Research Models for Studying Vascular Calcification


Abstract
:1. Introduction
2. In Vitro Models
2.1. Cell Types
2.2. Stimuli for Calcification In Vitro
2.3. Limitations of In Vitro Models
3. Ex Vivo Models
4. In Vivo Models
4.1. Naturally Occuring
4.2. Induction of a Disease State by Chirurgic Intervention and Substance Application
4.3. Genetically Modified Mouse Models
4.3.1. Phosphate Metabolism
4.3.2. Absence or Dysfuntion of Calcification Inhibitor Proteins
4.3.3. Pyrophosphate System
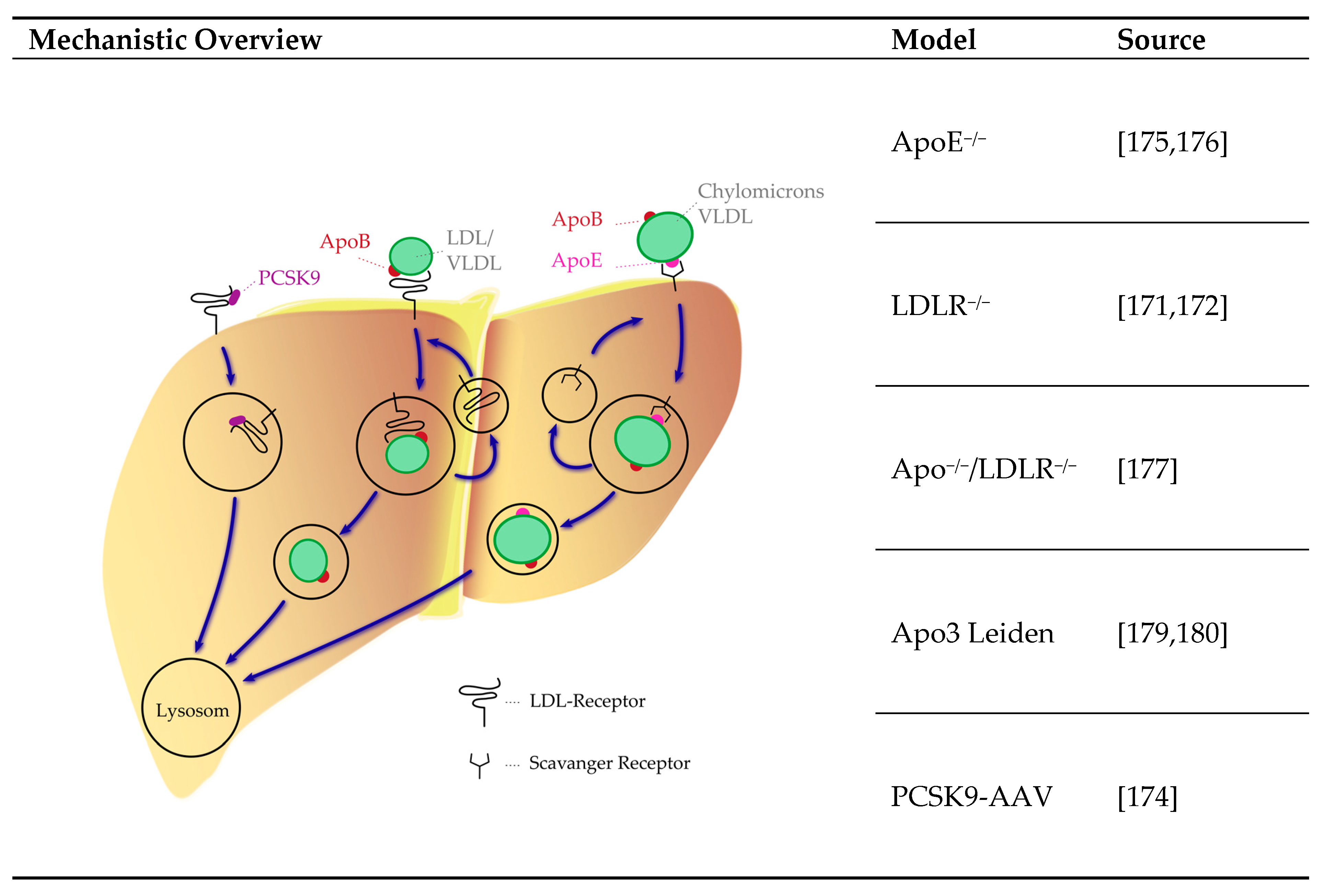
4.3.4. Lipoprotein System
4.4. Limitation of In Vivo Models
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Ascorbic Acid |
| ahsg | α2-Heremans-Schmidt Glycoprotein |
| AAV | Adeno-Associated Virus Vector |
| ALP | Alkaline Phosphatase |
| ApoE | Apolipoprotein E |
| Abcc6 | ATP Binding Cassette Subfamily C Member 6 |
| BUN | Blood Urea Nitrogen |
| BMP | Bone Morphogenic Protein |
| CKD | Chronic Kidney Disease |
| EC | Endothelial Cell |
| EndMT | Endothelial-to-Mesenchymal Transition |
| Enpp | Ectonucleotide Pyrophosphatase Phosphodiesterase |
| ECM | Extracellular Matrix |
| FBS | Fetal Bovine Serum |
| FGF23 | Fibroblast Growth Factor 23 |
| Galnt3 | GalNAc trans-ferase 3 |
| HDL | High Density Lipoprotein |
| hFTC | Hyperphosphatemic Tumoral Familial Calcinosis |
| Ppi | Inorganic Pyrophosphate |
| LDL | Low Density Lipoprotein |
| LDLR | Low Density Lipoprotein Receptor |
| LMNA | Gene encoding the Lamin A/C protein |
| MGP | Matrix Gla Protein |
| MMP | Matrix Metalloproteinases |
| Opn | Osteopontin |
| Opg | oPsteoprotegerin |
| PTH | Parathyroid Hormone |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin Type 9 |
| ROS | Reactive Oxygen Species |
| RANK | Receptor Activated Nuclear Factor κB |
| RANKL | Receptor Activated Nuclear Factor κB Ligand |
| SPP1 | Secreted Phosphoprotein 1 |
| NPT2a | Sodium-Dependent Phosphate Co-Transporters Type IIa |
| NPT2c | Sodium-Dependent Phosphate Co-Transporters Type IIc |
| SD | Streptozotocin-induced Diabetes |
| TNFRSF11B | Tumor Necrosis Factor Receptor Superfamily Member 11B |
| VC | Vascular Calcification |
| VSMC | Vascular Smooth Muscle Cell |
| VLDL | Very Low Density Lipoprotein |
References
- Demer, L.L.; Watson, K.E.; Bostrom, K. Mechanism of calcification in atherosclerosis. Trends Cardiovasc. Med. 1994, 4, 45–49. [Google Scholar] [CrossRef]
- Jiang, Z.M.; Wu, X.J.; Liu, Y.; Du, X.H.; Shen, S.J.; Xu, L.Y.; Sun, W.X. Changes of gene expression profiles across different phases of vascular calcification in rats. Genet. Mol. Res. GMR 2013, 12, 5945–5957. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Inflammation and vascular calcification. Blood Purif. 2005, 23, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, P.; Ho, C.Y.; Liu, Y.; Beltran, L.E.; Ahmad, S.; Jacob, A.P.; Furmanik, M.; Laycock, J.; Long, D.A.; Shroff, R.; et al. Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int. 2019, 95, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Avogaro, A.; Fadini, G.P. Circulating cellular players in vascular calcification. Curr. Pharm. Des. 2014, 20, 5889–5896. [Google Scholar] [CrossRef]
- Cianciolo, G.; Capelli, I.; Cappuccilli, M.; Schillaci, R.; Cozzolino, M.; La Manna, G. Calcifying circulating cells: An uncharted area in the setting of vascular calcification in CKD patients. Clin. Kidney J. 2016, 9, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.C.; Wu, C.C.; Yen, J.F.; Liu, W.C. Vascular calcification and renal bone disorders. Sci. World J. 2014, 2014, 637065. [Google Scholar] [CrossRef]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Ronnemaa, T.; Laakso, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Niskanen, L.; Siitonen, O.; Suhonen, M.; Uusitupa, M.I. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabetes Care 1994, 17, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Neal, C.X.; O’Neill, K.D.; Brown, K.; Westenfeld, R.; Jahnen-Dechent, W.; Ketteler, M. Fetuin-A and matrix gla protein (MGP) are important inhibitors of vascular calcification in CKD. J. Am. Soc. Nephrol. 2003, 14, 692A. [Google Scholar]
- Lomashvili, K.A.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014, 85, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Farzaneh-Far, A.; Shanahan, C.M.; Weissberg, P.L. The role of apoptosis in the initiation of vascular calcification. Z. Fur Kardiol. 2001, 90 (Suppl. 3), 43–46. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- New, S.E.; Aikawa, E. Role of extracellular vesicles in de novo mineralization: An additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1753–1758. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Sinha, A.; Nosoudi, N.; Grover, A.; Vyavahare, N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp. Cell Res. 2014, 323, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Tolle, M.; Reshetnik, A.; Schuchardt, M.; Hohne, M.; van der Giet, M. Arteriosclerosis and vascular calcification: Causes, clinical assessment and therapy. Eur. J. Clin. Investig. 2015, 45, 976–985. [Google Scholar] [CrossRef]
- Li, Q.; Sundberg, J.P.; Levine, M.A.; Terry, S.F.; Uitto, J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle 2015, 14, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef]
- Alesutan, I.; Musculus, K.; Castor, T.; Alzoubi, K.; Voelkl, J.; Lang, F. Inhibition of Phosphate-Induced Vascular Smooth Muscle Cell Osteo-/Chondrogenic Signaling and Calcification by Bafilomycin A1 and Methylamine. Kidney Blood Press. Res. 2015, 40, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.D.; Estepa, M.; Boehme, B.; Pieske, B.; Lang, F.; Eckardt, K.U.; Voelkl, J.; Alesutan, I. Inhibition of vascular smooth muscle cell calcification by vasorin through interference with TGFbeta1 signaling. Cell. Signal. 2019, 64, 109414. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, M.; Tolle, M.; Prufer, J.; Prufer, N.; Huang, T.; Jankowski, V.; Jankowski, J.; Zidek, W.; van der Giet, M. Uridine adenosine tetraphosphate activation of the purinergic receptor P2Y enhances in vitro vascular calcification. Kidney Int. 2012, 81, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prufer, J.; Schuchardt, M.; Tolle, M.; Prufer, N.; Hohne, M.; Zidek, W.; van der Giet, M. Harmful effects of the azathioprine metabolite 6-mercaptopurine in vascular cells: Induction of mineralization. PLoS ONE 2014, 9, e101709. [Google Scholar] [CrossRef]
- Mackenzie, N.C.; Zhu, D.; Longley, L.; Patterson, C.S.; Kommareddy, S.; MacRae, V.E. MOVAS-1 cell line: A new in vitro model of vascular calcification. Int. J. Mol. Med. 2011, 27, 663–668. [Google Scholar]
- Frauscher, B.; Kirsch, A.H.; Schabhuttl, C.; Schweighofer, K.; Ketszeri, M.; Pollheimer, M.; Dragun, D.; Schroder, K.; Rosenkranz, A.R.; Eller, K.; et al. Autophagy Protects from Uremic Vascular Media Calcification. Front Immunol 2018, 9, 1866. [Google Scholar] [CrossRef] [Green Version]
- Skafi, N.; Abdallah, D.; Soulage, C.; Reibel, S.; Vitale, N.; Hamade, E.; Faour, W.; Magne, D.; Badran, B.; Hussein, N.; et al. Phospholipase D: A new mediator during high phosphate-induced vascular calcification associated with chronic kidney disease. J. Cell. Physiol. 2019, 234, 4825–4839. [Google Scholar] [CrossRef]
- Firulli, A.B.; Han, D.; Kelly-Roloff, L.; Koteliansky, V.E.; Schwartz, S.M.; Olson, E.N.; Miano, J.M. A comparative molecular analysis of four rat smooth muscle cell lines. In In Vitro Cellular & Developmental Biology-Animal; Springer: Berlin/Heidelberg, Germany, 1998; Volume 34, pp. 217–226. [Google Scholar]
- Sutra, T.; Morena, M.; Bargnoux, A.S.; Caporiccio, B.; Canaud, B.; Cristol, J.P. Superoxide production: A procalcifying cell signalling event in osteoblastic differentiation of vascular smooth muscle cells exposed to calcification media. Free Radic. Res. 2008, 42, 789–797. [Google Scholar] [CrossRef]
- Rao, R.S.; Miano, J.M.; Olson, E.N.; Seidel, C.L. The A10 cell line: A model for neonatal, neointimal, or differentiated vascular smooth muscle cells? Cardiovasc. Res. 1997, 36, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Beazley, K.E.; Deasey, S.; Lima, F.; Nurminskaya, M.V. Transglutaminase 2-mediated activation of beta-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schor, A.M.; Allen, T.D.; Canfield, A.E.; Sloan, P.; Schor, S.L. Pericytes derived from the retinal microvasculature undergo calcification in vitro. J. Cell Sci. 1990, 97 Pt 3, 449–461. [Google Scholar] [PubMed]
- Wu, M.; Zhang, J.D.; Tang, R.N.; Crowley, S.D.; Liu, H.; Lv, L.L.; Ma, K.L.; Liu, B.C. Elevated PTH induces endothelial-to-chondrogenic transition in aortic endothelial cells. Am. J. Physiol. Ren. Physiol. 2017, 312, F436–F444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eghbali-Fatourechi, G.Z.; Lamsam, J.; Fraser, D.; Nagel, D.; Riggs, B.L.; Khosla, S. Circulating osteoblast-lineage cells in humans. N. Engl. J. Med. 2005, 352, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Otsuru, S.; Tamai, K.; Yamazaki, T.; Yoshikawa, H.; Kaneda, Y. Bone marrow-derived osteoblast progenitor cells in circulating blood contribute to ectopic bone formation in mice. Biochem. Biophys. Res. Commun. 2007, 354, 453–458. [Google Scholar] [CrossRef]
- Gossl, M.; Modder, U.I.; Atkinson, E.J.; Lerman, A.; Khosla, S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J. Am. Coll. Cardiol. 2008, 52, 1314–1325. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Hunt, P.; McElvain, M.; Black, T.; Kaufman, S.; Choi, E.S. Osteoblast precursor cells are found in CD34+ cells from human bone marrow. Stem Cells 1997, 15, 368–377. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Vigili de Kreutzenberg, S.; Agostini, C.; Cabrelle, A.; Binotto, G.; Rattazzi, M.; Bertacco, E.; et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ. Res. 2011, 108, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Yip, C.Y.; Chen, J.H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef] [Green Version]
- Voelkl, J.; Lang, F.; Eckardt, K.U.; Amann, K.; Kuro, O.M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. CMLS 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [Green Version]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging 2019, 11, 5445–5462. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Cheng, W.; Huang, T.; Yuan, J.; Wang, X.; Song, M. Vascular Adventitia Calcification and Its Underlying Mechanism. PLoS ONE 2015, 10, e0132506. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.J.; Ashton, B.A.; Walsh, S.; Beresford, J.N.; Grant, M.E.; Canfield, A.E. Vascular pericytes express osteogenic potential in vitro and in vivo. J. Bone Min. Res. 1998, 13, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Canfield, A.E.; Doherty, M.J.; Wood, A.C.; Farrington, C.; Ashton, B.; Begum, N.; Harvey, B.; Poole, A.; Grant, M.E.; Boot-Handford, R.P. Role of pericytes in vascular calcification: A review. Z. Fur Kardiol. 2000, 89 (Suppl 2), 20–27. [Google Scholar] [CrossRef]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R.; Bostrom, K.I. A role for the endothelium in vascular calcification. Circ. Res. 2013, 113, 495–504. [Google Scholar] [CrossRef]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.-J.; Ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef]
- Bogdanova, M.; Kostina, A.; Zihlavnikova Enayati, K.; Zabirnyk, A.; Malashicheva, A.; Stenslokken, K.O.; Sullivan, G.J.; Kaljusto, M.L.; Kvitting, J.P.; Kostareva, A.; et al. Inflammation and Mechanical Stress Stimulate Osteogenic Differentiation of Human Aortic Valve Interstitial Cells. Front. Physiol. 2018, 9, 1635. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Z.Z.; Chen, H.; Zhang, G.J.; Kong, Y.H.; Kang, X.X. Oxidized low-density lipoprotein and beta-glycerophosphate synergistically induce endothelial progenitor cell ossification. Acta Pharmacol. Sin. 2011, 32, 1491–1497. [Google Scholar] [CrossRef]
- Jaiswal, N.; Haynesworth, S.E.; Caplan, A.I.; Bruder, S.P. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J. Cell. Biochem. 1997, 64, 295–312. [Google Scholar] [CrossRef]
- Kanemaru, K.; Seya, K.; Miki, I.; Motomura, S.; Furukawa, K. Calcification of aortic smooth muscle cells isolated from spontaneously hypertensive rats. J. Pharmacol. Sci. 2008, 106, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, R.T. The role of ascorbic acid in mesenchymal differentiation. Nutr. Rev. 1992, 50, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Wouters, O.Y.; de Rond, S.; Boersema, M.; Gjaltema, R.A.F.; Bank, R.A. Ascorbic acid promotes a TGFβ1-induced myofibroblast phenotype switch. Physiol. Rep. 2017, 5, e13324. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.; Ivanova, S.; Roomi, M.W.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Extracellular Matrix-Mediated Control of Aortic Smooth Muscle Cell Growth and Migration by a Combination of Ascorbic Acid, Lysine, Proline, and Catechins. J. Cardiovasc. Pharmacol. 2007, 50, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Bell, J.; Juliao, S.; Li, L.; May, J.M. Ascorbic acid uptake and regulation of type I collagen synthesis in cultured vascular smooth muscle cells. J. Vasc. Res. 2009, 46, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Roszkowska, M.; Strzelecka-Kiliszek, A.; Bessueille, L.; Buchet, R.; Magne, D.; Pikula, S. Collagen promotes matrix vesicle-mediated mineralization by vascular smooth muscle cells. J. Inorg. Biochem. 2018, 186, 1–9. [Google Scholar] [CrossRef]
- Gayrard, N.; Muyor, K.; Notarnicola, C.; Duranton, F.; Jover, B.; Argilés, À. Optimisation of cell and ex vivo culture conditions to study vascular calcification. PLoS ONE 2020, 15, e0230201. [Google Scholar] [CrossRef]
- Chen, N.X.; Duan, D.; O’Neill, K.D.; Moe, S.M. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2006, 21, 3435–3442. [Google Scholar] [CrossRef] [Green Version]
- Hjortnaes, J.; Goettsch, C.; Hutcheson, J.D.; Camci-Unal, G.; Lax, L.; Scherer, K.; Body, S.; Schoen, F.J.; Kluin, J.; Khademhosseini, A.; et al. Simulation of early calcific aortic valve disease in a 3D platform: A role for myofibroblast differentiation. J. Mol. Cell. Cardiol. 2016, 94, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Akiyoshi, T.; Ota, H.; Iijima, K.; Son, B.K.; Kahyo, T.; Setou, M.; Ogawa, S.; Ouchi, Y.; Akishita, M. A novel organ culture model of aorta for vascular calcification. Atherosclerosis 2016, 244, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuffaha, R.; Voelkl, J.; Pieske, B.; Lang, F.; Alesutan, I. Role of PKB/SGK-dependent phosphorylation of GSK-3alpha/beta in vascular calcification during cholecalciferol overload in mice. Biochem. Biophys. Res. Commun. 2018, 503, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Hernandez-Martinez, E.; Merida-Herrero, E.; Gonzalez-Parra, E. Impact of acetate- or citrate-acidified bicarbonate dialysate on ex vivo aorta wall calcification. Sci. Rep. 2019, 9, 11374. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Quan, Z.; Luo, D.; Chen, W.; Peng, D. Spironolactone dosedependently alleviates the calcification of aortic rings cultured in hyperphosphatemic medium with or without hyperglycemia by suppressing phenotypic transition of VSMCs through downregulation of Pit1. Mol. Med. Rep. 2019, 19, 3622–3632. [Google Scholar] [PubMed]
- Schuchardt, M.; Siegel, N.V.; Babic, M.; Reshetnik, A.; Lutzenberg, R.; Zidek, W.; van der Giet, M.; Tolle, M. A Novel Long-Term ex vivo Model for Studying Vascular Calcification Pathogenesis: The Rat Isolated-Perfused Aorta. J. Vasc. Res. 2020, 57, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Kaspareit-Rittinghausen, J.; Rapp, K.; Deerberg, F.; Wcislo, A.; Messow, C. Hereditary polycystic kidney disease associated with osteorenal syndrome in rats. Vet. Pathol. 1989, 26, 195–201. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X.; Seifert, M.F.; Sinders, R.M.; Duan, D.; Chen, X.; Liang, Y.; Radcliff, J.S.; White, K.E.; Gattone, V.H., 2nd. A rat model of chronic kidney disease-mineral bone disorder. Kidney Int. 2009, 75, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Hildreth, C.M.; Phillips, J.K.; Avolio, A.P. Aortic stiffness is associated with vascular calcification and remodeling in a chronic kidney disease rat model. Am. J. Physiol.-Renal Physiol. 2011, 300, F1431–F1436. [Google Scholar] [CrossRef] [Green Version]
- Rings, R.W.; Wagner, J.E. Incidence of cardiac and other soft tissue mineralized lesions in DBA-2 mice. Lab. Anim. Sci. 1972, 22, 344–352. [Google Scholar]
- Chauntin, A.; Ferris, E. Experimental renal insufficiency produced by partial nephrectomy. Arch. Intern. Med. 1932, 49, 767–787. [Google Scholar] [CrossRef]
- Gagnon, R.F.; Duguid, W.P. A reproducible model for chronic renal failure in the mouse. Urol. Res. 1983, 11, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular calcification in animal models of CKD: A review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Oura, H.; Okada, T. Metabolic effects of dietary purine in rats. J. Nutr. Sci. Vitaminol. 1982, 28, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, I.; Hamano, T.; Mikami, S.; Fujii, N.; Takabatake, Y.; Nagasawa, Y.; Kawada, N.; Ito, T.; Rakugi, H.; Imai, E.; et al. Fully phosphorylated fetuin-A forms a mineral complex in the serum of rats with adenine-induced renal failure. Kidney Int. 2009, 75, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Shobeiri, N.; Pang, J.; Adams, M.A.; Holden, R.M. Cardiovascular disease in an adenine-induced model of chronic kidney disease: The temporal link between vascular calcification and haemodynamic consequences. J. Hypertens. 2013, 31, 160–168. [Google Scholar] [CrossRef]
- Katsumata, K.; Kusano, K.; Hirata, M.; Tsunemi, K.; Nagano, N.; Burke, S.K.; Fukushima, N. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int. 2003, 64, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.A.; Roublick, A.M.; Williamson, M.K. Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int. 2006, 70, 1577–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neven, E.; Dauwe, S.; De Broe, M.E.; D’Haese, P.C.; Persy, V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. 2007, 72, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, C.; Davis, J.; Miller, G.; Shatzen, E.; Cattley, R.; Li, X.; Martin, D.; Yao, W.; Lane, N.; Shalhoub, V. The calcimimetic AMG 641 abrogates parathyroid hyperplasia, bone and vascular calcification abnormalities in uremic rats. Eur. J. Pharmacol. 2009, 616, 306–313. [Google Scholar] [CrossRef]
- Neven, E.; Dams, G.; Postnov, A.; Chen, B.; De Clerck, N.; De Broe, M.E.; D’Haese, P.C.; Persy, V. Adequate phosphate binding with lanthanum carbonate attenuates arterial calcification in chronic renal failure rats. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2009, 24, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Lomashvili, K.A.; Monier-Faugere, M.C.; Wang, X.; Malluche, H.H.; O’Neill, W.C. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 2009, 75, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persy, V.; Postnov, A.; Neven, E.; Dams, G.; De Broe, M.; D’Haese, P.; De Clerck, N. High-resolution X-ray microtomography is a sensitive method to detect vascular calcification in living rats with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2110–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamagaki, K.; Yuan, Q.; Ohkawa, H.; Imazeki, I.; Moriguchi, Y.; Imai, N.; Sasaki, S.; Takeda, K.; Fukagawa, M. Severe hyperparathyroidism with bone abnormalities and metastatic calcification in rats with adenine-induced uraemia. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2006, 21, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana, A.C.; Degaspari, S.; Catanozi, S.; Delle, H.; de Sa Lima, L.; Silva, C.; Blanco, P.; Solez, K.; Scavone, C.; Noronha, I.L. Thalidomide suppresses inflammation in adenine-induced CKD with uraemia in mice. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2013, 28, 1140–1149. [Google Scholar] [CrossRef]
- Ferrari, G.O.; Ferreira, J.C.; Cavallari, R.T.; Neves, K.R.; dos Reis, L.M.; Dominguez, W.V.; Oliveira, E.C.; Graciolli, F.G.; Passlick-Deetjen, J.; Jorgetti, V.; et al. Mineral bone disorder in chronic kidney disease: Head-to-head comparison of the 5/6 nephrectomy and adenine models. BMC Nephrol. 2014, 15, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, Y.; Ohnishi, A.; Watanabe, M.; Ishibashi, K.; Kusano, E. Establishment of acute kidney injury mouse model by 0.75% adenine ingestion. Ren. Fail. 2011, 33, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wei, Q.; Shao, H.; Sun, Z.; Liu, N. A rat model of diabetic artery calcification. J. Endocrinol. Investig. 2012, 35, 497–503. [Google Scholar]
- Ejerblad, S.; Eriksson, I.; Johansson, H. Uraemic arterial disease. An experimental study with special reference to the effect of parathyroidectomy. Scand. J. Urol. Nephrol. 1979, 13, 161–169. [Google Scholar] [CrossRef]
- Krog, M.; Ejerblad, S.; Eriksson, I.; Johansson, H. Arterial calcifications in uraemic rats treated with 1-alpha-hydroxycholecalciferol and parathyroidectomy. Scand. J. Urol. Nephrol. 1984, 18, 227–239. [Google Scholar] [CrossRef]
- Hirata, M.; Katsumata, K.; Endo, K.; Fukushima, N.; Ohkawa, H.; Fukagawa, M. In subtotally nephrectomized rats 22-oxacalcitriol suppresses parathyroid hormone with less risk of cardiovascular calcification or deterioration of residual renal function than 1,25(OH)2 vitamin D3. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2003, 18, 1770–1776. [Google Scholar] [CrossRef] [Green Version]
- Henley, C.; Colloton, M.; Cattley, R.C.; Shatzen, E.; Towler, D.A.; Lacey, D.; Martin, D. 1,25-Dihydroxyvitamin D3 but not cinacalcet HCl (Sensipar/Mimpara) treatment mediates aortic calcification in a rat model of secondary hyperparathyroidism. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2005, 20, 1370–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, I.; Aguilera-Tejero, E.; Mendoza, F.J.; Almaden, Y.; Perez, J.; Martin, D.; Rodriguez, M. Calcimimetic R-568 decreases extraosseous calcifications in uremic rats treated with calcitriol. J. Am. Soc. Nephrol. Jasn 2006, 17, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Wu-Wong, J.R.; Noonan, W.; Ma, J.; Dixon, D.; Nakane, M.; Bolin, A.L.; Koch, K.A.; Postl, S.; Morgan, S.J.; Reinhart, G.A. Role of phosphorus and vitamin D analogs in the pathogenesis of vascular calcification. J. Pharmacol. Exp. Ther. 2006, 318, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Suzuki, Y.; Matsushita, M.; Fujii, H.; Miyaura, C.; Aizawa, S.; Kogo, H. Prevention of aortic calcification by etidronate in the renal failure rat model. Eur. J. Pharmacol. 2007, 558, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Cardus, A.; Panizo, S.; Parisi, E.; Fernandez, E.; Valdivielso, J.M. Differential effects of vitamin D analogs on vascular calcification. J. Bone Min. Res. 2007, 22, 860–866. [Google Scholar] [CrossRef]
- Mendoza, F.J.; Lopez, I.; Montes de Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney Int. 2008, 73, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Lopez, I.; Mendoza, F.J.; Aguilera-Tejero, E.; Perez, J.; Guerrero, F.; Martin, D.; Rodriguez, M. The effect of calcitriol, paricalcitol, and a calcimimetic on extraosseous calcifications in uremic rats. Kidney Int. 2008, 73, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Haut, L.L.; Alfrey, A.C.; Guggenheim, S.; Buddington, B.; Schrier, N. Renal toxicity of phosphate in rats. Kidney Int. 1980, 17, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Staniforth, M.E.; Liapis, H.; Finch, J.; Burke, S.K.; Dusso, A.S.; Slatopolsky, E. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003, 64, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizobuchi, M.; Ogata, H.; Hatamura, I.; Koiwa, F.; Saji, F.; Shiizaki, K.; Negi, S.; Kinugasa, E.; Ooshima, A.; Koshikawa, S.; et al. Up-regulation of Cbfa1 and Pit-1 in calcified artery of uraemic rats with severe hyperphosphataemia and secondary hyperparathyroidism. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2006, 21, 911–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graciolli, F.G.; Neves, K.R.; dos Reis, L.M.; Graciolli, R.G.; Noronha, I.L.; Moyses, R.M.; Jorgetti, V. Phosphorus overload and PTH induce aortic expression of Runx2 in experimental uraemia. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2009, 24, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Corriere, M.A.; Matrisian, L.M.; Guzman, R.J. Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1510–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assmann, A.; Zwirnmann, K.; Heidelberg, F.; Schiffer, F.; Horstkotter, K.; Munakata, H.; Gremse, F.; Barth, M.; Lichtenberg, A.; Akhyari, P. The degeneration of biological cardiovascular prostheses under pro-calcific metabolic conditions in a small animal model. Biomaterials 2014, 35, 7416–7428. [Google Scholar] [CrossRef] [PubMed]
- Bas, A.; Lopez, I.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J. Bone Min. Res. 2006, 21, 484–490. [Google Scholar] [CrossRef]
- Tang, F.T.; Chen, S.R.; Wu, X.Q.; Wang, T.Q.; Chen, J.W.; Li, J.; Bao, L.P.; Huang, H.Q.; Liu, P.Q. Hypercholesterolemia accelerates vascular calcification induced by excessive vitamin D via oxidative stress. Calcif. Tissue Int. 2006, 79, 326–339. [Google Scholar] [CrossRef]
- Atkinson, J.; Poitevin, P.; Chillon, J.M.; Lartaud, I.; Levy, B. Vascular Ca overload produced by vitamin D3 plus nicotine diminishes arterial distensibility in rats. Am. J. Physiol. 1994, 266 Pt 2, H540–H547. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Zhao, X.; Pu, X.P. Proteome analysis of the left ventricle in the vitamin D(3) and nicotine-induced rat vascular calcification model. J. Proteom. 2011, 74, 480–489. [Google Scholar] [CrossRef]
- Niederhoffer, N.; Bobryshev, Y.V.; Lartaud-Idjouadiene, I.; Giummelly, P.; Atkinson, J. Aortic calcification produced by vitamin D3 plus nicotine. J. Vasc. Res. 1997, 34, 386–398. [Google Scholar] [CrossRef]
- Shuvy, M.; Abedat, S.; Beeri, R.; Valitzki, M.; Stein, Y.; Meir, K.; Lotan, C. Electromagnetic fields promote severe and unique vascular calcification in an animal model of ectopic calcification. Exp. Toxicol. Pathol. Off. J. Ges. Fur Toxikol. Pathol. 2014, 66, 345–350. [Google Scholar] [CrossRef]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular calcification: The killer of patients with chronic kidney disease. J. Am. Soc. Nephrol. JASN 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; Liu, S.; Tang, W.; Zhou, J.; Wang, Y.; Yao, X.; Quarles, L.D. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J. Am. Soc. Nephrol. JASN 2007, 18, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesse, M.; Frohlich, L.F.; Zeitz, U.; Lanske, B.; Erben, R.G. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. J. Int. Soc. Matrix Biol. 2007, 26, 75–84. [Google Scholar] [CrossRef]
- Razzaque, M.S.; Sitara, D.; Taguchi, T.; St-Arnaud, R.; Lanske, B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 720–722. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, M.; Nakatani, T.; Lanske, B.; Razzaque, M.S. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. 2009, 75, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3562–3571. [Google Scholar] [CrossRef] [Green Version]
- Kuro, O.M. Molecular Mechanisms Underlying Accelerated Aging by Defects in the FGF23-Klotho System. Int. J. Nephrol. 2018, 2018, 9679841. [Google Scholar] [CrossRef]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Folsom, L.J.; Imel, E.A. Hyperphosphatemic familial tumoral calcinosis: Genetic models of deficient FGF23 action. Curr. Osteoporos. Rep. 2015, 13, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Gattineni, J.; Baum, M. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): Implications for disorders of phosphate metabolism. Pediatr. Nephrol. 2010, 25, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tang, W.; Zhou, J.; Stubbs, J.R.; Luo, Q.; Pi, M.; Quarles, L.D. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J. Am. Soc. Nephrol. JASN 2006, 17, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Pike, J.W. Mechanistic homeostasis of vitamin D metabolism in the kidney through reciprocal modulation of Cyp27b1 and Cyp24a1 expression. J. Steroid Biochem. Mol. Biol. 2020, 196, 105500. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology 2009, 150, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Topaz, O.; Shurman, D.L.; Bergman, R.; Indelman, M.; Ratajczak, P.; Mizrachi, M.; Khamaysi, Z.; Behar, D.; Petronius, D.; Friedman, V.; et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet. 2004, 36, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Esapa, C.T.; Head, R.A.; Jeyabalan, J.; Evans, H.; Hough, T.A.; Cheeseman, M.T.; McNally, E.G.; Carr, A.J.; Thomas, G.P.; Brown, M.A.; et al. A mouse with an N-Ethyl-N-nitrosourea (ENU) Induced Trp589Arg Galnt3 mutation represents a model for hyperphosphataemic familial tumoural calcinosis. PLoS ONE 2012, 7, e43205. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, S.; Gray, A.K.; Padgett, L.R.; Allen, M.R.; Clinkenbeard, E.L.; Sarpa, N.M.; White, K.E.; Econs, M.J. Genetic rescue of glycosylation-deficient Fgf23 in the Galnt3 knockout mouse. Endocrinology 2014, 155, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hum, J.M.; O’Bryan, L.M.; Tatiparthi, A.K.; Cass, T.A.; Clinkenbeard, E.L.; Cramer, M.S.; Bhaskaran, M.; Johnson, R.L.; Wilson, J.M.; Smith, R.C.; et al. Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho. J. Am. Soc. Nephrol. JASN 2017, 28, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho in health and disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Clin. Investig. 2007, 117, 2684–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, A.; Traghella, I.; Mazzone, A.; Sbrana, S.; Vassalle, C. Chapter Two―Vascular and valvular calcification biomarkers. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 95, pp. 73–103. [Google Scholar]
- Back, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceicao, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement from the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2018, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Schinke, T.; Trindl, A.; Muller-Esterl, W.; Sablitzky, F.; Kaiser, S.; Blessing, M. Cloning and targeted deletion of the mouse fetuin gene. J. Biol. Chem. 1997, 272, 31496–31503. [Google Scholar] [CrossRef] [Green Version]
- Schafer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Muller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Speer, M.Y.; McKee, M.D.; Guldberg, R.E.; Liaw, L.; Yang, H.Y.; Tung, E.; Karsenty, G.; Giachelli, C.M. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: Evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 2002, 196, 1047–1055. [Google Scholar] [CrossRef]
- Galvin, K.M.; Donovan, M.J.; Lynch, C.A.; Meyer, R.I.; Paul, R.J.; Lorenz, J.N.; Fairchild-Huntress, V.; Dixon, K.L.; Dunmore, J.H.; Gimbrone, M.A., Jr.; et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000, 24, 171–174. [Google Scholar] [CrossRef] [PubMed]
- El-Maadawy, S.; Kaartinen, M.T.; Schinke, T.; Murshed, M.; Karsenty, G.; McKee, M.D. Cartilage formation and calcification in arteries of mice lacking matrix Gla protein. Connect. Tissue Res. 2003, 44 (Suppl. 1), 272–278. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J. Cell Biol. 2004, 165, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Shanahan, C.M. Molecular mechanisms mediating vascular calcification: Role of matrix Gla protein. Nephrology 2006, 11, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grotzinger, J.; Yamamoto, K.; Renne, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.A.; Nguyen, T.M.; Williamson, M.K. Biochemical characterization of the serum fetuin-mineral complex. J. Biol. Chem. 2003, 278, 22153–22160. [Google Scholar] [CrossRef] [Green Version]
- Collin-Osdoby, P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ. Res. 2004, 95, 1046–1057. [Google Scholar] [CrossRef]
- Min, H.; Moronyb, S.; Sarosib, I.; Dunstanb, C.; Capparellib, C.; Scullyb, S.; Vanb, G.; Kaufmanb, S.; Kostenuikb, P.; Laceyb, D. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J. Exp. Med. 2000, 192, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Ishida, W.; Hamamoto, T.; Kusanagi, K.; Yagi, K.; Kawabata, M.; Takehara, K.; Sampath, T.K.; Kato, M.; Miyazono, K. Smad6 is a Smad1/5-induced smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J. Biol. Chem. 2000, 275, 6075–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, K.; Kamiya, Y.; Imamura, T.; Miyazono, K.; Miyazawa, K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 2007, 282, 20603–20611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleisch, H.; Bisaz, S. Mechanism of calcification: Inhibitory role of pyrophosphate. Nature 1962, 195, 911. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, N.C.; Zhu, D.; Milne, E.M.; van’t Hof, R.; Martin, A.; Darryl Quarles, L.; Millan, J.L.; Farquharson, C.; MacRae, V.E. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Watanabe, R.; Fujita, N.; Sato, Y.; Kobayashi, T.; Morita, M.; Oike, T.; Miyamoto, K.; Kuro, O.M.; Michigami, T.; Fukumoto, S.; et al. Enpp1 is an anti-aging factor that regulates Klotho under phosphate overload conditions. Sci. Rep. 2017, 7, 7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Guo, H.; Chou, D.W.; Berndt, A.; Sundberg, J.P.; Uitto, J. Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy. Dis. Models Mech. 2013, 6, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Kauffenstein, G.; Pizard, A.; Le Corre, Y.; Vessieres, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.G.; Bergen, A.A.; et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acin-Perez, R.; Enriquez, J.A.; Lopez-Otin, C.; Andres, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Varadi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.; Borst, P.; et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Varadi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Snook, A.E.; Uitto, J.; Li, Q. Adenovirus-Mediated ABCC6 Gene Therapy for Heritable Ectopic Mineralization Disorders. J. Investig. Derm. 2019, 139, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andres-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; Lopez-Otin, C.; Andres, V. Vascular Smooth Muscle-Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharm. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research—Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl Ester Transfer Protein. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef]
- Awan, Z.; Denis, M.; Bailey, D.; Giaid, A.; Prat, A.; Goltzman, D.; Seidah, N.G.; Genest, J. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis 2011, 219, 455–462. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Fisher, E.A.; Breslow, J.L. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc. Natl. Acad. Sci. USA 2005, 102, 2069–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettsch, C.; Hutcheson, J.D.; Hagita, S.; Rogers, M.A.; Creager, M.D.; Pham, T.; Choi, J.; Mlynarchik, A.K.; Pieper, B.; Kjolby, M.; et al. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis 2016, 251, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: Potential role of chondrocyte-like cells. Arter. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Langheinrich, A.C.; Michniewicz, A.; Sedding, D.G.; Walker, G.; Beighley, P.E.; Rau, W.S.; Bohle, R.M.; Ritman, E.L. Correlation of vasa vasorum neovascularization and plaque progression in aortas of apolipoprotein E(−/−)/low-density lipoprotein(−/−) double knockout mice. Arter. Thromb. Vasc. Biol. 2006, 26, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Massy, Z.A.; Ivanovski, O.; Nguyen-Khoa, T.; Angulo, J.; Szumilak, D.; Mothu, N.; Phan, O.; Daudon, M.; Lacour, B.; Drueke, T.B.; et al. Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J. Am. Soc. Nephrol. JASN 2005, 16, 109–116. [Google Scholar] [CrossRef]
- van Vlijmen, B.J.; van den Maagdenberg, A.M.; Gijbels, M.J.; van der Boom, H.; HogenEsch, H.; Frants, R.R.; Hofker, M.H.; Havekes, L.M. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J. Clin. Investig. 1994, 93, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Lutgens, E.; Daemen, M.; Kockx, M.; Doevendans, P.; Hofker, M.; Havekes, L.; Wellens, H.; de Muinck, E.D. Atherosclerosis in APOE*3-Leiden transgenic mice: From proliferative to atheromatous stage. Circulation 1999, 99, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen & Co Ltd.: London UK, 1959. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Cell Type | Source |
|---|---|---|
| Tunica Externa | Myofibroblasts | [20] |
| Tunica Media | Primary VSMC | [21,22,23,24,25] |
| MOVAS | [26,27,28] | |
| A7r5 | [29,30] | |
| A10 | [31,32] | |
| Tunica Intima | Pericytes | [33] |
| Endothelial Cells | [34] | |
| Circulating | Mesenchymal origin | [35,36] |
| Hematopoietic origin | [37,38,39] | |
| Heart | Valvular Interstitial Cells | [40] |
| Supplement | Common Concentration | Source |
|---|---|---|
| Serum/FBS | 0%–20% | [21,24,25,33] |
| Glucose | 5–25 mM | [21,60] |
| Inorganic Phosphate: Sodium hydrogen phosphate | 1.6 mM | [32] |
| Organic Phosphate: β-glycerophosphate | 1.25–10 mM | [21,27,52] |
| Calcium | 2.5 mM CaCl | [43] |
| Ascorbic Acid | 10 µg/mL–50 µg/mL | [24,25,27,40] |
| Sodium pyruvate | 10 mM | [21] |
| Insulin | 10−7 M | [21] |
| Model Type | Predominant Localization of Calcification | |
|---|---|---|
| Intimal | Medial | |
| Naturally Occurring | DBA2 mice CY+ rat with autosomal dominant polycystic kidney disease Lewis polycystic kidney disease rat | |
| Operation | Kidney reduction (electrocautery, nephrectomy) | |
| Feeding/Substance Application | Cholesterol Rich Chow PCSK9-AAV | Adenine Vitamin D Phosphate Streptozotocin |
| Genetic Modification | Lipoprotein System ApoE−/− Lldlr−/− ApoE3 Leiden PCSK9-AAV | Phosphate Metabolism Klotho−/− FGF23−/− Galnt−/− Tcal/Tcal Pyrophosphate Metabolism Abcc−/− Enpp1−/− Lmna−/− Osteogenic Signaling Fetuin A−/− Opg−/− Mgp−/− Opn−/− Madh6−/− |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, J.; Babic, M.; Tölle, M.; van der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. https://doi.org/10.3390/ijms21062204
Herrmann J, Babic M, Tölle M, van der Giet M, Schuchardt M. Research Models for Studying Vascular Calcification. International Journal of Molecular Sciences. 2020; 21(6):2204. https://doi.org/10.3390/ijms21062204
Chicago/Turabian StyleHerrmann, Jaqueline, Milen Babic, Markus Tölle, Markus van der Giet, and Mirjam Schuchardt. 2020. "Research Models for Studying Vascular Calcification" International Journal of Molecular Sciences 21, no. 6: 2204. https://doi.org/10.3390/ijms21062204