DNA Methylation Profiles in a Group of Workers Occupationally Exposed to Nanoparticles

 ,
,  , and
, and
Abstract
1. Introduction
2. Results
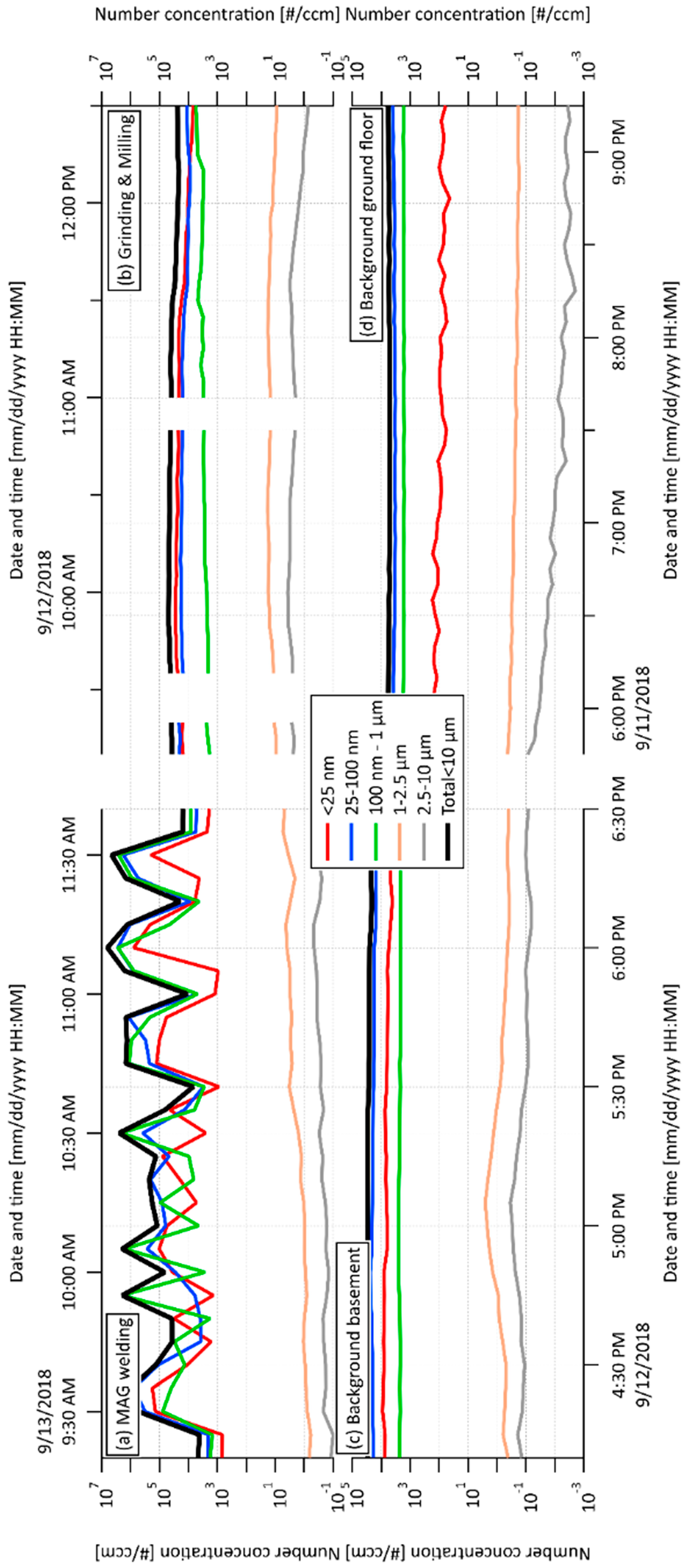
2.1. Exposure Online Monitoring by Scanning Mobility Particle Sizer (SMPS) and Aerodynamic Particle Sizer (APS) and Proportions of Particulate Matter (PM) Fractions
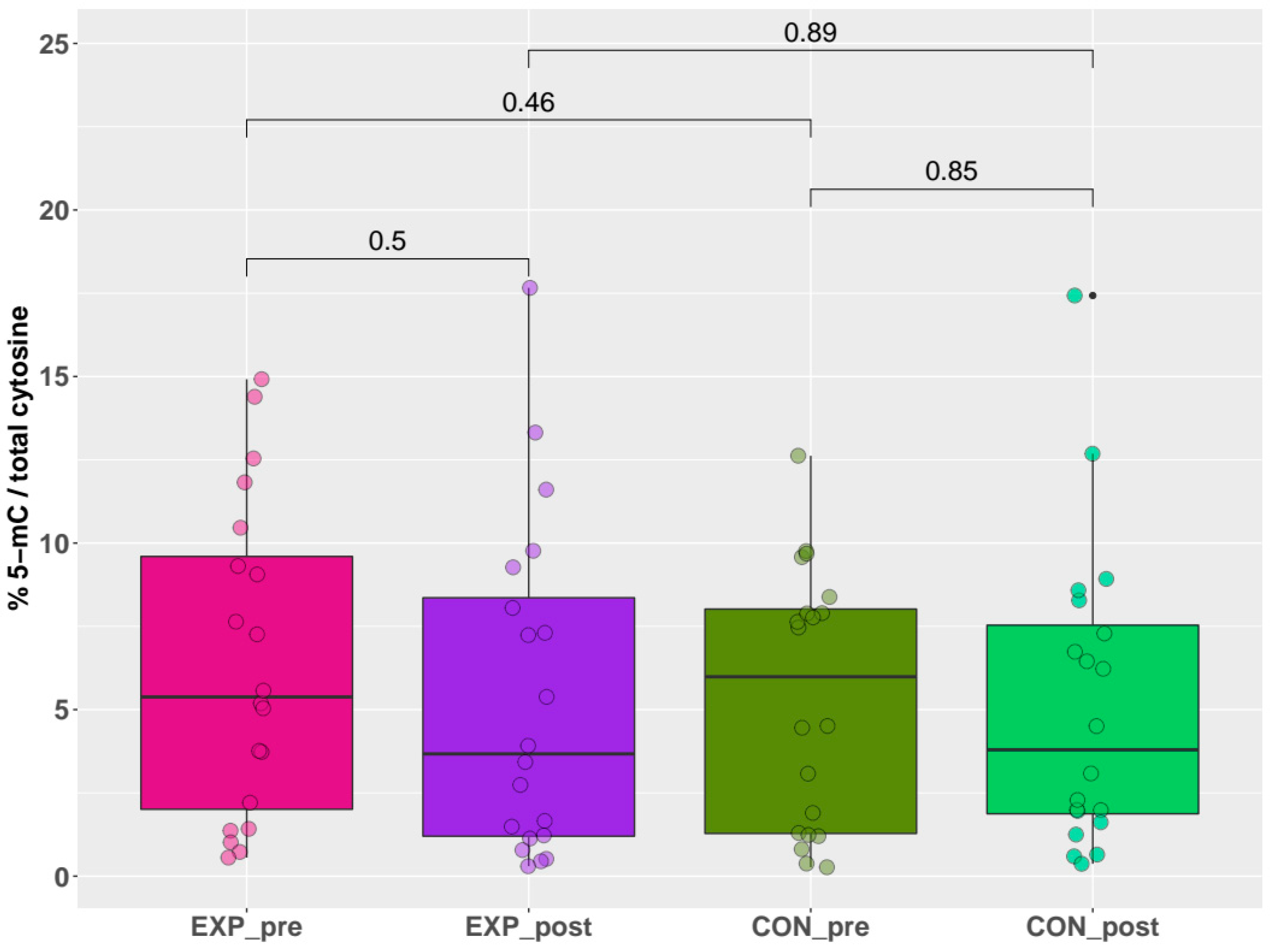
2.2. Global DNA Methylation
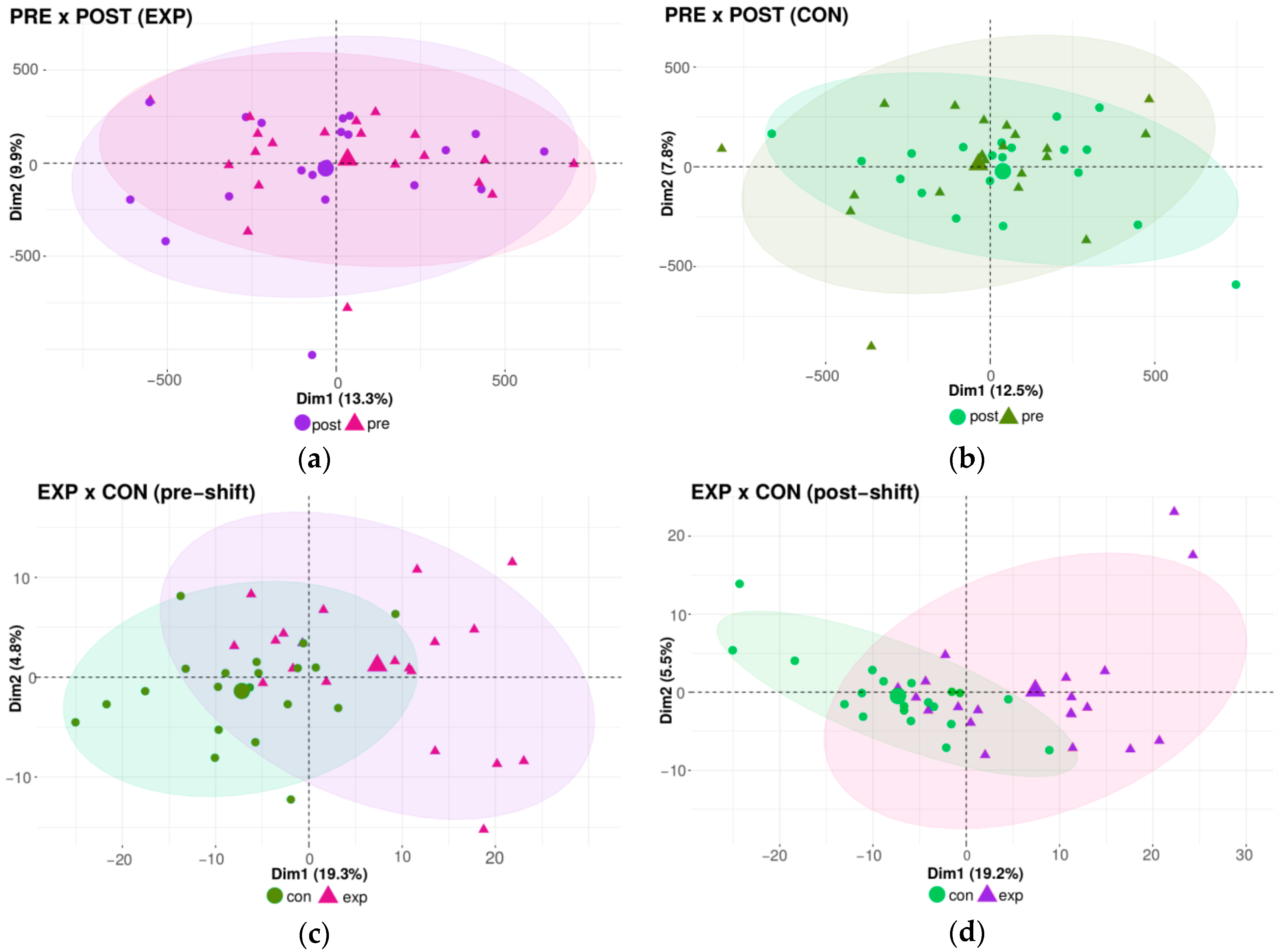
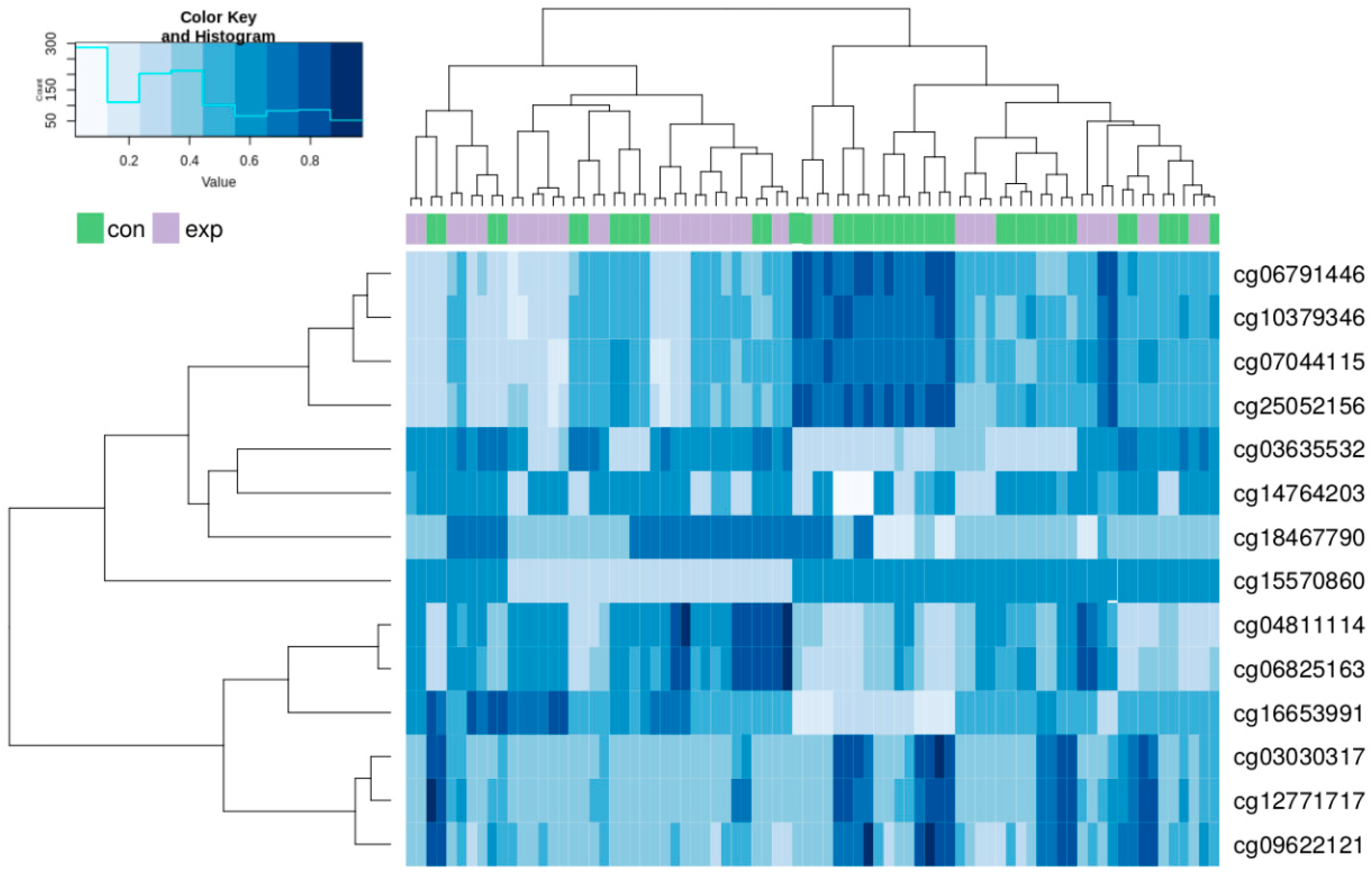
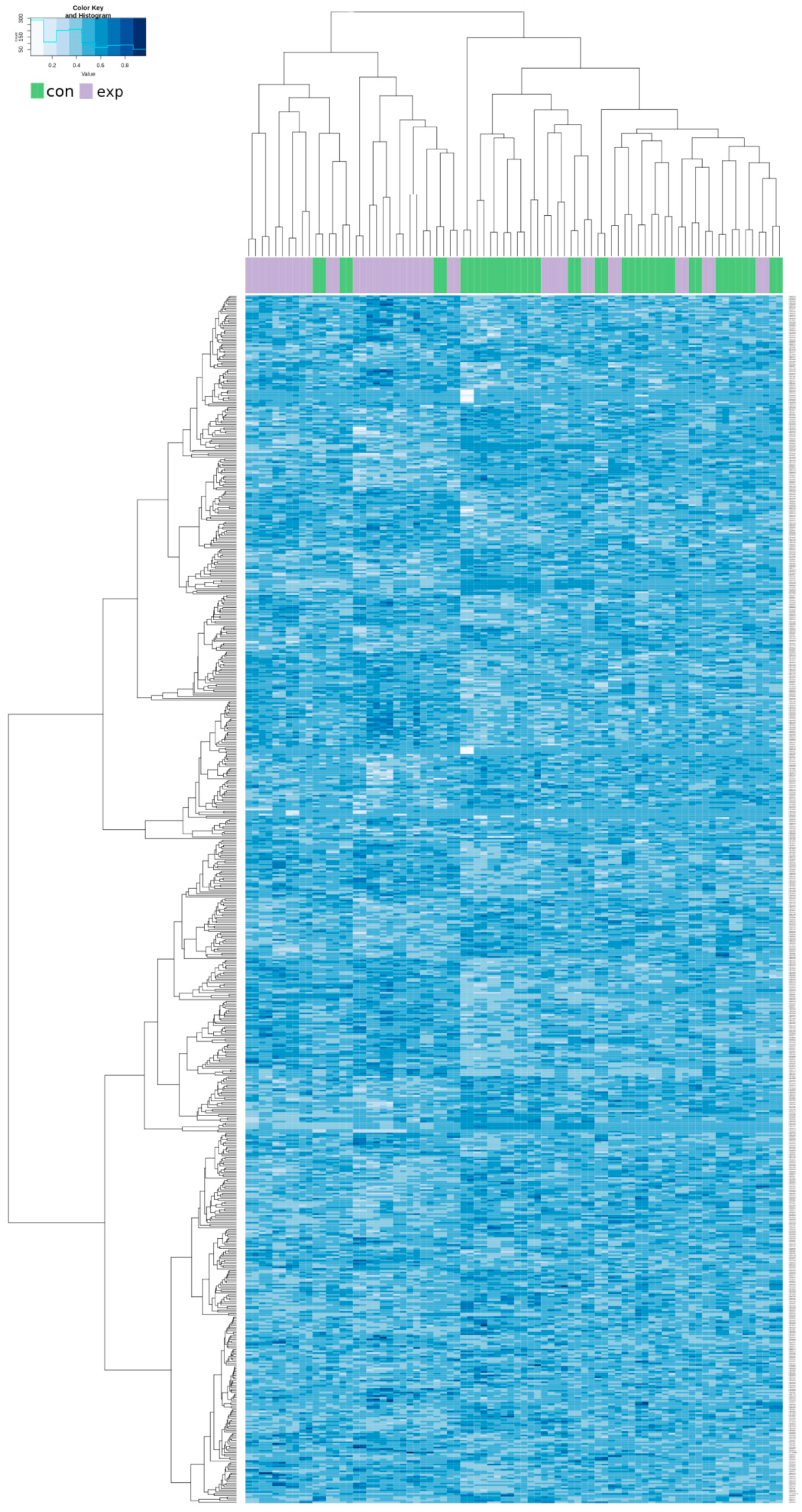
2.3. Genome-Wide DNA Methylation Microarray Analysis
3. Discussion
4. Materials and Methods
4.1. Study Population and Sampling
4.2. Exposure Monitoring Measurements
4.3. DNA Isolation and Quality Assessment
4.4. Quantitative DNA Methylation Analysis
4.5. Qualitative Infinium HD Methylation Assay
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 16HBE14o- | Human bronchial epithelial cells |
| 5-mC | 5-methyl-cytosine |
| APS | Aerodynamic particle sizer |
| ARVCF | ARVCF delta catenin family member |
| BEAS-2B | Human bronchial epithelial lung cells |
| BH | Benjamini-Hochberg methods |
| BICC1 | BicC family RNA binding protein 1 |
| BMI | Body mass index |
| CLDN10 | Claudin 10 |
| CPC | Condensation particle counter |
| DNMT | DNA methyltransferase |
| DYNLL1 | Dynein light chain LC8 – type 1 |
| FDR | False discovery rate |
| FCGBP | Fc fragment of IgG binding protein |
| FEV1 | Forced expiratory volume in 1 s |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FVC | Forced vital capacity |
| HaCaT | Human keratinocytes cells |
| HepG2 | Human liver cancer cells |
| HCG27 | HLA complex group 27 |
| HRP | Horseradish peroxidase |
| ITO | Indium tin oxide |
| LGR6 | Leucine rich repeat containing G |
| MAG | Metal active gas |
| MN | Micronuclei |
| MRC5 | Human fetal lung cells |
| MWCNT | Multi walled carbon nanotubes |
| NDRG4 | NDRG family member 4 |
| NHEJ | Non-homologous end joining repair |
| NM | Nanomaterials |
| NP | Nanoparticles |
| PCA | Principal component analysis |
| PM | Particulate matter |
| RADIL | Rap associating with DIL domain |
| SGCZ | Sarcoglycan zeta |
| SHISA3 | Shisa family member 3 |
| SMPS | Scanning mobility particle sizer |
| SNPs | Single nucleotide polymorphisms |
| SPION | Supermagnetic iron nanoparticles |
| SWCNT | Single walled carbon nanotubes |
| TLE2 | TLE family member 2, transcriptional corepressor |
| TMEM18 | Transmembrane protein 18 |
| TMEM9B | TMEM9 domain family member B |
| TSS | Transcription start site |
| UGT2B15/B17 | UDP glucuronosyltransferase family 2 member B15/B17 |
| WGBS | Whole-genome bisulfite sequencing |
| WTIP | WT1 interacting protein |
Appendix A

References
- The European Commission. Commission recommendation of 18 October 2011 on the definition of nanomaterial (2011/696/EU). Off. J. Eur. Union 2011, L 275, 38–40. [Google Scholar]
- Gong, C.; Tao, G.; Yang, L.; Liu, J.; Liu, Q.; Zhuang, Z. SiO2 nanoparticles induce global genomic hypomethylation in HaCaT cells. Biochem. Biophys. Commun. 2010, 397, 397–400. [Google Scholar] [CrossRef]
- Patil, N.A.; Gade, W.N.; Deobagkar, D.D. Epigenetic modulation upon exposure of lung fibroblasts to TiO2 and ZnO nanoparticles: Alterations in DNA methylation. Int. J. Nanomed. 2016, 11, 4509–4519. [Google Scholar]
- Li, J.; Tian, M.; Cui, L.; Dwyer, J.; Fullwood, N.J.; Shen, H.; Martin, F.L. Low-dose carbon-based nanoparticle-induced effects in A549 lung cells determined by biospectroscopy are associated with increases in genomic methylation. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Öner, D.; Ghosh, M.; Bové, H.; Moisse, M.; Boeckx, B.; Duca, R.C.; Poels, K.; Luyts, K.; Putzeys, E.; Landuydt, K.V.; et al. Differences in MWCNT- and SWCNT- induced DNA methylation alterations in association with the nuclear deposition. Part. Fibre Toxicol. 2018, 15, 1–19. [Google Scholar] [CrossRef]
- Sierra, M.I.; Rubio, L.; Bayón, G.F.; Cobo, I.; Menendez, P.; Morales, P.; Mangas, C.; Urdinguio, R.C.; Lopez, V.; Valdes, A.; et al. DNA methylation changes in human lung epithelia cells exposed to multi-walled carbon nanotubes. Nanotoxicology 2017, 11, 857–870. [Google Scholar] [CrossRef]
- Lu, X.; Miousse, I.R.; Pirela, S.V.; Melnyk, S.; Koturbash, I.; Demokritou, P. Short-term exposure to engineered nanomaterials affects cellular epigenome. Nanotoxicology 2016, 10, 140–150. [Google Scholar] [CrossRef]
- Bonadio, R.S.; Arcanjo, A.C.; Lima, E.C.; Vasconcelos, A.T.; Silva, R.C.; Horst, F.H.; Azevedo, R.B.; Poҫas-Fonseca, M.J.; Longo, J.P.F. DNA methylation alteration induced by transient exposure of MCF-7 cells to maghemite nanoparticles. Nanomedicine 2017, 12, 2637–2649. [Google Scholar] [CrossRef]
- Bonadio, R.S.; Cunha, M.C.P.C.D.; Longo, J.P.F.; Azevedo, R.B.; PoÇas-Fonseca, M.J. Exposure to maghemite nanoparticles induces epigenetic alterations in human submandibular gland cells. J. Nanosci. Nanotechnol. 2020, 20, 1454–1462. [Google Scholar] [CrossRef]
- Brzóska, K.; Grądzka, I.; Kruszewski, M. Silver, gold, and iron oxide nanoparticles alter miRNA expression but do not affect DNA methylation in HepG2 cells. Materials 2019, 12, 1038. [Google Scholar] [CrossRef]
- Tabish, A.M.; Poels, K.; Byun, H.-M.; Iuyts, K.; Baccarelli, A.A.; Martens, J.; Kerkhofs, S.; Seys, S.; Hoet, P.; Godderis, L. Changes in DNA methylation in mouse lungs after a single intra-tracheal administration of nanomaterials. PLoS ONE 2017, 12, e0169886. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, Y.; Ye, H.; Huang, K.; Lv, Z.; Ke, Y. Different effects of titanium dioxide nanoparticles instillation in young and adult mice on DNA methylation related with lung inflammation and fibrosis. Ecotoxicol. Environ. Saf. 2019, 176, 1–10. [Google Scholar] [CrossRef]
- Ognik, K.; Cholewinńska, E.; Juśkiewicz, J.; Zduńczyk, Z.; Tutaj, K.; Szlązak, R. The effect of copper nanoparticles and copper (II) salt on redox reactions and epigenetic changes in a rat model. J. Anim. Physiol. Anim. Nutr. 2019, 103, 675–686. [Google Scholar] [CrossRef]
- Liou, S.-H.; Wu, W.-T.; Liao, H.-Y.; Chen, C.-Y.; Tsai, C.-Y.; Jung, W.-T.; Lee, H.-L. Global DNA methylation and oxidative stress biomarkers in workers exposed to metal oxide nanoparticles. J. Hazard. Mater. 2017, 331, 329–335. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. DNA methylation: A profile of methods and applications. Biotechniques 2002, 33, 636–649. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef]
- Noehammer, C.; Pulverer, W.; Hassler, H.R.; Hofner, M.; Wielscher, M.; Vierlinger, K.; Liloglou, T.; McCarthy, D.; Jensen, T.J.; Nygren, A.; et al. Strategies for validation and testing of DNA methylation biomarkers. Epigenomics 2014, 6, 603–622. [Google Scholar] [CrossRef]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef]
- Pelclova, D.; Zdimal, V.; Schwarz, J.; Dvorackova, S.; Komarc, M.; Ondracek, J.; Kostejn, M.; Kacer, P.; Vlckova, S.; Fenclova, Z.; et al. Markers of oxidative stress in the exhaled breath condensate of workers handling nanocomposites. Nanomaterials 2018, 8, 611. [Google Scholar] [CrossRef]
- Pelclova, D.; Zdimal, V.; Komarc, M.; Vlckova, S.; Fenclova, Z.; Ondracek, J.; Schwarz, J.; Kostejn, M.; Kacer, P.; Vlckova, S.; et al. Deep airway inflammation and respiratory disorders in nanocomposite workers. Nanomaterials 2018, 8, 731. [Google Scholar] [CrossRef]
- Rossnerova, A.; Pelclova, D.; Zdimal, V.; Rossner, P., Jr.; Elzeinova, F.; Vrbova, K.; Topinka, J.; Schwarz, J.; Ondracek, J.; Kostejn, M.; et al. Micronucleus levels in nanocomposites production workers: Interpretation of results from two years of monitoring. In Proceedings of the NANOCON 2018: 10th Anniversary International Conference on Nanomaterials—Research & Application, Brno, Czech Republic, 17–19 October 2018; pp. 554–559. [Google Scholar]
- Rossnerova, A.; Pelclova, D.; Zdimal, V.; Rossner, P., Jr.; Elzeinova, F.; Vrbova, K.; Topinka, J.; Schwarz, J.; Ondracek, J.; Kostejn, M.; et al. The repeated cytogenetic analysis of subjects occupationally exposed to nanoparticles: A pilot study. Mutagenesis 2019, 34, 253–256. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Pelclova, D.; Dlouha, D.; Mikuska, P.; Dvorackova, S.; Vlckova, S.; Fenclova, Z.; Ondracek, J.; Kostejn, M.; Schwarz, J.; et al. Leukocyte telomere length is not affected by long-term occupational exposure to nano metal oxides. Ind. Health 2019, 57, 741–744. [Google Scholar] [CrossRef]
- Novotna, B.; Pelclova, D.; Rossnerova, A.; Zdimal, V.; Lischkova, L.; Vlckova, S.; Fenclova, Z.; Klusackova, P.; Zavodna, T.; Topinka, J.; et al. The genotoxic effects in the leukocytes of workers handling nanocomposite materials. Mutagenesis 2020. submitted. [Google Scholar]
- Rossnerova, A.; Tulupova, E.; Tabashidze, N.; Schmuczerova, J.; Dostal, M.; Rossner, P., Jr.; Gmuender, H.; Sram, R.J. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat. Res. 2013, 741–742, 18–26. [Google Scholar] [CrossRef]
- Rossnerova, A.; Pokorna, M.; Svecova, V.; Sram, R.J.; Topinka, J.; Zölzer, F.; Rossner, P., Jr. Adaptation of the human population to the environment: Current knowledge, clues from Czech cytogenetic and “omics” biomonitoring studies and possible mechanisms. Mutat. Res. 2017, 773, 188–203. [Google Scholar] [CrossRef]
- Vandegehuchte, M.B.; Janssen, C.R. Epigenetics in an ecotoxicological context. Mutat. Res. 2014, 764–765, 36–45. [Google Scholar] [CrossRef]
- Mirbahai, L.; Chipman, J.K. Epigenetic memory of environmental organisms: A reflection of lifetime stressor exposures. Mutat. Res. 2014, 764–765, 10–17. [Google Scholar] [CrossRef]
- Giuliani, C.; Bacalini, M.G.; Sazzini, M.; Pirazzini, C.; Franceschi, C.; Garagnani, P.; Luiselli, D. The epigenetic side of human adaptation: Hypotheses, evidences and theories. Ann. Hum. Biol. 2015, 42, 1–9. [Google Scholar] [CrossRef]
- Vineis, P.; Chatziioannou, A.; Cunliffe, V.T.; Flanagan, J.M.; Hanson, M.; Kirsch-Volders, M.; Kryptopoulos, S. Epigenetic memory in response to environmental stressors. FASEB J. 2017, 31, 2241–2251. [Google Scholar] [CrossRef]
- D’Urso, A.; Brickner, J.H. Epigenetic transcriptional memory. Curr. Genet. 2017, 63, 435–439. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Archana, G.; Rakshit, M.; Ng, K.E.; Tay, C.Y. Human keratinocytes adapt to ZnO nanoparticles induced toxicity via complex paracrine crosstalk and Nrf2.proteasomal signal transduction. Nanotoxicology 2018, 12, 1215–1229. [Google Scholar] [CrossRef]
- Fabrizio, P.; Garvis, S.; Palladino, F. Histone methylation and memory of environmental stress. Cells 2019, 8, 339. [Google Scholar] [CrossRef]
- Langie, S.A.; Szarc Vel Szic, K.; Declerck, K.; Traen, S.; Koppen, G.; Van Camp, G.; Schoeters, G.; Vanden Berghe, W.; De Boever, P. Whole-genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Alijagic, A.; Gaglio, D.; Napodano, E.; Russo, R.; Costa, C.; Benada, O.; Kofroňová, O.; Pinsino, A. Titanium dioxide nanoparticles temporarity influence the sea urchin immunological state suppressing inflammatory-relate gene transcription and boosting antioxidant metabolic activity. J. Hazard. Mater. 2020, 384, 1–11. [Google Scholar] [CrossRef]
- Sevane, N.; Martinez, R.; Bruford, M.W. Genome-wide differential DNA methylation in tropically adapted Creole cattle and their Iberian ancestors. Anim. Genet. 2019, 50, 15–26. [Google Scholar] [CrossRef]
- Dodeller, F.; Gottar, M.; Huesken, D.; Ioutgeno, V.; Cenni, B. The lysosomal transmembrane protein 9B regulates the activity of inflammatory signaling pathways. J. Biol. Chem. 2008, 283, 21487–21494. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Almén, M.S.; Jacobsson, J.A.; Ameur, A.; Moschonis, G.; Dedoussis, G.; Marcus, C.; Gyllensten, U.; Fredriksson, R.; Schiöth, H.B. Determination of obesity associated gene variants related to TMEM18 through ultra-deep targeted re-sequencing in a case-control cohort for pediatric obesity. Genet. Res. 2015, 97, 1–9. [Google Scholar] [CrossRef]
- Zlatohlavek, L.; Maratka, V.; Tumova, E.; Ceska, R.; Lanska, V.; Vrablik, M.; Hubacek, J.A. Body adiposity changes after lifestyle interventions in children/adolescents and the NYD-SP18 and TMEM18 variants. Med. Sci. Monit. 2018, 24, 7493–7498. [Google Scholar] [CrossRef]
- Rodhe, K.; Keller, M.; Klös, M.; Schleinitz, D.; Dietrich, A.; Schön, M.R.; Gärtner, D.; Lohmann, T.; Dreßler, M.; Stumvoll, M.; et al. Adipose tissue depot specific promoter methylation of TMEM18. Mol. Med. 2014, 92, 881–888. [Google Scholar]
- Joseph, P.V.; Jaime-Lara, R.B.; Wang, Y.; Xiang, L.; Henderson, W.A. Comprehensive and systematic analysis of gene expression patterns associated with body mass index. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Tsai, M.H.; Chen, W.C.; Yu, S.L.; Chen, C.C.; Jao, T.M.; Huang, C.Y.; Tzeng, S.T.; Yen, S.J.; Yang, Y.C. DNA hypermethylation of SHISA3 in colorectal cancer: An independent predictor of poor prognosis. Ann. Surg. Oncol. 2015, 3, 1481–1489. [Google Scholar] [CrossRef]
- Oeser, S.G.; Bingham, J.P.; Collier, A.C. Regulation of hepatic UGT2B15 by methylation in adults of Asian descent. Pharmaceutics 2018, 10, 6. [Google Scholar] [CrossRef]
- Seaborne, R.A.; Strauss, J.; Cocks, M.; Shepherd, S.; O’Brien, T.D.; van Someren, K.A.; Bell, P.G.; Murgatroyd, C.; Morton, J.P.; Stewart, C.E.; et al. Human skeletal muscle possesses an epigenetic memory of hypertrophy. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- West, K.L.; Kelliher, J.L.; Xu, Z.; An, L.; Reed, M.R.; Eoff, R.L.; Wang, J.; Huen, M.S.Y.; Leung, J.W.C. LC8/DYNLL1 is a 53BP1 effector and regulates checkpoint activation. Nucleic Acids Res. 2019, 47, 6236–6249. [Google Scholar] [CrossRef]
- Orr, S.E.; Gokulan, K.; Boudreau, M.; Cerniglia, C.E.; Khare, S. Alteration in the mRNA expression of genes associated with gastrointestinal permeability and ileal TNF-α secretion due to the exposure of silver nanoparticles in Sprague-Dawley rats. J. Nanobiotechnol. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Rossnerova, A.; Pelclova, D.; Zdimal, V.; Elzeinova, F.; Margaryan, H.; Chvojkova, I.; Topinka, J.; Schwarz, J.; Ondracek, J.; Kostejn, M.; et al. Males-females differences in the spectrum of chromosomal aberrations in the group of nanocomposites production workers. In Proceedings of the NANOCON 2019: 11th Anniversary International Conference on Nanomaterials—Research & Application, Brno, Czech Republic, 16–18 October 2019. in press. [Google Scholar]
- Talbot, N.; Kubelova, L.; Makes, O.; Ondracek, J.; Cusack, M.; Schwarz, J.; Vodicka, P.; Zikova, N.; Zdimal, V. Transformations of aerosol particles from an outdoor to indoor environment. Aerosol Air Qual. Res. 2017, 17, 653–665. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Iimma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number Concentrations of PM Fractions per cm3 | ||||||
|---|---|---|---|---|---|---|
| Processes | 1. | 2. | 3. | 4. | 5. | Total |
| Backgrounds | <25 nm | 25–100 nm | 100 nm–1 µm | 1−2.5 µm | 2.5−10 µm | <10 µm |
| MAG welding | 1680 | 3840 | 3790 | 1.1 | 0.215 | 9311 |
| Grinding and Milling | 9700 | 16400 | 3040 | 16 | 2.53 | 29159 |
| Background, basement | 574 | 6260 | 2370 | 0.42 | 0.078 | 9204 |
| Background, ground floor | 92.5 | 3550 | 1680 | 0.232 | 0.006 | 5323 |
| # Chrom. | CpG Locus (cg number) | Island Relation | Gene | Relevance or Phenotype | log FC | p-Value | p-Value Adjusted |
|---|---|---|---|---|---|---|---|
| #1 | cg04811114 | Open Sea | LGR6 | Signaling pathways | 1.645 | 1.30 × 10−7 | 0.002 |
| cg06825163 | Open Sea | “ | Breast carcinoma | 1.647 | 2.20 × 10−7 | 0.003 | |
| cg00588342 | Open Sea | “ | 0.294 | 1.44 × 10−5 | 0.027 | ||
| cg26347746 | Open Sea | “ | 0.863 | 2.61 × 10−7 | 0.003 | ||
| cg25270774 | Open Sea | “ | 1.233 | 3.25 × 10−7 | 0.003 | ||
| cg05044291 | Open Sea | “ | 1.282 | 3.11 × 10−7 | 0.003 | ||
| #6 | cg12771717 | N Shore | HCG27* | Regulation | −1.813 | 1.77 × 10−6 | 0.009 |
| cg03030317 | N Shore | “ | Ankylosing | −1.699 | 2.34 × 10−7 | 0.003 | |
| cg09622121 | N Shore | “ | spondylitis | −1.651 | 3.21 × 10−7 | 0.003 | |
| cg23595396 | N Shore | “ | Alopecia | −1.059 | 7.77 × 10−7 | 0.006 | |
| cg24023453 | N Shore | “ | Asthma | −0.833 | 1.24 × 10−6 | 0.008 | |
| Lung carcinoma | |||||||
| Blood cells count | |||||||
| #7 | cg18467790 | N Shelf | RADIL | Hypothyroidism | 3.111 | 3.97 × 10−8 | 0.001 |
| #10 | cg25052156 | N Shore | FGFR2 | Signaling pathways | −1.584 | 4.21 × 10−9 | <0.001 |
| cg06791446 | N Shore | “ | Kinase activity | −1.579 | 3.42 × 10−8 | 0.001 | |
| cg10379346 | N Shore | “ | Reg. cell prolif. | −1.558 | 4.18 × 10−8 | 0.001 | |
| cg16653991 | Open Sea | “ | Apoptosis | 1.659 | 7.81 × 10−8 | 0.001 | |
| cg11430259 | N Shore | “ | Lung carcinoma | −1.442 | 3.74 × 10−7 | 0.004 | |
| cg02210151 | N Shore | “ | Brest carcinoma | −1.152 | 4.66 × 10−8 | 0.001 | |
| cg22633036 | N Shore | “ | Colorectal cancer | −1.117 | 2.63 × 10−7 | 0.003 | |
| cg13437682 | N Shore | “ | Blood pressure | −1.025 | 1.99 × 10−8 | <0.001 | |
| cg16961769 | Open Sea | “ | −0.843 | 1.41 × 10−6 | 0.008 | ||
| cg12669518 | Open Sea | “ | −0.820 | 1.11 × 10−7 | 0.002 | ||
| cg17681491 | N Shore | “ | −0.791 | 1.35 × 10−8 | <0.001 | ||
| cg14968358 | Open Sea | “ | −0732 | 3.37 × 10−5 | 0.043 | ||
| cg23248910 | Open Sea | “ | −0.719 | 3.71 × 10−7 | 0.003 | ||
| cg13707729 | Open Sea | “ | −0.486 | 1.02 × 10−5 | 0.023 | ||
| cg17723924 | Open Sea | “ | −0.405 | 5.75 × 10−6 | 0.017 | ||
| cg25833171 | Open Sea | “ | −0.351 | 2.71 × 10−5 | 0.038 | ||
| cg12990750 | Open Sea | “ | −0.317 | 4.31 × 10−5 | 0.048 | ||
| cg07344086 | Open Sea | “ | −0.261 | 2.53 × 10−5 | 0.037 | ||
| cg25409939 | Open Sea | “ | 0.627 | 6.14 × 10−6 | 0.018 | ||
| cg03552039 | Open Sea | “ | 0.634 | 3.85 × 10−7 | 0.003 | ||
| cg17280705 | Open Sea | “ | 0.638 | 1.37 × 10−5 | 0.027 | ||
| cg08195415 | Open Sea | “ | 0.665 | 5.48 × 10−6 | 0.017 | ||
| cg08899523 | Open Sea | “ | 1.452 | 6.86 × 10−8 | 0.001 | ||
| cg07044115 | Open Sea | out | −1.778 | 1.07 × 10−8 | <0.001 | ||
| #11 | cg15570860 | S Shore | TMEM9B | Signaling pathways | −3.843 | 1.55 × 10−7 | 0.002 |
| cg16733419 | N Shelf | “ | Proinf. cytokines↑ | −0.339 | 2.25 × 10−5 | 0.034 | |
| Hemoglobin level | |||||||
| BMI | |||||||
| #19 | cg03635532 | CpG Island | FCGBP | Lung function | 2.373 | 7.94 × 10−10 | <0.001 |
| cg18588295 | CpG Island | “ | Triglyceride change | 0.317 | 8.07 × 10−6 | 0.021 | |
| cg08054032 | S Shore | “ | 0.557 | 1.10 × 10−8 | <0.001 | ||
| cg14764203 | Open Sea | out | 1.530 | 5.99 × 10−6 | 0.018 |
| # Chrom. | CpG Locus (cg number) | Island Relation | Gene | Relevance or Phenotype | log FC | p-Value | p-Value Adjusted |
|---|---|---|---|---|---|---|---|
| #2 | cg18049933 | N Shore | LOC100996579 | uncharacterized | 0.588 | 4.20 × 10−5 | 0.048 |
| cg15237618 | N Shore | “ | 0.658 | 1.97 × 10−5 | 0.032 | ||
| cg23987493 | N Shore | “ | 0.671 | 7.68 × 10−6 | 0.021 | ||
| cg17611880 | N Shore | TMEM18 | Transcription rec. | 0.473 | 3.89 × 10−5 | 0.046 | |
| cg18263335 | N Shore | “ | BMI | 0.852 | 2.37 × 10−6 | 0.012 | |
| cg27237671 | N Shore | “ | Body fat distrib. | 0.863 | 7.28 × 10−6 | 0.020 | |
| #4 | cg22541001 | S Shore | SHISA3 | Signaling pathways | 0.370 | 3.87 × 10−7 | 0.004 |
| cg13587180 | S Shelf | “ | Tumor suppressor | 0.448 | 1.12 × 10−6 | 0.007 | |
| cg11065575 | S Shelf | “ | Cytokine level | 0.541 | 4.30 × 10−5 | 0.048 | |
| Type II diabetes | |||||||
| cg13365324 | Open Sea | UGT2B15 | Blood cell distrib. | 1.243 | 5.61 × 10−6 | 0.018 | |
| cg07973162 | Open Sea | and B17 | Cholesterol | −1.171 | 7.00 × 10−6 | 0.020 | |
| cg07952421 | Open Sea | “ | Triglyceride | −1.103 | 1.88 × 10−5 | 0.032 | |
| Xenobiotics detox. | |||||||
| #8 | cg27405903 | Open Sea | SGCZ | Cognitive function | 0.457 | 3.36 × 10−6 | 0.014 |
| cg05986192 | Open Sea | “ | BMI | 0.641 | 2.07 × 10−6 | 0.011 | |
| cg17481116 | Open Sea | “ | 0.785 | 1.37 × 10−6 | 0.008 | ||
| #10 | cg08466030 | Open Sea | BICC1 | Gen expr. regul. | 0.397 | 2.70 × 10−5 | 0.038 |
| cg27040468 | Open Sea | “ | Signaling pathways | 0.660 | 1.07 × 10−6 | 0.007 | |
| cg12342675 | Open Sea | “ | Uric acid level | 0.987 | 8.73 × 10−7 | 0.006 | |
| #12 | cg27279351 | CpG Island | DYNLL1 | Intrac. transport | −0.305 | 1.62 × 10−7 | 0.002 |
| cg19946631 | N Shore | “ | Cellular senescence | −0.299 | 4.68 × 10−6 | 0.016 | |
| cg25284772 | N Shore | “ | Reticulocyte count | −0.241 | 1.24 × 10−6 | 0.008 | |
| Blood pressure | |||||||
| #13 | cg24545961 | S Shore | CLDN10 | Signaling pathways | −1.498 | 5.45 × 10−8 | 0.001 |
| cg25702335 | S Shore | “ | −1.488 | 1.14 × 10−7 | 0.002 | ||
| cg24529736 | S Shore | “ | −0.801 | 1.26 × 10−7 | 0.002 | ||
| cg05709657 | S Shore | “ | −0.741 | 1.07 × 10−5 | 0.023 | ||
| #16 | cg04484415 | N Shore | NDRG4 | Signaling pathways | 0.541 | 4.28 × 10−5 | 0.048 |
| cg05725404 | N Shore | “ | Apoptosis | 0.714 | 1.10 × 10−5 | 0.024 | |
| cg17457090 | N Shore | “ | QT interval | 0.616 | 3.47 × 10−5 | 0.043 | |
| Colorectal cancer | |||||||
| #19 | cg00857137 | CpG Island | TLE2 | Signaling pathways | 0.377 | 4.87 × 10−6 | 0.017 |
| cg26717563 | N Shore | “ | Blood cells count | 0.472 | 2.64 × 10−6 | 0.012 | |
| cg19334452 | CpG Island | “ | 0.533 | 1.66 × 10−6 | 0.009 | ||
| cg11374335 | N Shore | WTIP | Cellular senescence | 0.383 | 2.90 × 10−11 | <0.001 | |
| cg06177396 | N Shore | “ | Transcr. regulator | 0.408 | 8.65 × 10−12 | <0.001 | |
| cg04928251 | N Shore | “ | Metal ion binding | 0.547 | 1.00 × 10−10 | <0.001 | |
| “ | BMI | ||||||
| Triglyceride | |||||||
| Blood cells count | |||||||
| #22 | cg07821417 | N Shelf | ARVCF | Blood cells count | 0.317 | 2.00 × 10−5 | 0.032 |
| cg16324072 | S Shelf | “ | Blood metab. level | 0.337 | 4.72 × 10−6 | 0.016 | |
| cg13823643 | S Shore | “ | Serum metab. level | 0.373 | 6.16 × 10−7 | 0.005 |
| Characteristics Group | N | Mean ± SD | Median (Range) | p |
|---|---|---|---|---|
| Age (years) | ||||
| All | 40 | 42.1 ± 11.9 | 41 (21−72) | |
| Exposed | 20 | 39.3 ± 11 | 36.5 (24−65) | 0.129 |
| Controls | 20 | 45.0 ± 12.4 | 46 (21−72) | |
| Gender (M/F) | ||||
| All | 29/11 | N/A | N/A | |
| Exposed | 14/6 | N/A | N/A | 0.731 |
| Controls | 15/5 | N/A | N/A | |
| BMI (kg/m2) | ||||
| All | 40 | 26.4 ± 5.1 | 26 (19−38.9) | |
| Exposed | 20 | 26.8 ± 5.3 | 26.4 (19−36.7) | 0.655 |
| Controls | 20 | 26 ± 5 | 24.9 (20.2−38.9) |
| Characteristics Group | Mean ± SD | Median (Range) | p |
|---|---|---|---|
| NP exposure record | |||
| Exposed (n = 20) | |||
| Long-term (years) | 14.5 ± 9.2 | 12 (3−32) | |
| Common daily (min) | 115.5 ± 68.3 | 105 (60−270) | 0.028 |
| Short-term (min) | 154.5 ± 34.1 | 150 (120−240) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossnerova, A.; Honkova, K.; Pelclova, D.; Zdimal, V.; Hubacek, J.A.; Chvojkova, I.; Vrbova, K.; Rossner, P., Jr.; Topinka, J.; Vlckova, S.; et al. DNA Methylation Profiles in a Group of Workers Occupationally Exposed to Nanoparticles. Int. J. Mol. Sci. 2020, 21, 2420. https://doi.org/10.3390/ijms21072420
Rossnerova A, Honkova K, Pelclova D, Zdimal V, Hubacek JA, Chvojkova I, Vrbova K, Rossner P Jr., Topinka J, Vlckova S, et al. DNA Methylation Profiles in a Group of Workers Occupationally Exposed to Nanoparticles. International Journal of Molecular Sciences. 2020; 21(7):2420. https://doi.org/10.3390/ijms21072420
Chicago/Turabian StyleRossnerova, Andrea, Katerina Honkova, Daniela Pelclova, Vladimir Zdimal, Jaroslav A. Hubacek, Irena Chvojkova, Kristyna Vrbova, Pavel Rossner, Jr., Jan Topinka, Stepanka Vlckova, and et al. 2020. "DNA Methylation Profiles in a Group of Workers Occupationally Exposed to Nanoparticles" International Journal of Molecular Sciences 21, no. 7: 2420. https://doi.org/10.3390/ijms21072420
APA StyleRossnerova, A., Honkova, K., Pelclova, D., Zdimal, V., Hubacek, J. A., Chvojkova, I., Vrbova, K., Rossner, P., Jr., Topinka, J., Vlckova, S., Fenclova, Z., Lischkova, L., Klusackova, P., Schwarz, J., Ondracek, J., Ondrackova, L., Kostejn, M., Klema, J., & Dvorackova, S. (2020). DNA Methylation Profiles in a Group of Workers Occupationally Exposed to Nanoparticles. International Journal of Molecular Sciences, 21(7), 2420. https://doi.org/10.3390/ijms21072420