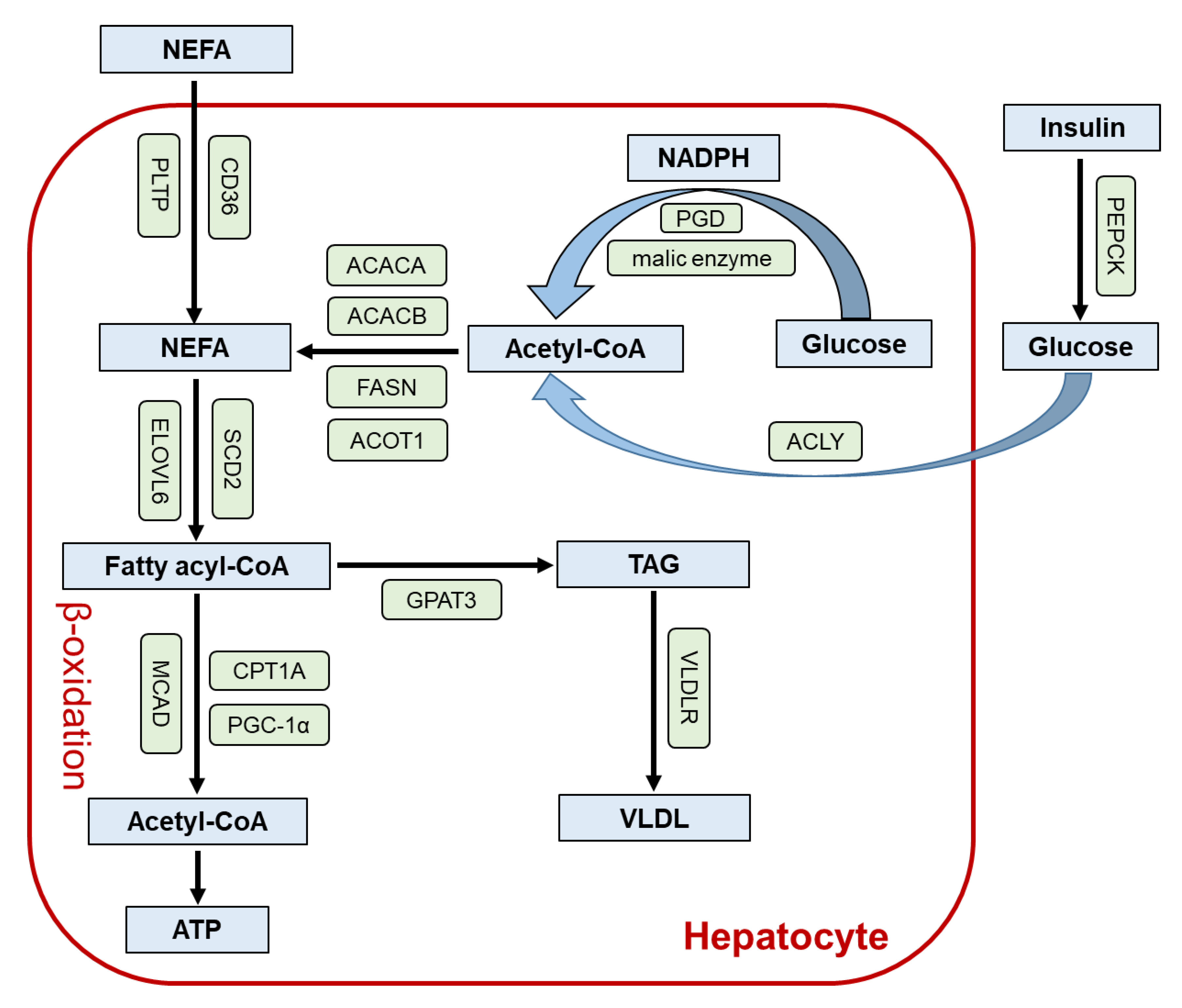
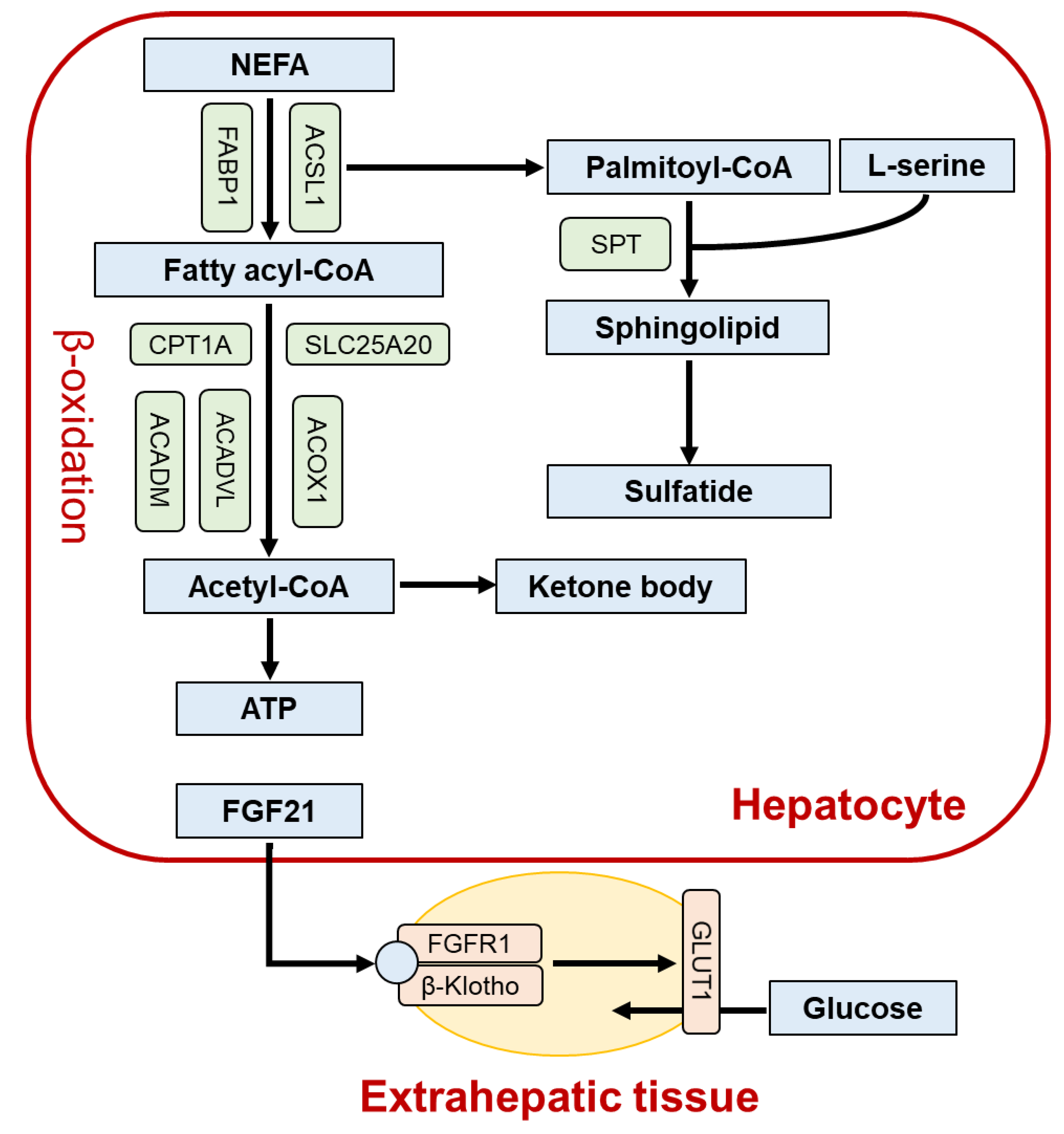
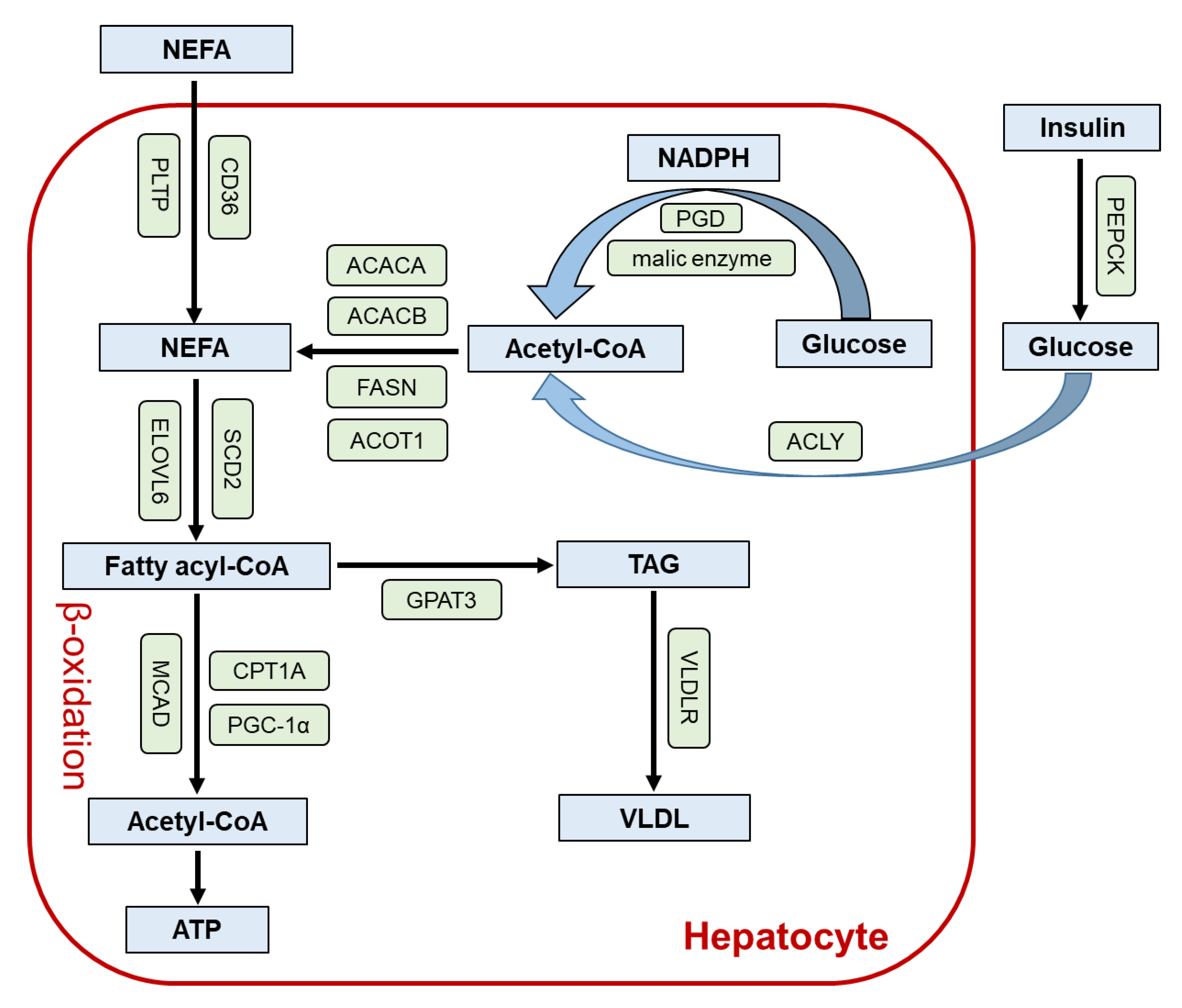
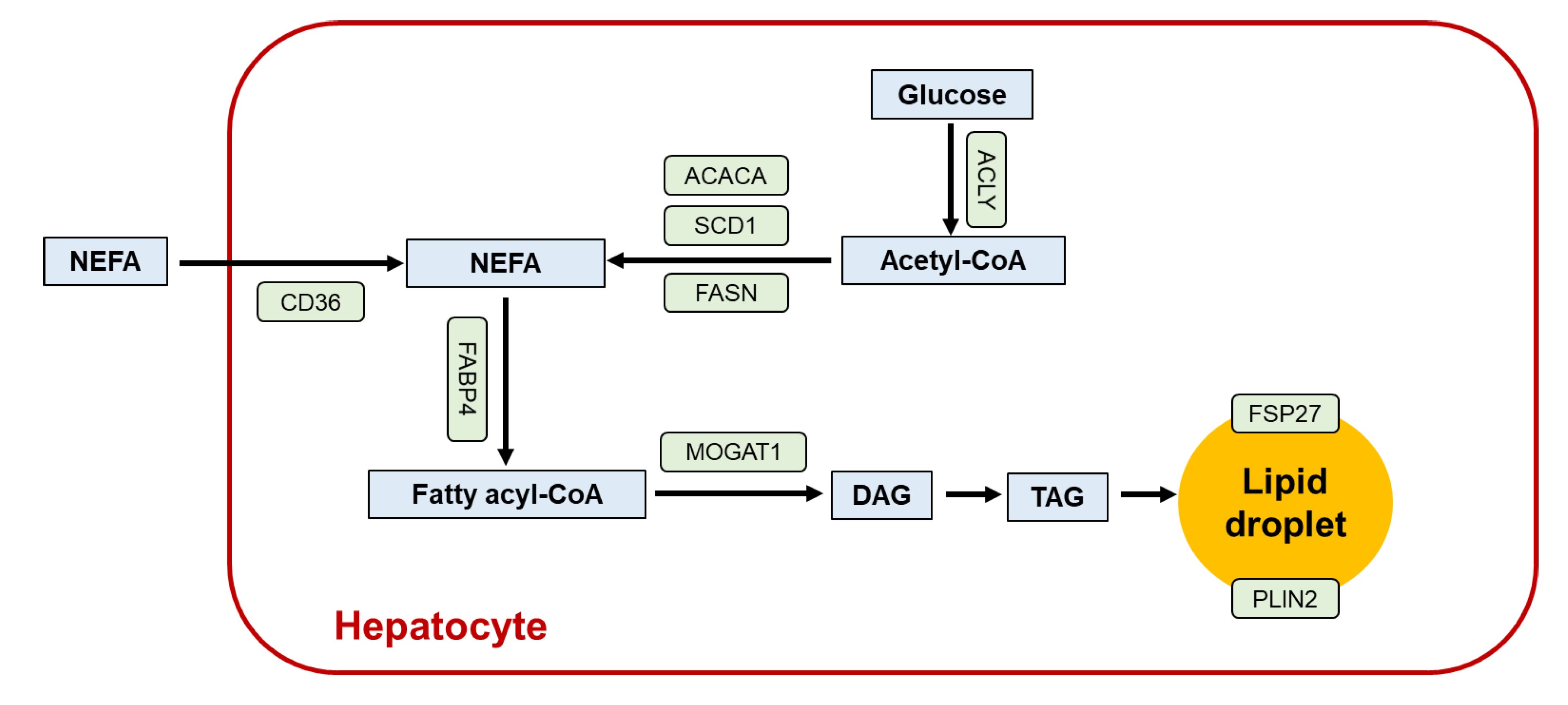
2. Role of PPARα in the Liver
3. Lessons from Liver-Specific Ppara-Null Mice
4. Role of PPARβ/δ in the Liver
5. Lessons from Liver-Specific Ppard-Null Mice
6. Role of PPARγ in the Liver
7. Lessons from Liver-specific Pparg-null Mice
8. The Other Factors Modulating Hepatic PPAR Function
9. Conclusions—Issues to Be Solved for Contributing to Human Health
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-coenzyme A carboxylase |
| AMPK | adenosine monophosphate-activated protein kinase |
| ATP | adenosine triphosphate |
| CCl4 | carbon tetrachloride |
| CD | cluster of differentiation |
| cDNA | complementary DNA |
| CoA | coenzyme A |
| ER | endoplasmic reticulum |
| FA | fatty acid |
| FABP | fatty acid-binding protein |
| FGF21 | fibroblast growth factor 21 |
| FSP27 | fat-specific protein 27 |
| HFD | high fat diet |
| HSC | hepatic stellate cell |
| IL | interleukin |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-κB | nuclear factor kappa B |
| PC | phosphatidylcholine |
| PP | peroxisome proliferator |
| PPAR | peroxisome proliferator-activated receptor |
| PUFA | polyunsaturated fatty acid |
| ROS | reactive oxygen species |
| SPT | serine palmitoyltransferase |
| SREBP | sterol regulatory element binding protein |
| TAG | triacylglycerol |
| TZD | thiazolidinedione |
| VLDL | very-low-density lipoprotein |
References
- Reddy, J.K.; Krishnakantha, T.P. Hepatic peroxisome proliferation: Induction by two novel compounds structurally unrelated to clofibrate. Science 1975, 190, 787–789. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Krey, G.; Keller, H.; Mahfoudi, A.; Medin, J.; Ozato, K.; Dreyer, C.; Wahli, W. Xenopus peroxisome proliferator activated receptors: Genomic organization, response element recognition, heterodimer formation with retinoid X receptor and activation by fatty acids. J. Steroid Biochem. Mol. Biol. 1993, 47, 65–73. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-alpha agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, J.T.; Rogers, L.C.; Paumi, C.M.; Hantgan, R.R.; Thomas, L.R.; Clay, C.E.; High, K.; Chen, Y.Q.; Willingham, M.C.; Smitherman, P.K.; et al. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim. Biophys. Acta 2005, 1736, 228–236. [Google Scholar] [CrossRef]
- Altmann, R.; Hausmann, M.; Spöttl, T.; Gruber, M.; Bull, A.W.; Menzel, K.; Vogl, D.; Herfarth, H.; Schölmerich, J.; Falk, W.; et al. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem. Pharmacol. 2007, 74, 612–622. [Google Scholar] [CrossRef]
- Takeuchi, S.; Matsuda, T.; Kobayashi, S.; Takahashi, T.; Kojima, H. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma and quantitative analysis of in vivo induction pathway. Toxicol. Appl. Pharmacol. 2006, 217, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Cunningham, M.L.; Hummer, B.T.; Lau, C.; Meek, B.; Peters, J.M.; Popp, J.A.; Rhomberg, L.; Seed, J.; Klaunig, J.E. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit. Rev. Toxicol. 2014, 44, 1–49. [Google Scholar] [CrossRef]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Temelkova-Kurktschiev, T.; Hanefeld, M.; Chinetti, G.; Zawadzki, C.; Haulon, S.; Kubaszek, A.; Koehler, C.; Leonhardt, W.; Staels, B.; Laakso, M. Ala12Ala genotype of the peroxisome proliferator-activated receptor gamma2 protects against atherosclerosis. J. Clin. Endocrinol. Metab. 2004, 89, 4238–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Kusakabe, T.; Noguchi, T.; Takiguchi, S.; Suzuki, T.; Yamano, S.; Gonzalez, F.J. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008, 7, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Yang, Y.; Lu, Y.; Kamijo, Y.; Yamada, Y.; Nakamura, K.; Koyama, M.; Yamaguchi, S.; Sugiyama, E.; Tanaka, N.; et al. Decreased Fatty Acid β-Oxidation Is the Main Cause of Fatty Liver Induced by Polyunsaturated Fatty Acid Deficiency in Mice. Tohoku J. Exp. Med. 2017, 242, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Rivier, M.; Castiel, I.; Safonova, I.; Ailhaud, G.; Michel, S. Peroxisome proliferator-activated receptor-alpha enhances lipid metabolism in a skin equivalent model. J. Investig. Dermatol. 2000, 114, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yang, Y.; Zhang, X.; Nakajima, T.; Tanaka, N.; Sugiyama, E.; Kamijo, Y.; Lu, Y.; Moriya, K.; Koike, K.; et al. Age-dependent PPARα activation induces hepatic sulfatide accumulation in transgenic mice carrying the hepatitis C virus core gene. Glycoconj. J. 2016, 33, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Kamijo, Y.; Yuzhe, H.; Kimura, T.; Tanaka, N.; Sugiyama, E.; Nakamura, K.; Kyogashima, M.; Hara, A.; Aoyama, T. Peroxisome proliferator-activated receptor α mediates enhancement of gene expression of cerebroside sulfotransferase in several murine organs. Glycoconj. J. 2013, 30, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Nakajima, T.; Kamijo, Y.; Tanaka, N.; Wang, L.; Hara, A.; Sugiyama, E.; Tanaka, E.; Gonzalez, F.J.; Aoyama, T. Hepatic Cerebroside Sulfotransferase Is Induced by PPARα Activation in Mice. PPAR Res. 2012, 2012, 174932. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Kutsukake, Y.; Uemura, K.I.; Taketomi, T. Anticoagulant activity of sulfatide and its anti-thrombotic effect in rabbit. J. Biochem. 1993, 113, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Adams, A.C. Inventing new medicines: The FGF21 story. Mol. Metab. 2013, 3, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Takahashi, S.; Zhang, Y.; Krausz, K.W.; Smith, P.B.; Patterson, A.D.; Gonzalez, F.J. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 2015, 1852, 1242–1252. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, T.; Tanaka, N.; Komatsu, M.; Ichijo, T.; Sano, K.; Horiuchi, A.; Joshita, S.; Umemura, T.; Matsumoto, A.; Yoshizawa, K.; et al. Development from simple steatosis to liver cirrhosis and hepatocellular carcinoma: A 27-year follow-up case. Clin. J. Gastroenterol. 2008, 1, 116–121. [Google Scholar] [CrossRef]
- Hu, X.; Tanaka, N.; Guo, R.; Lu, Y.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. PPARα protects against trans-fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 2017, 39, 77–85. [Google Scholar] [CrossRef]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Tanaka, N.; Kimura, T.; Fujimori, N.; Nagaya, T.; Komatsu, M.; Tanaka, E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J. Gastroenterol. 2019, 25, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–7231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Yazaki, M.; Tanaka, N.; Sano, K.; Hashimoto, E.; Takei, Y.; Song, Y.Z.; Tanaka, E.; Kiyosawa, K.; Saheki, T.; et al. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J. Hepatol. 2008, 49, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kimura, T.; Yazaki, M.; Tanaka, N.; Yang, Y.; Nakajima, T.; Horiuchi, A.; Fang, Z.Z.; Joshita, S.; Matsumoto, A.; et al. Steatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARα. Biochim. Biophys. Acta 2015, 1852, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef] [Green Version]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-specific PPARα expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smati, S.; Régnier, M.; Fougeray, T.; Polizzi, A.; Fougerat, A.; Lasserre, F.; Lukowicz, C.; Tramunt, B.; Guillaume, M.; Burnol, A.F.; et al. Regulation of hepatokine gene expression in response to fasting and feeding: Influence of PPAR-α and insulin-dependent signalling in hepatocytes. Diabetes Metab. 2019, in press. [Google Scholar] [CrossRef]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Pena, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARbeta/delta: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, E913. [Google Scholar] [CrossRef] [Green Version]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Barroso, E.; Rodriguez-Calvo, R.; Serrano-Marco, L.; Astudillo, A.M.; Balsinde, J.; Palomer, X.; Vázquez-Carrera, M. The PPARbeta/delta activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1alpha-Lipin 1-PPARalpha pathway leading to increased fatty acid oxidation. Endocrinology 2011, 152, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luquet, S.; Gaudel, C.; Holst, D.; Lopez-Soriano, J.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of PPAR delta in lipid absorption and metabolism: A new target for the treatment of type 2 diabetes. Biochim. Biophys. Acta 2005, 1740, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Lee, C.H.; Barak, Y.; He, W.; Rosenfeld, J.; Liao, D.; Han, J.; Kang, H.; Evans, R.M. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Xie, X.; Fan, Y.; Tian, J.; Guan, Y.; Wang, X.; Zhu, Y.; Wang, N. Peroxisome proliferator-activated receptor-delta induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008, 48, 432–441. [Google Scholar] [CrossRef]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. PPARdelta activation attenuates hepatic steatosis in Ldlr-/- mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Serrano, L.; Rodriguez-Calvo, R.; Davidson, M.M.; Merlos, M.; El Kochairi, I.; Michalik, L.; Wahli, W.; et al. PPARbeta/delta activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim. Biophys. Acta 2011, 1811, 59–67. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Balandaram, G.; Kramer, L.R.; Kang, B.H.; Murray, I.A.; Perdew, G.H.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta suppresses liver tumorigenesis in hepatitis B transgenic mice. Toxicology 2016, 363, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hellemans, K.; Michalik, L.; Dittie, A.; Knorr, A.; Rombouts, K.; De Jong, J.; Heirman, C.; Quartier, E.; Schuit, F.; Wahli, W.; et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology 2003, 124, 184–201. [Google Scholar] [CrossRef]
- Iwaisako, K.; Haimerl, M.; Paik, Y.H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor delta agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Yeon, J.E.; Ko, E.J.; Yoon, E.L.; Suh, S.J.; Kang, K.; Kim, H.R.; Kang, S.H.; Yoo, Y.J.; Je, J.; et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 12787–12799. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Veiga, F.M.; Rachid, T.L.; de Oliveira, L.; Graus-Nunes, F.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. GW0742 (PPAR-beta agonist) attenuates hepatic endoplasmic reticulum stress by improving hepatic energy metabolism in high-fat diet fed mice. Mol. Cell. Endocrinol. 2018, 474, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Wang, D.; Katkuri, S.; Wang, H.; Dey, S.K.; DuBois, R.N. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat. Med. 2004, 10, 245–247. [Google Scholar] [CrossRef]
- Peters, J.M.; Gonzalez, F.J.; Muller, R. Establishing the Role of PPARbeta/delta in Carcinogenesis. Trends Endocrinol. Metab. 2015, 26, 595–607. [Google Scholar] [CrossRef] [Green Version]
- Kaupang, A.; Kase, E.T.; Vo, C.X.; Amundsen, M.; Vik, A.; Hansen, T.V. Synthesis of 5-trifluoromethyl-2-sulfonylpyridine PPARbeta/delta antagonists: Effects on the affinity and selectivity towards PPARbeta/delta. Bioorganic Med. Chem. 2016, 24, 247–260. [Google Scholar] [CrossRef]
- Kaupang, A.; Paulsen, S.M.; Steindal, C.C.; Ravna, A.W.; Sylte, I.; Halvorsen, T.G.; Thoresen, G.H.; Hansen, T.V. Synthesis, biological evaluation and molecular modeling studies of the PPARbeta/delta antagonist CC618. Eur. J. Med. Chem. 2015, 94, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L. GOLDEN-505 Investigator Study Group. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadra, K.; Quignodon, L.; Sardella, C.; Joye, E.; Mucciolo, A.; Chrast, R.; Desvergne, B. PPARgamma in placental angiogenesis. Endocrinology 2010, 151, 4969–4981. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.Q.; Yeldandi, V.; Yeldandi, A.V.; Reddy, J.K. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Ko, E.H.; Kim, J.E.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S.; et al. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARgamma2 is an initial event to induce fatty liver in response to a diet high in butter: PPARgamma2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Mullican, S.E.; DiSpirito, J.R.; Peed, L.C.; Lazar, M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc. Natl. Acad. Sci. USA 2013, 110, 18656–18661. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Takahashi, S.; Matsubara, T.; Jiang, C.; Sakamoto, W.; Chanturiya, T.; Gonzalez, F.J. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J. Biol. Chem. 2015, 290, 3092–3105. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Horiuchi, A.; Yokoyama, T.; Kaneko, G.; Horigome, N.; Yamaura, T.; Nagaya, T.; Komatsu, M.; Sano, K.; Miyagawa, S.; et al. Clinical characteristics of de novo nonalcoholic fatty liver disease following pancreaticoduodenectomy. J. Gastroenterol. 2011, 46, 758–768. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Kimura, T.; Kitabatake, H.; Fujimori, N.; Komatsu, M.; Horiuchi, A.; Yamaura, T.; Umemura, T.; Sano, K.; et al. Mechanism of the development of nonalcoholic steatohepatitis after pancreaticoduodenectomy. BBA Clin. 2015, 3, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.; Scherer, P.E. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metab. 2013, 2, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors: New targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 2003, 14, 459–468. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papavassiliou, A.G. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma ‘on/off’ switches and induction of CBP in colon adenocarcinomas: Correlation with COX-2 expression. Int. J. Colorectal Dis. 2007, 22, 57–68. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, H.F., Jr.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [Green Version]
- Bae, M.A.; Rhee, S.D.; Jung, W.H.; Ahn, J.H.; Song, B.J.; Cheon, H.G. Selective inhibition of activated stellate cells and protection from carbon tetrachloride-induced liver injury in rats by a new PPARgamma agonist KR62776. Arch. Pharm. Res. 2010, 33, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Zhao, D.; Zhang, Y.; Chen, Y.; Zhang, S.; Li, Q.; Zeng, P.; Li, X.; Zhang, W.; Duan, Y.; et al. Rosiglitazone ameliorates bile duct ligation-induced liver fibrosis by down-regulating NF-kappaB-TNF-alpha signaling pathway in a PPARgamma-dependent manner. Biochem. Biophys. Res. Commun. 2019, 519, 854–860. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Lopez-Parra, M.; Garcia-Alonso, V.; Titos, E.; Martinez-Clemente, M.; Gonzalez-Periz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Titos, E.; Rius, B.; Gonzalez-Periz, A.; Garcia-Alonso, V.; Lopez-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Clària, J. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Toti, S.; Lodovici, M.; Giovannelli, L.; Cinci, L.; D’Ambrosio, M.; Luceri, C. Dietary Extra-Virgin Olive Oil Polyphenols Do Not Attenuate Colon Inflammation in Transgenic HLAB-27 Rats but Exert Hypocholesterolemic Effects through the Modulation of HMGCR and PPAR-alpha Gene Expression in the Liver. Lifestyle Genom. 2018, 11, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Avila, J.A.; Gonzalez-Aguilar, G.A.; Alvarez-Parrilla, E.; de la Rosa, L.A. Modulation of PPAR Expression and Activity in Response to Polyphenolic Compounds in High Fat Diets. Int. J. Mol. Sci. 2016, 17, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Jia, Y. The effect of bioactive compounds in tea on lipid metabolism and obesity through regulation of peroxisome proliferator-activated receptors. Curr. Opin. Lipidol. 2015, 26, 3–9. [Google Scholar] [CrossRef]
- Li, J.; Mihalcioiu, M.; Li, L.; Zakikhani, M.; Camirand, A.; Kremer, R. Vitamin D prevents lipid accumulation in murine muscle through regulation of PPARgamma and perilipin-2 expression. J. Steroid Biochem. Mol. Biol. 2018, 177, 116–124. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; D’Urso, M.C.; di Blasio, G.; Brignone, M.S.; De Simone, R.; Minghetti, L. Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav. Immun. 2016, 55, 225–235. [Google Scholar] [CrossRef]
- Jansson, E.A.; Are, A.; Greicius, G.; Kuo, I.C.; Kelly, D.; Arulampalam, V.; Pettersson, S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Vallee, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/beta-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Baker, C.; Christian, P.; Naples, M.; Tong, X.; Zhang, K.; Santha, M.; Adeli, K. Hepatic mitochondrial and ER stress induced by defective PPARalpha signaling in the pathogenesis of hepatic steatosis. American journal of physiology. Endocrinol. Metab. 2014, 306, E1264–E1273. [Google Scholar]
- Lu, Y.; Liu, X.; Jiao, Y.; Xiong, X.; Wang, E.; Wang, X.; Zhang, Z.; Zhang, H.; Pan, L.; Guan, Y.; et al. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARalpha. J. Clin. Investig. 2014, 124, 3501–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. https://doi.org/10.3390/ijms21062061
Wang Y, Nakajima T, Gonzalez FJ, Tanaka N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. International Journal of Molecular Sciences. 2020; 21(6):2061. https://doi.org/10.3390/ijms21062061
Chicago/Turabian StyleWang, Yaping, Takero Nakajima, Frank J. Gonzalez, and Naoki Tanaka. 2020. "PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice" International Journal of Molecular Sciences 21, no. 6: 2061. https://doi.org/10.3390/ijms21062061
APA StyleWang, Y., Nakajima, T., Gonzalez, F. J., & Tanaka, N. (2020). PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. International Journal of Molecular Sciences, 21(6), 2061. https://doi.org/10.3390/ijms21062061