The Genetics of Alzheimer’s Disease in the Chinese Population


Abstract
:1. Introduction
2. Genetics of AD
2.1. EOAD Susceptibility Genes
2.1.1. APP
2.1.2. PSEN1
2.1.3. PSEN2
2.2. LOAD Susceptibility Genes
2.2.1. APOE Gene
2.2.2. TREM2
2.2.3. DAPK1
2.2.4. Other Genes Implicated in LOAD
3. Challenges and Future Insights
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Peprah, K.; McCormack, S. Medical Cannabis for the Treatment of Dementia: A Review of Clinical Effectiveness and Guidelines; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2019. [Google Scholar]
- Sherzai, D.; Sherzai, A. Preventing Alzheimer’s: Our most urgent health care priority. Am. J. Lifestyle Med. 2019, 13, 451–461. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S. Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries. Clin. Interv. Aging 2016, 11, 1467–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.Y.; Wang, W.; Wu, J.J.; Liu, L.; Theodoratou, E.; Car, J.; Middleton, L.; Russ, T.C.; Deary, I.J.; Campbell, H.; et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: A systematic review and analysis. Lancet (Lond. Engl.) 2013, 381, 2016–2023. [Google Scholar] [CrossRef]
- Jia, L.; Quan, M.; Fu, Y.; Zhao, T.; Li, Y.; Wei, C.; Tang, Y.; Qin, Q.; Wang, F.; Qiao, Y.; et al. Group for the Project of Dementia Situation in, C. Dementia in China: Epidemiology, clinical management, and research advances. Lancet Neurol. 2020, 19, 81–92. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Jia, R.X.; Liang, J.H.; Li, J.; Qian, S.; Li, J.Y.; Xu, Y. Dementia in China (2015–2050) estimated using the 1% population sampling survey in 2015. Geriatr. Gerontol. Int. 2019, 19, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar]
- Jia, J.; Wei, C.; Chen, S.; Li, F.; Tang, Y.; Qin, W.; Zhao, L.; Jin, H.; Xu, H.; Wang, F.; et al. The cost of Alzheimer’s disease in China and re-estimation of costs worldwide. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 483–491. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet (Lond. Engl.) 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-onset Alzheimer disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-onset Alzheimer disease. Neurol. Clin. 2017, 35, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Katsumata, Y.; Fardo, D.W.; Bachstetter, A.D.; Artiushin, S.C.; Wang, W.X.; Wei, A.; Brzezinski, L.J.; Nelson, B.G.; Huang, Q.; Abner, E.L.; et al. Alzheimer disease pathology-associated polymorphism in a complex variable number of tandem repeat region within the MUC6 gene, near the AP2A2 gene. J. Neuropathol. Exp. Neurol. 2020, 79, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2008, 10, 231–239. [Google Scholar] [CrossRef] [Green Version]
- R, A.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar]
- Huq, A.J.; Fransquet, P.; Laws, S.M.; Ryan, J.; Sebra, R.; Masters, C.L.; Winship, I.M.; James, P.A.; Lacaze, P. Genetic resilience to Alzheimer’s disease in APOE epsilon4 homozygotes: A systematic review. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; He, Z.; Zhang, D.; Wang, G.T.; Renton, A.E.; Vardarajan, B.N.; Nothnagel, M.; Goate, A.M.; Mayeux, R.; Leal, S.M. A rare variant nonparametric linkage method for nuclear and extended pedigrees with application to late-onset alzheimer disease via WGS data. Am. J. Hum. Genet. 2019, 105, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Amlie-Wolf, A.; Tang, M.; Way, J.; Dombroski, B.; Jiang, M.; Vrettos, N.; Chou, Y.F.; Zhao, Y.; Kuzma, A.; Mlynarski, E.E.; et al. Inferring the molecular mechanisms of noncoding Alzheimer’s disease-associated genetic variants. J. Alzheimer’s Dis. 2019, 72, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Liu, L.; Hara, N. Genetic analysis of Alzheimer’s disease: The impact of rare variants and their significance. Brain Nerve Shinkei Kenkyu Shinpo 2019, 71, 1071–1079. [Google Scholar]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Muller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Annu. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Ancolio, K.; Dumanchin, C.; Barelli, H.; Warter, J.M.; Brice, A.; Campion, D.; Frebourg, T.; Checler, F. Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val-715→Met betaAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 4119–4124. [Google Scholar] [CrossRef] [Green Version]
- Campion, D.; Brice, A.; Hannequin, D.; Charbonnier, F.; Dubois, B.; Martin, C.; Michon, A.; Penet, C.; Bellis, M.; Calenda, A.; et al. No founder effect in three novel Alzheimer’s disease families with APP 717 Val→Ile mutation. Clerget-darpoux. French Alzheimer’s Disease Study Group. J. Med. Genet. 1996, 33, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, J.R.; Hake, A.M.; Quaid, K.A.; Farlow, M.R.; Ghetti, B. Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch. Neurol. 2000, 57, 885–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goate, A. Segregation of a missense mutation in the amyloid beta-protein precursor gene with familial Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9 (Suppl. 3), 341–347. [Google Scholar] [CrossRef]
- Kaden, D.; Harmeier, A.; Weise, C.; Munter, L.M.; Althoff, V.; Rost, B.R.; Hildebrand, P.W.; Schmitz, D.; Schaefer, M.; Lurz, R.; et al. Novel APP/Abeta mutation K16N produces highly toxic heteromeric Abeta oligomers. EMBO Mol. Med. 2012, 4, 647–659. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, J.; Qin, W.; Wu, L.; Li, D.; Li, H. A Novel AbetaPP M722K mutation affects amyloid-beta secretion and tau phosphorylation and may cause early-onset familial Alzheimer’s disease in Chinese individuals. J. Alzheimer’s Dis. 2015, 47, 157–165. [Google Scholar] [CrossRef]
- Thajeb, P.; Wang, P.; Chien, C.L.; Harrigan, R. Novel polymorphisms of the amyloid precursor protein (APP) gene in Chinese/Taiwanese patients with Alzheimer’s disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2009, 16, 259–263. [Google Scholar] [CrossRef]
- Jiang, B.; Zhou, J.; Li, H.L.; Chen, Y.G.; Cheng, H.R.; Ye, L.Q.; Liu, D.S.; Chen, D.F.; Tao, Q.Q.; Wu, Z.Y. Mutation screening in Chinese patients with familial Alzheimer’s disease by whole-exome sequencing. Neurobiol. Aging 2019, 76, 215.e15–215.e21. [Google Scholar] [CrossRef]
- Gao, Y.; Ren, R.J.; Zhong, Z.L.; Dammer, E.; Zhao, Q.H.; Shan, S.; Zhou, Z.; Li, X.; Zhang, Y.Q.; Cui, H.L.; et al. Mutation profile of APP, PSEN1, and PSEN2 in Chinese familial Alzheimer’s disease. Neurobiol. Aging 2019, 77, 154–157. [Google Scholar] [CrossRef]
- Peng, X.L.; Hou, L.; Xu, S.H.; Hua, Y.; Zhou, S.J.; Zhang, Y.; Zheng, Y.P.; Fu, Y.H.; Xu, Q.; Zhang, L.S.; et al. Novel APP K724M mutation causes Chinese early-onset familial Alzheimer’s disease and increases amyloid-beta42 to amyloid-beta40 ratio. Neurobiol. Aging 2014, 35, 2657.e1–2657.e6. [Google Scholar] [CrossRef]
- Jiao, B.; Tang, B.; Liu, X.; Xu, J.; Wang, Y.; Zhou, L.; Zhang, F.; Yan, X.; Zhou, Y.; Shen, L. Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol. Aging 2014, 35, 1957.e1–1957.e6. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, M.Y.; Liu, J.S.; Wu, Y.S.; Peng, C.H.; Chang, Y.Y. A novel APP mutation (D678H) in a Taiwanese patient exhibiting dementia and cerebral microvasculopathy. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2014, 21, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Chen, W.T.; Hong, C.J.; Lin, Y.T.; Chang, W.H.; Huang, H.T.; Liao, J.Y.; Chang, Y.J.; Hsieh, Y.F.; Cheng, C.Y.; Liu, H.C.; et al. Amyloid-beta (Abeta) D7H mutation increases oligomeric Abeta42 and alters properties of Abeta-zinc/copper assemblies. PLoS ONE 2012, 7, e35807. [Google Scholar]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Fang, B.; Jia, L.; Jia, J. Chinese Presenilin-1 V97L mutation enhanced Abeta42 levels in SH-SY5Y neuroblastoma cells. Neurosci. Lett. 2006, 406, 33–37. [Google Scholar] [CrossRef]
- Jia, J.; Xu, E.; Shao, Y.; Sun, Y.; Li, D. One novel presenilin-1 gene mutation in a Chinese pedigree of familial Alzheimer’s disease. J. Alzheimer’s Dis. 2005, 7, 119–124; discussion 173–180. [Google Scholar] [CrossRef]
- Qiu, Q.; Jia, L.; Wang, Q.; Zhao, L.; Jin, H.; Li, T.; Quan, M.; Xu, L.; Li, B.; Li, Y.; et al. Identification of a novel PSEN1 Gly111Val missense mutation in a Chinese pedigree with early-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155.e1–155.e4. [Google Scholar] [CrossRef]
- Fang, B.Y.; Jia, J.P. The effect of two newly Chinese presenilin-1 mutations on the sensitivity to trophic factor withdrawal in human neuroblastoma cells. Zhonghua Yi Xue Za Zhi 2007, 87, 336–340. [Google Scholar]
- Xu, E.; Jia, J.; Sun, W. Mutation site of presenilin-1 gene in familial Alzheimer’s disease. Zhonghua Yi Xue Za Zhi 2002, 82, 1518–1520. [Google Scholar] [PubMed]
- Qiu, Q.; Shen, L.; Jia, L.; Wang, Q.; Li, F.; Li, Y.; Jia, J. A Novel PSEN1 M139L mutation found in a Chinese pedigree with early-onset Alzheimer’s disease increases Abeta42/Abeta40 ratio. J. Alzheimer’s Dis. 2019, 69, 199–212. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.Y.; Ki, C.S.; Kim, S.H. Presenilin 1 gene mutation (M139I) in a patient with an early-onset Alzheimer’s disease: Clinical characteristics and genetic identification. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc.Clin. Neurophysiol. 2010, 31, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wei, J.; Liao, S.; Wang, L.; Jiang, H.; Tang, B. A novel presenilin 1 mutation (Ser169del) in a Chinese family with early-onset Alzheimer’s disease. Neurosci. Lett. 2010, 468, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Rogaeva, E.A.; Fafel, K.C.; Song, Y.Q.; Medeiros, H.; Sato, C.; Liang, Y.; Richard, E.; Rogaev, E.I.; Frommelt, P.; Sadovnick, A.D.; et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology 2001, 57, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Yang, Z.H.; Zhang, Y.; Yang, J.; Shang, D.D.; Zhang, S.Y.; Wu, J.; Ji, Y.; Zhao, L.; Shi, C.H.; et al. Two novel mutations and a de novo mutation in PSEN1 in early-onset Alzheimer’s disease. Aging Dis. 2019, 10, 908–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Zhang, J.; Shi, Y.; Wang, W.; Ren, Z.; Xia, M.; Zhang, Y.; Yang, M. Gene mutations in a Han Chinese Alzheimer’s disease cohort. Brain Behav. 2019, 9, e01180. [Google Scholar] [CrossRef] [Green Version]
- Aldudo, J.; Bullido, M.J.; Valdivieso, F. DGGE method for the mutational analysis of the coding and proximal promoter regions of the Alzheimer’s disease presenilin-1 gene: Two novel mutations. Hum. Mutat. 1999, 14, 433–439. [Google Scholar] [CrossRef]
- Dong, J.; Qin, W.; Wei, C.; Tang, Y.; Wang, Q.; Jia, J. A novel PSEN1 K311R mutation discovered in Chinese families with late-onset Alzheimer’s disease affects amyloid-beta production and tau phosphorylation. J. Alzheimer’s Dis. 2017, 57, 613–623. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Li, G.D.; Dai, S.X.; Bi, R.; Zhang, D.F.; Li, Z.F.; Xu, X.F.; Zhou, T.C.; Yu, L.; Yao, Y.G. Identification of PSEN1 mutations p.M233L and p.R352C in Han Chinese families with early-onset familial Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1602.e3–1602.e6. [Google Scholar] [CrossRef]
- Lou, F.; Luo, X.; Li, M.; Ren, Y.; He, Z. Very early-onset sporadic Alzheimer’s disease with a de novo mutation in the PSEN1 gene. Neurobiol. Aging 2017, 53, 193.e1–193.e5. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wang, Y.; Liu, S.; Liu, M.; Liu, S.; Zhou, Y.; Wang, J.; Cai, L.; Huo, Y.R.; Gao, S.; et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement. Geriatr. Cognit. Disord. 2015, 39, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Chen, S.; Shi, Y.; Huang, Y.; Xu, J.; Zhao, T.; He, S.; Wu, Y.; Xu, C.; Zang, W.; et al. Probable novel PSEN2 Pro123Leu mutation in a Chinese Han family of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3334.e13–3334.e18. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Yu, S.; Zhang, Z.; Yi, X.; Ye, L.; Tang, W.; Qiu, C.; Wen, H.; Sun, Y.; Gao, J.; et al. Novel mutation in the PSEN2 gene (N141Y) associated with early-onset autosomal dominant Alzheimer’s disease in a Chinese Han family. Neurobiol. Aging 2014, 35, 2420.e1–2420.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, D.F.; Jiang, H.Y.; Fan, Y.; Ma, L.; Shen, Z.; Bi, R.; Xu, M.; Tan, L.; Shan, B.; et al. Mutation and association analyses of dementia-causal genes in Han Chinese patients with early-onset and familial Alzheimer’s disease. J. Psychiatr. Res. 2019, 113, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Hardy, J. The presenilins and Alzheimer’s disease. Hum. Mol. Genet. 1997, 6, 1639–1646. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Qian, C.; Liu, Z.; Lu, X.; Lin, H. A study on mutation of exon 5 of presenilin-1 in Alzheimer’s disease. Zhonghua Yi Xue Yi Chuan Xue Za Zhi = Zhonghua Yixue Yichuanxue Zazhi = Chin. J. Med. Genet. 1999, 16, 349–352. [Google Scholar]
- Shao, Y.; Li, M.; Wu, M.; Shi, K.; Fang, B.; Wang, J. FAD-linked Presenilin-1 V97L mutation impede tranport regulation and intracellular Ca(2+) homeostasis under ER stress. Int. J. Clin. Exp. Med. 2015, 8, 20742–20750. [Google Scholar]
- Deng, B.; Lian, Y.; Wang, X.; Zeng, F.; Jiao, B.; Wang, Y.R.; Liang, C.R.; Liu, Y.H.; Bu, X.L.; Yao, X.Q.; et al. Identification of a novel mutation in the presenilin 1 gene in a Chinese Alzheimer’s disease family. Neurotox. Res. 2014, 26, 211–215. [Google Scholar] [CrossRef]
- Wang, H.; Sun, R.; Shi, Y.; Xia, M.; Zhao, J.; Yang, M.; Ma, L.; Sun, Y.; Li, G.; Zhang, H.; et al. Probable novel PSEN1 Gln222Leu mutation in a Chinese family with early-onset Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 764–769. [Google Scholar] [CrossRef]
- Bernardi, L.; Tomaino, C.; Anfossi, M.; Gallo, M.; Geracitano, S.; Puccio, G.; Colao, R.; Frangipane, F.; Mirabelli, M.; Smirne, N.; et al. Late onset familial Alzheimer’s disease: Novel presenilin 2 mutation and PS1 E318G polymorphism. J. Neurol. 2008, 255, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Ferrara, M.; Alinani, A.; Sutton, R.B.; Fama, F.; Picco, A.; Rodriguez, G.; Nobili, F.; Momeni, P. Screening of early and late onset Alzheimer’s disease genetic risk factors in a cohort of dementia patients from Liguria, Italy. Curr. Alzheimer Res. 2015, 12, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Genome-wide association studies in Alzheimer’s disease. Hum. Mol. Genet. 2009, 18, R137–R145. [Google Scholar] [CrossRef] [PubMed]
- Mak, Y.T.; Chiu, H.; Woo, J.; Kay, R.; Chan, Y.S.; Hui, E.; Sze, K.H.; Lum, C.; Kwok, T.; Pang, C.P. Apolipoprotein E genotype and Alzheimer’s disease in Hong Kong elderly Chinese. Neurology 1996, 46, 146–149. [Google Scholar] [CrossRef]
- Hu, C.J.; Sung, S.M.; Liu, H.C.; Hsu, W.C.; Lee, L.S.; Lee, C.C.; Tsai, C.H.; Chang, J.G. Genetic risk factors of sporadic Alzheimer’s disease among Chinese in Taiwan. J. Neurol. Sci. 2000, 181, 127–131. [Google Scholar] [CrossRef]
- Jiao, B.; Liu, X.; Tang, B.; Hou, L.; Zhou, L.; Zhang, F.; Zhou, Y.; Guo, J.; Yan, X.; Shen, L. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer’s disease from mainland China. Neurobiol. Aging 2014, 35, 2422.e9–2422.e11. [Google Scholar] [CrossRef]
- Bonham, L.W.; Sirkis, D.W.; Fan, J.; Aparicio, R.E.; Tse, M.; Ramos, E.M.; Wang, Q.; Coppola, G.; Rosen, H.J.; Miller, B.L.; et al. Identification of a rare coding variant in TREM2 in a Chinese individual with Alzheimer’s disease. Neurocase 2017, 23, 65–69. [Google Scholar] [CrossRef]
- Jiang, T.; Hou, J.K.; Gao, Q.; Yu, J.T.; Zhou, J.S.; Zhao, H.D.; Zhang, Y.D. TREM2 p.H157Y variant and the risk of Alzheimer’s disease: A meta-analysis involving 14,510 subjects. Curr. Neurovasc. Res. 2016, 13, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cheng, L.; Zhang, Y.; Bai, W.; Zhou, W.; Wang, T.; Han, Z.; Zong, J.; Jin, S.; Zhang, J.; et al. Rs4878104 contributes to Alzheimer’s disease risk and regulates DAPK1 gene expression. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.C.; Zhang, W.; Yu, J.T.; Zhang, Q.; Tian, Y.; Lu, R.C.; Yu, N.N.; Chi, Z.F.; Tan, L. Association of DAPK1 genetic variations with Alzheimer’s disease in Han Chinese. Brain Res. 2011, 1374, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kauwe, J.S.; Wang, J.; Mayo, K.; Morris, J.C.; Fagan, A.M.; Holtzman, D.M.; Goate, A.M. Alzheimer’s disease risk variants show association with cerebrospinal fluid amyloid beta. Neurogenetics 2009, 10, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel-Smith, M.K.; Hunkapiller, J.; Bhangale, T.R.; Srinivasan, K.; Maloney, J.A.; Atwal, J.K.; Sa, S.M.; Yaylaoglu, M.B.; Foreman, O.; Ortmann, W.; et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat. Med. 2014, 20, 1452–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Chen, Y.; Mok, K.Y.; Zhao, Q.; Chen, K.; Hardy, J.; Li, Y.; Fu, A.K.Y.; Guo, Q.; Ip, N.Y. Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 1697–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, X.Q.; Zhao, Q.H.; Huang, Y.; Li, X.; Ren, R.J.; Chen, S.D.; Wang, G.; Guo, Q.H. Genetic features of MAPT, GRN, C9orf72 and CHCHD10 gene mutations in chinese patients with frontotemporal dementia. Curr. Alzheimer Res. 2017, 14, 1102–1108. [Google Scholar] [CrossRef]
- Xiao, T.; Jiao, B.; Zhang, W.; Pan, C.; Wei, J.; Liu, X.; Zhou, Y.; Zhou, L.; Tang, B.; Shen, L. Identification of CHCHD10 mutation in Chinese patients with Alzheimer disease. Mol. Neurobiol. 2017, 54, 5243–5247. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and abeta secretion. Cell 2017, 168, 427.e21–441.e21. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (N. Y.) 1993, 261, 921–923. [Google Scholar] [CrossRef]
- St Clair, D.; Rennie, M.; Slorach, E.; Norrman, J.; Yates, C.; Carothers, A. Apolipoprotein E epsilon 4 allele is a risk factor for familial and sporadic presenile Alzheimer’s disease in both homozygote and heterozygote carriers. J. Med. Genet. 1995, 32, 642–644. [Google Scholar] [CrossRef]
- Saunders, A.M. Apolipoprotein E and Alzheimer disease: An update on genetic and functional analyses. J. Neuropathol. Exp. Neurol. 2000, 59, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Pan, X.; Fei, G.; Wang, C.; Wan, W.; Sang, S.; Wang, H.; Wang, Z.; Zhong, C. Decreased function of delayed recall in non-demented elderly subjects with apolipoprotein E epsilon4 allele. Front. Aging Neurosci. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, V.; Spalletta, G.; Souza, R.P.; Graff, A.; Bastos-Rodrigues, L.; Camargos Bicalho, M.A. Definition of late onset Alzheimer’s disease and anticipation effect of genome-wide significant risk variants: Pilot study of the APOE e4 allele. Neuropsychobiology 2019, 77, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Baum, L.; Ng, H.K.; Chan, L.Y.; Sastre, I.; Artiga, M.J.; Valdivieso, F.; Bullido, M.J.; Chiu, H.F.; Pang, C.P. Apolipoprotein E promoter and alpha2-macroglobulin polymorphisms are not genetically associated with Chinese late onset Alzheimer’s disease. Neurosci. Lett. 1999, 269, 173–177. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Tulloch, J.; Leong, L.; Chen, S.; Keene, C.D.; Millard, S.P.; Shutes-David, A.; Lopez, O.L.; Kofler, J.; Kaye, J.A.; Woltjer, R.; et al. APOE DNA methylation is altered in Lewy body dementia. Alzheimer’sDement. J. Alzheimer’s Assoc. 2018, 14, 889–894. [Google Scholar] [CrossRef]
- Prokopenko, I.; Miyakawa, G.; Zheng, B.; Heikkinen, J.; Petrova Quayle, D.; Udeh-Momoh, C.; Claringbould, A.; Neumann, J.; Haytural, H.; Kaakinen, M.A.; et al. Alzheimer’s disease pathology explains association between dementia with Lewy bodies and APOE-epsilon4/TOMM40 long poly-T repeat allele variants. Alzheimer’s Dement. (N. Y.) 2019, 5, 814–824. [Google Scholar]
- Sun, R.; Yang, S.; Zheng, B.; Liu, J.; Ma, X. Apolipoprotein E Polymorphisms and Parkinson Disease With or Without Dementia: A Meta-Analysis Including 6453 Participants. J. Geriatr. Psychiatry Neurol. 2019, 32, 3–15. [Google Scholar] [CrossRef]
- Larsson, V.; Torisson, G.; Londos, E. Relative survival in patients with dementia with Lewy bodies and Parkinson’s disease dementia. PLoS ONE 2018, 13, e0202044. [Google Scholar] [CrossRef]
- Dumolt, J.H.; Ma, M.; Mathew, J.; Patel, M.S.; Rideout, T.C. Gestational hypercholesterolemia alters fetal hepatic lipid metabolism and microRNA expression in Apo-E deficient mice. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E831–E838. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Zhao, D.; Yang, X.; Zhang, J.; He, H.; Yu, C. Geniposide against atherosclerosis by inhibiting the formation of foam cell and lowering reverse lipid transport via p38/MAPK signaling pathways. Eur. J. Pharmacol. 2019, 864, 172728. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hashimoto, T.; Tai, H.C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain J. Neurol. 2012, 135 Pt 7, 2155–2168. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nystrom, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Zhang, T.J.; Jiang, H.; Lefton, K.B.; Robinson, G.O.; Vassar, R.; Sullivan, P.M.; Holtzman, D.M. Murine versus human apolipoprotein E4: Differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol. Commun. 2015, 3, 70. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Gao, L.; Jiang, Y.; Shang, S.; Chen, C.; Dang, L.; Zhao, B.; Zhang, J.; Wang, J.; Huo, K.; et al. Apolipoprotein E epsilon4 allele is associated with plasma amyloid beta and amyloid beta transporter levels: A cross-sectional study in a rural area of Xi’an, China. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2020, 28, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Investig. 2000, 106, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Liu, C.; Yu, J. Genome-wide association studies for cerebrospinal fluid soluble TREM2 in Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 297. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, M.; Lopera, F.; Siniard, A.L.; Corneveaux, J.J.; Schrauwen, I.; Carvajal, J.; Munoz, C.; Ramirez-Restrepo, M.; Gaiteri, C.; Myers, A.J.; et al. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2077.e11–2077.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.T.; Jiang, T.; Wang, Y.L.; Wang, H.F.; Zhang, W.; Hu, N.; Tan, L.; Sun, L.; Tan, M.S.; Zhu, X.C. Triggering receptor expressed on myeloid cells 2 variant is rare in late-onset Alzheimer’s disease in Han Chinese individuals. Neurobiol. Aging 2014, 35, 937.e1–937.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Guo, Q.; Zhou, Y.; Chen, K.; Xu, Y.; Ding, D.; Hong, Z.; Zhao, Q. Lack of association between triggering receptor expressed on myeloid cells 2 polymorphism rs75932628 and late-onset Alzheimer’s disease in a Chinese Han population. Psychiatr. Genet. 2018, 28, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Gong, L.; Zheng, L.; Chen, K.; Zhou, Y.; Gu, Y.; Xu, Y.; Guo, Q.; Hong, Z.; et al. A novel homozygous mutation in TREM2 found in a Chinese early-onset dementia family with mild bone involvement. Neurobiol. Aging 2020, 86, 201.e1–201.e7. [Google Scholar] [CrossRef]
- Hou, X.H.; Bi, Y.L.; Tan, M.S.; Xu, W.; Li, J.Q.; Shen, X.N.; Dou, K.X.; Tan, C.C.; Tan, L.; Alzheimer’s Disease Neuroimaging Initiative; et al. Genome-wide association study identifies Alzheimer’s risk variant in MS4A6A influencing cerebrospinal fluid sTREM2 levels. Neurobiol. Aging 2019, 84, 241.e13–241.e20. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef]
- Joob, B.; Wiwanitkit, V. Cyclooxygenase-2 gene polymorphisms and risk of Alzheimer’s disease: A possible biomoleular explanation. Neurol. India 2019, 67, 1142. [Google Scholar] [CrossRef]
- Dunn, A.R.; O’Connell, K.M.S.; Kaczorowski, C.C. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci.Biobehav. Rev. 2019, 103, 73–80. [Google Scholar] [CrossRef]
- Nacmias, B.; Piaceri, I.; Bagnoli, S.; Tedde, A.; Piacentini, S.; Sorbi, S. Genetics of Alzheimer’s disease and frontotemporal dementia. Curr. Mol. Med. 2014, 14, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Grupe, A.; Rowland, C.; Nowotny, P.; Kauwe, J.S.; Smemo, S.; Hinrichs, A.; Tacey, K.; Toombs, T.A.; Kwok, S.; et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum. Mol. Genet. 2006, 15, 2560–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Lew, F.; et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.M.; You, M.H.; Chen, C.H.; Suh, J.; Tanzi, R.E.; Lee, T.H. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum. Mol. Genet. 2016, 25, 2498–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulaiman Alsaadi, M. Role of DAPK1 in neuronal cell death, survival and diseases in the nervous system. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2019, 74, 11–17. [Google Scholar] [CrossRef] [PubMed]
- You, M.H.; Kim, B.M.; Chen, C.H.; Begley, M.J.; Cantley, L.C.; Lee, T.H. Death-associated protein kinase 1 phosphorylates NDRG2 and induces neuronal cell death. Cell Death Differ. 2017, 24, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.M.; You, M.H.; Chen, C.H.; Lee, S.; Hong, Y.; Kimchi, A.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014, 5, e1237. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, B.L.; Sun, F.R.; Li, J.Q.; Cao, X.P.; Tan, L. The role of UNC5C in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 178. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; Nebot, I.; Gorostidi, A.; Ortega-Cubero, S.; Hernandez, I.; Rojas-Garcia, R.; Garcia-Redondo, A.; Povedano, M.; Llado, A.; Alvarez, V.; et al. Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain J. Neurol. 2015, 138 Pt 12, e400. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tan, L.; Chen, Q.; Tan, M.S.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Wang, H.F.; Zhang, Y.D.; Yu, J.T. A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol. Aging 2016, 42, 217.e1–217.e3. [Google Scholar] [CrossRef]
- Pericak-Vance, M.A.; Bebout, J.L.; Gaskell, P.C., Jr.; Yamaoka, L.H.; Hung, W.Y.; Alberts, M.J.; Walker, A.P.; Bartlett, R.J.; Haynes, C.A.; Welsh, K.A.; et al. Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am. J. Hum. Genet. 1991, 48, 1034–1050. [Google Scholar] [PubMed]
- Lalli, M.A.; Bettcher, B.M.; Arcila, M.L.; Garcia, G.; Guzman, C.; Madrigal, L.; Ramirez, L.; Acosta-Uribe, J.; Baena, A.; Wojta, K.J.; et al. Whole-genome sequencing suggests a chemokine gene cluster that modifies age at onset in familial Alzheimer’s disease. Mol. Psychiatry 2015, 20, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
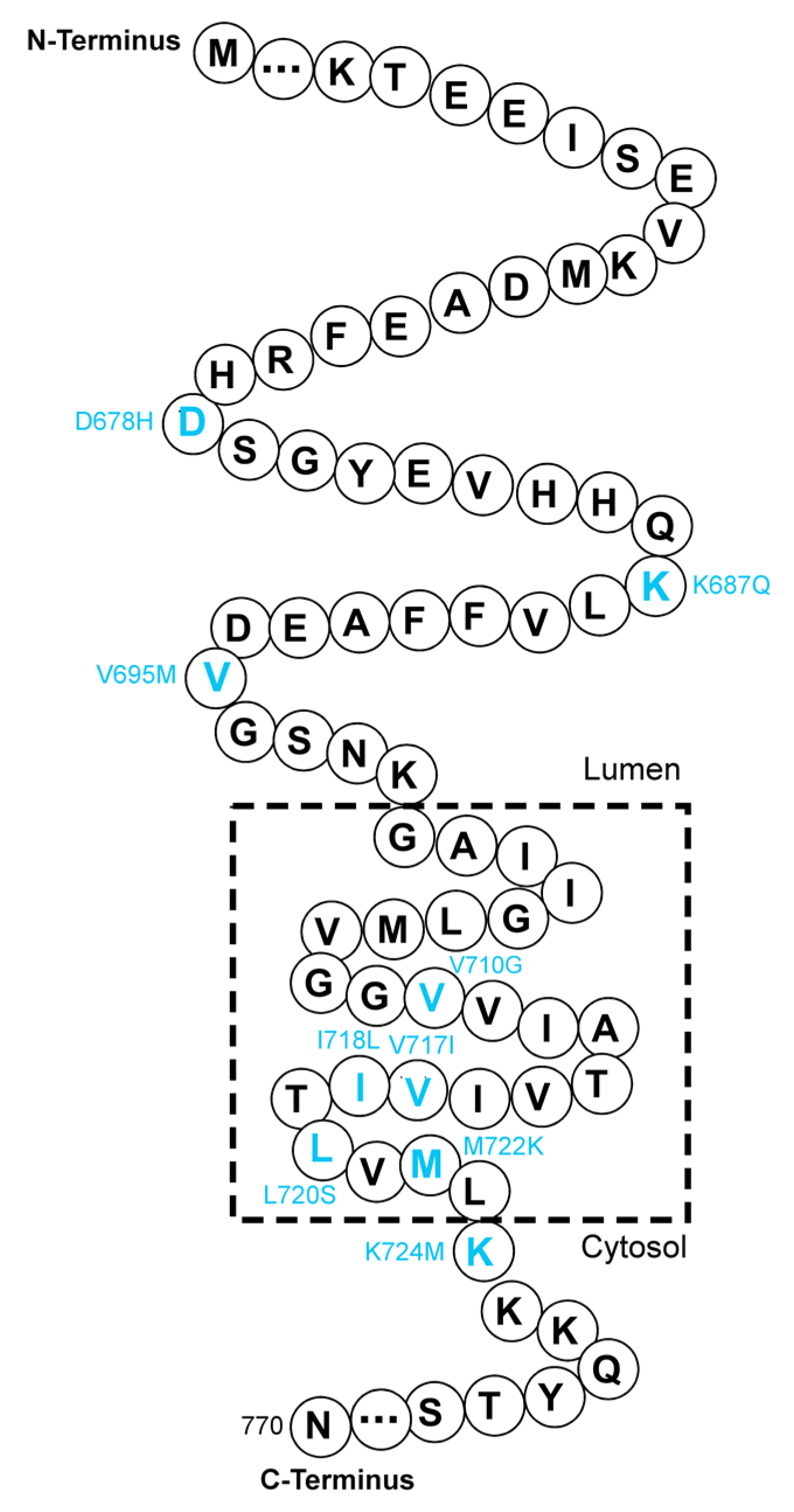



{kind=link}
{kind=link}
{kind=link}
| Gene | Location (Exon) | Mutation(s) | Pathogenicity | References |
|---|---|---|---|---|
| APP | 6 | D244G | Uncertain | [39] |
| 7 | T297M | Uncertain | [39] | |
| D322G | Uncertain | [39] | ||
| 16 | D678H | Increasing Aβ42/Aβ40 ratio; promoting Aβ aggregation | [44,46] | |
| K687Q | Uncertain | [39] | ||
| 17 | V695M | Uncertain | [40] | |
| V710G | Uncertain | [38] | ||
| V717I | Increasing Aβ42 generation | [42,47] | ||
| I718L | Uncertain | [38] | ||
| L720S | Uncertain | [38] | ||
| M722K | Increasing Aβ42/Aβ40 ratio and tau phosphorylation | [37] | ||
| K724M | Increasing Aβ42/Aβ40 ratio | [41] | ||
| PSEN1 | 4 | V97L | Elevating Aβ42 generation | [48,49] |
| V103G | Uncertain | [40] | ||
| F105C | Uncertain | [42] | ||
| G111V | Elevating Aβ42/Aβ40 ratio by reducing Aβ40 generation. | [45,50] | ||
| 5 | A136G | Moderately decreasing Aβ; causing dendritic spine loss | [51,52] | |
| M139L | Increasing Aβ42/Aβ40 ratio | [40,53] | ||
| M139I | Increasing Aβ42/Aβ40 ratio | [39,54] | ||
| T147I | Uncertain | [39] | ||
| R157S | Uncertain | [39] | ||
| 6 | I167del | Decreasing Aβ42 production in vitro | [42] | |
| S169del | Decreasing Aβ42 production in vitro | [55] | ||
| L173W | Increasing Aβ42/Aβ40 ratio | [33,39] | ||
| F177S | Decreasing Aβ42 in CSF | [39,56] | ||
| F177V | Uncertain | [40] | ||
| 7 | G206V | Uncertain | [57] | |
| H214R | Uncertain | [57] | ||
| L226R | Uncertain | [58] | ||
| M233L | Increasing Aβ42/Aβ40 ratio | [55,59] | ||
| L248P | Uncertain | [42] | ||
| Y256N | Uncertain | [57] | ||
| 8 | R269H | Uncertain | [39] | |
| 9 | K311R | Increasing Aβ42/Aβ40 ratio and phosphorylated tau | [45,60] | |
| 10 | R352C | Slightly decreasing Aβ generation | [61] | |
| 11 | V391G | Uncertain | [62] | |
| 12 | A434T | Uncertain | [42] | |
| PSEN2 | 3 | G34S | Uncertain | [45] |
| 4 | R62H | No effect on Aβ42/Aβ40 ratio | [45] | |
| K82R | Uncertain | [63] | ||
| 5 | P123L | Uncertain | [64] | |
| V139M | Uncertain | [39] | ||
| N141Y | Uncertain | [65] | ||
| N141D | Uncertain | [66] | ||
| V150M | Uncertain | [40] | ||
| R163C | Uncertain | [40] | ||
| 6 | H169N | Uncertain | [63] | |
| 7 | V214L | Uncertain | [63] | |
| S236S | Uncertain | [45] | ||
| M298T | Uncertain | [45] | ||
| 11 | A379D | Uncertain | [66] |
| Gene | Chromosome | Mutation(s)/SNPs | Pathogenicity | References |
|---|---|---|---|---|
| APOE | 19q13.2 | APOE ε4 | Strongly promoting the deposition of Aβ | [76,77] |
| TREM2 | 6p21.1 | A130V | Uncertain | [78] |
| A139T | Uncertain | [79] | ||
| H157Y | Increasing the shedding of soluble TREM2 and decreasing the phagocytosis | [80] | ||
| DAPK1 | 9q34.1 | rs4878104 | Associated with increased total Aβ levels | [81,82,83] |
| UNC5C | 4q22.3 | rs137875858 | Increasing cell death | [78,84] |
| GCH1 | 14q22.2 | rs72713460 | Uncertain | [85] |
| KCNJ15 | 21q22.13 | rs928771 | Uncertain | [85] |
| CHCHD10 | 22q11.23 | 63 C > T (no AA change) | Uncertain | [86] |
| P24L | Uncertain | [86] | ||
| A35D | Uncertain | [87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, C.-L.; Zhang, T.; Lee, T.H. The Genetics of Alzheimer’s Disease in the Chinese Population. Int. J. Mol. Sci. 2020, 21, 2381. https://doi.org/10.3390/ijms21072381
Gan C-L, Zhang T, Lee TH. The Genetics of Alzheimer’s Disease in the Chinese Population. International Journal of Molecular Sciences. 2020; 21(7):2381. https://doi.org/10.3390/ijms21072381
Chicago/Turabian StyleGan, Chen-Ling, Tao Zhang, and Tae Ho Lee. 2020. "The Genetics of Alzheimer’s Disease in the Chinese Population" International Journal of Molecular Sciences 21, no. 7: 2381. https://doi.org/10.3390/ijms21072381
APA StyleGan, C.-L., Zhang, T., & Lee, T. H. (2020). The Genetics of Alzheimer’s Disease in the Chinese Population. International Journal of Molecular Sciences, 21(7), 2381. https://doi.org/10.3390/ijms21072381