Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Localization of IL-1β and RARγ in Fractured Rat Ribs
2.2. Modulation of the Expression of Chondrocyte-Specific Genes’ mRNA by IL-1β and Retinoid Receptor Agonist in ATDC5 Cells
2.3. The Effect of the RAR Inverse Agonist AGN194310 on IL-1β Modulated Genes
2.4. Analysis of the Actions of the RAR Inverse Agonist AGN194310 on IL-1β-Induced Mitogen-Activated Protein Kinase (MAPK)
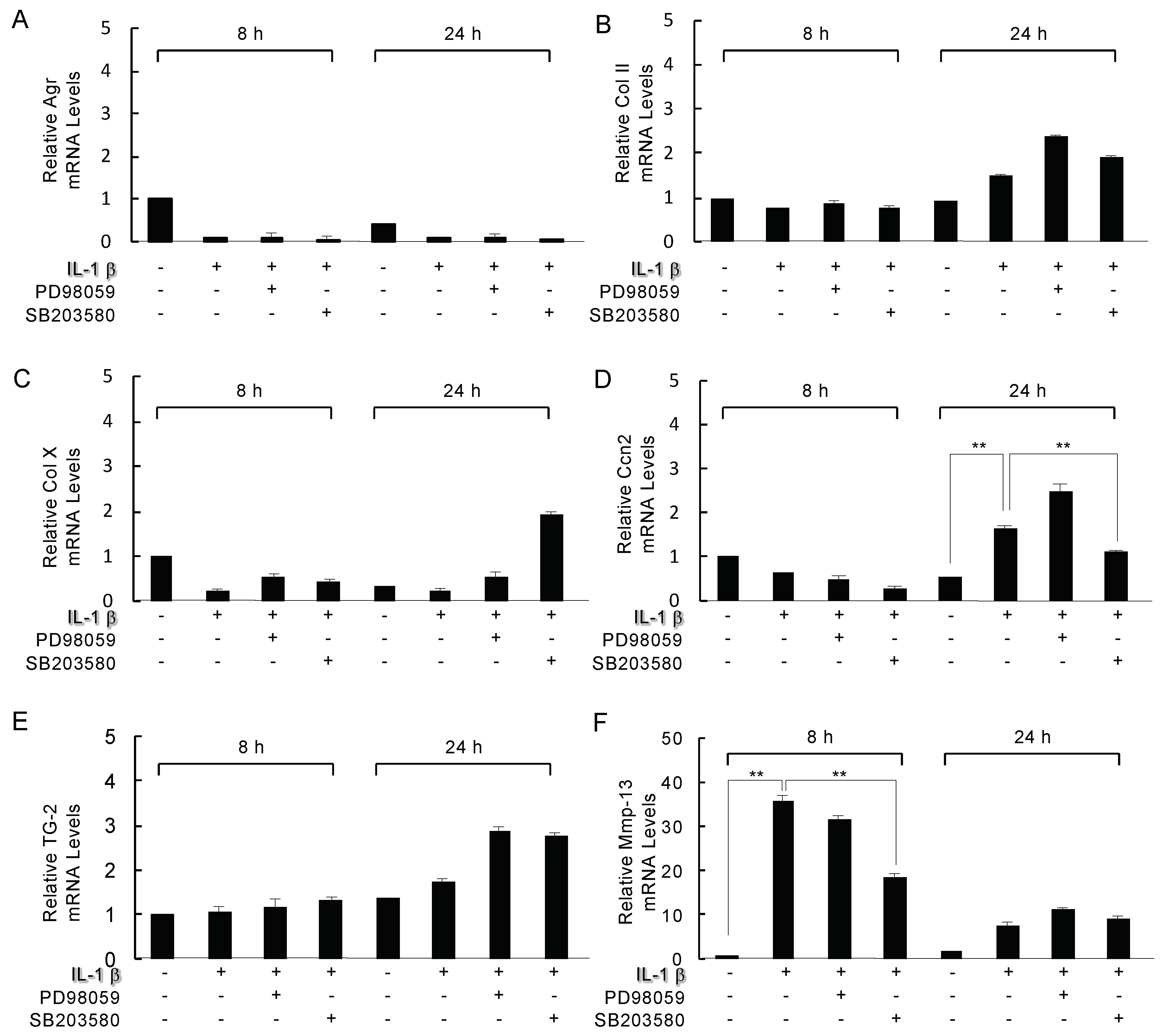
2.5. Analysis of the Inhibition of Mitogen-Activated Protein Kinase (MAPK) on IL-1β Modulated Genes
2.6. The Localization of MMP-13, CCN2, p-p38 MAPK, and α-SMA in Fractured Rat Ribs
3. Discussion
4. Materials and Methods
4.1. Experimental Model for Fracture Healing
4.2. Immunohistochemistry
4.3. Cell Cultures
4.4. Immunoblot Analysis
4.5. p38 MAPK Assay
4.6. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IL-1β | Interleukin-1β |
| RAR | retinoid receptor |
| MMP | matrix metalloproteinase |
| CTGF/CCN2 | connective tissue growth factor |
| ECM | extracellular matrix |
References
- Dennis, S.C.; Berkland, C.J.; Bonewald, L.F.; Detamore, M.S. Endochondral ossification for enhancing bone regeneration: Converging native extracellular matrix biomaterials and developmental engineering in vivo. Tissue Eng. Part B Rev. 2015, 21, 247–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Ghali, O.; Lencel, P.; Broux, O.; Chauveau, C.; Devedjian, J.C.; Hardouin, P.; Magne, D. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. 2009, 84, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Wehling, N.; Palmer, G.D.; Pilapil, C.; Liu, F.; Wells, J.W.; Muller, P.E.; Evans, C.H.; Porter, R.M. Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum. 2009, 60, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Mumme, M.; Scotti, C.; Papadimitropoulos, A.; Todorov, A.; Hoffmann, W.; Bocelli-Tyndall, C.; Jakob, M.; Wendt, D.; Martin, I.; Barbero, A. Interleukin-1beta modulates endochondral ossification by human adult bone marrow stromal cells. Eur. Cells Mater. 2012, 24, 224–236. [Google Scholar] [CrossRef]
- Murakami, S.; Lefebvre, V.; de Crombrugghe, B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha. J. Biol. Chem. 2000, 275, 3687–3692. [Google Scholar] [CrossRef] [Green Version]
- Kon, T.; Cho, T.J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef]
- Williams, J.A.; Kondo, N.; Okabe, T.; Takeshita, N.; Pilchak, D.M.; Koyama, E.; Ochiai, T.; Jensen, D.; Chu, M.L.; Kane, M.A.; et al. Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse. Dev. Biol. 2009, 328, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2018, 109, 267–275. [Google Scholar] [CrossRef]
- Williams, J.A.; Kane, M.; Okabe, T.; Enomoto-Iwamoto, M.; Napoli, J.L.; Pacifici, M.; Iwamoto, M. Endogenous retinoids in mammalian growth plate cartilage: Analysis and roles in matrix homeostasis and turnover. J. Biol. Chem. 2010, 285, 36674–36681. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Uchibe, K.; Son, J.; Larmour, C.; Pacifici, M.; Enomoto-Iwamoto, M.; Iwamoto, M. Genetic and pharmacological inhibition of retinoic acid receptor gamma function promotes endochondral bone formation. J. Orthop. Res. 2017, 35, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Koyama, E.; Okui, T.; Masui, M.; Kunisada, Y.; Ibaragi, S.; Yoshioka, N.; Kurio, N.; Yoshida, S.; Sasaki, A.; et al. Retinoic Receptor Signaling Regulates Hypertrophic Chondrocyte-specific Gene Expression. In Vivo 2019, 33, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The early fracture hematoma and its potential role in fracture healing. Tissue Eng. Part B Rev. 2010, 16, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Baht, G.S.; Vi, L.; Alman, B.A. The Role of the Immune Cells in Fracture Healing. Curr. Osteoporos Rep. 2018, 16, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, A.; Bergdolt, S.; Wiegner, R.; Radermacher, P.; Huber-Lang, M.; Ignatius, A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur. Cells Mater. 2016, 32, 152–162. [Google Scholar] [CrossRef]
- Ai-Aql, Z.S.; Alagl, A.S.; Graves, D.T.; Gerstenfeld, L.C.; Einhorn, T.A. Molecular mechanisms controlling bone formation during fracture healing and distraction osteogenesis. J. Dent. Res. 2008, 87, 107–118. [Google Scholar] [CrossRef]
- Lange, J.; Sapozhnikova, A.; Lu, C.; Hu, D.; Li, X.; Miclau, T.; Marcucio, R.S. Action of IL-1beta during fracture healing. J. Orthop. Res. 2010, 28, 778–784. [Google Scholar]
- Gerstenfeld, L.C.; Cullinane, D.M.; Barnes, G.L.; Graves, D.T.; Einhorn, T.A. Fracture healing as a post-natal developmental process: Molecular, spatial, and temporal aspects of its regulation. J. Cell. Biochem. 2003, 88, 873–884. [Google Scholar] [CrossRef]
- Purton, L.E.; Dworkin, S.; Olsen, G.H.; Walkley, C.R.; Fabb, S.A.; Collins, S.J.; Chambon, P. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 2006, 203, 1283–1293. [Google Scholar] [CrossRef]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.T.; Purton, L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobick, B.E.; Kulyk, W.M. Regulation of cartilage formation and maturation by mitogen-activated protein kinase signaling. Birth Defects Res. C Embryo Today 2008, 84, 131–154. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Suh, J.H.; Kim, H.N.; Kim, A.Y.; Park, S.Y.; Shin, C.S.; Choi, J.Y.; Kim, J.B. Berberine promotes osteoblast differentiation by Runx2 activation with p38 MAPK. J. Bone Miner. Res. 2008, 23, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.Y.; Zhang, X.Z.; Liu, L.; Wan, Z.M.; Li, R.X.; Guo, Y. Involvement of p38MAPK/NF-kappaB signaling pathways in osteoblasts differentiation in response to mechanical stretch. Ann. Biomed. Eng. 2012, 40, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lin, Z.W.; Wang, G.; Zhang, H.; Liu, B.; Xu, Q.J. MicroRNA-495 downregulates AQP1 and facilitates proliferation and differentiation of osteoblasts in mice with tibial fracture through activation of p38 MAPK signaling pathway. Sci. Rep. 2019, 9, 16171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Pan, Q.; Mao, Z.; Wang, P.; Zhang, R.; Ma, X.; Chen, J.; You, H. Kaempferol inhibits interleukin1beta stimulated matrix metalloproteinases by suppressing the MAPKassociated ERK and P38 signaling pathways. Mol. Med. Rep. 2018, 18, 2697–2704. [Google Scholar] [PubMed] [Green Version]
- Reimold, A.M.; Grusby, M.J.; Kosaras, B.; Fries, J.W.; Mori, R.; Maniwa, S.; Clauss, I.M.; Collins, T.; Sidman, R.L.; Glimcher, M.J.; et al. Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature 1996, 379, 262–265. [Google Scholar] [CrossRef]
- Zhen, X.; Wei, L.; Wu, Q.; Zhang, Y.; Chen, Q. Mitogen-activated protein kinase p38 mediates regulation of chondrocyte differentiation by parathyroid hormone. J. Biol. Chem. 2001, 276, 4879–4885. [Google Scholar] [CrossRef] [Green Version]
- Stanton, L.A.; Sabari, S.; Sampaio, A.V.; Underhill, T.M.; Beier, F. p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem. J. 2004, 378, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, P.; Chen, Q.; Wei, X.; Zhao, T.; Wang, Z.; Wei, L. Mitogen-activated protein kinase p38 induces HDAC4 degradation in hypertrophic chondrocytes. Biochim. Biophys. Acta 2015, 1853, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockel, S.J.; Kudirka, C.J.; Guzi, J.A.; Bernier, M.S. Regulation of Sox9 activity by crosstalk with nuclear factor-kappaB and retinoic acid receptors. Arthritis Res. Ther. 2008, 10, R3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novack, D.V. Role of NF-kB in the skeleton. Cell Res. 2011, 21, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatomi, C.; Nakatomi, M.; Matsubara, T.; Komori, T.; Doi-Inoue, T.; Ishimaru, N.; Weih, F.; Iwamoto, T.; Matsuda, M.; Kokabu, S.; et al. Constitutive activation of the alternative NF-κB pathway disturbs endochondral ossification. Bone 2019, 121, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Liu, G.; Zhou, X.; Zhang, W.; Wang, C.; Hu, D.; Xue, D.; Pan, Z. Concentration-dependent, dual roles of IL-10 in the osteogenesis of human BMSCs via P38/MAPK and NF-κB signaling pathways. FASEB J. 2018, 32, 4917–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaki, N.; Takaishi, H.; Kamekura, S.; Kimura, T.; Okada, Y.; Minqi, L.; Amizuka, N.; Chung, U.I.; Nakamura, K.; Kawaguchi, H.; et al. Impaired bone fracture healing in matrix metalloproteinase-13 deficient mice. Biochem. Biophys. Res. Commun. 2007, 354, 846–851. [Google Scholar] [CrossRef]
- Zijlstra, A.; Aimes, R.T.; Zhu, D.; Regazzoni, K.; Kupriyanova, T.; Seandel, M.; Deryugina, E.I.; Quigley, J.P. Collagenolysis-dependent angiogenesis mediated by matrix metalloproteinase-13 (collagenase-3). J. Biol. Chem. 2004, 279, 27633–27645. [Google Scholar] [CrossRef] [Green Version]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [Green Version]
- Shimo, T.; Nakanishi, T.; Nishida, T.; Asano, M.; Kanyama, M.; Kuboki, T.; Tamatani, T.; Tezuka, K.; Takemura, M.; Matsumura, T.; et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J. Biochem. 1999, 126, 137–145. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [Green Version]
- Bahney, C.S.; Hu, D.P.; Miclau, T., 3rd; Marcucio, R.S. The multifaceted role of the vasculature in endochondral fracture repair. Front. Endocrinol. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Miclau, T.; Hu, D.; Marcucio, R.S. Ischemia leads to delayed union during fracture healing: A mouse model. J. Orthop. Res. 2007, 25, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.P.; Ferro, F.; Yang, F.; Taylor, A.J.; Chang, W.; Miclau, T.; Marcucio, R.S.; Bahney, C.S. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 2017, 144, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Yudoh, K.; Nakamura, H.; Kato, T.; Inoue, K.; Chiba, J.; Nishioka, K.; Masuko-Hongo, K. Distinct signaling pathways are involved in hypoxia- and IL-1-induced VEGF expression in human articular chondrocytes. J. Orthop. Res. 2006, 24, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le, B.F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endo thelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef]
- Horikiri, Y.; Shimo, T.; Kurio, N.; Okui, T.; Matsumoto, K.; Iwamoto, M.; Sasaki, A. Sonic hedgehog regulates osteoblast function by focal adhesion kinase signaling in the process of fracture healing. PLoS ONE 2013, 8, e76785. [Google Scholar] [CrossRef] [Green Version]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [Green Version]
- Shimo, T.; Matsumoto, K.; Takabatake, K.; Aoyama, E.; Takebe, Y.; Ibaragi, S.; Okui, T.; Kurio, N.; Takada, H.; Obata, K.; et al. The Role of Sonic Hedgehog Signaling in Osteoclastogenesis and Jaw Bone Destruction. PLoS ONE 2016, 11, e0151731. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimo, T.; Takebe, H.; Okui, T.; Kunisada, Y.; Ibaragi, S.; Obata, K.; Kurio, N.; Shamsoon, K.; Fujii, S.; Hosoya, A.; et al. Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing. Int. J. Mol. Sci. 2020, 21, 2365. https://doi.org/10.3390/ijms21072365
Shimo T, Takebe H, Okui T, Kunisada Y, Ibaragi S, Obata K, Kurio N, Shamsoon K, Fujii S, Hosoya A, et al. Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing. International Journal of Molecular Sciences. 2020; 21(7):2365. https://doi.org/10.3390/ijms21072365
Chicago/Turabian StyleShimo, Tsuyoshi, Hiroaki Takebe, Tatsuo Okui, Yuki Kunisada, Soichiro Ibaragi, Kyoichi Obata, Naito Kurio, Karnoon Shamsoon, Saki Fujii, Akihiro Hosoya, and et al. 2020. "Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing" International Journal of Molecular Sciences 21, no. 7: 2365. https://doi.org/10.3390/ijms21072365
APA StyleShimo, T., Takebe, H., Okui, T., Kunisada, Y., Ibaragi, S., Obata, K., Kurio, N., Shamsoon, K., Fujii, S., Hosoya, A., Irie, K., Sasaki, A., & Iwamoto, M. (2020). Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing. International Journal of Molecular Sciences, 21(7), 2365. https://doi.org/10.3390/ijms21072365