1. Introduction
Inflammation is considered a critical component of cancer progression. The presence of inflammatory cytokines in the tumor microenvironment has been linked to an aggressive phenotype in cancer cells [
1]. In particular, it has been proposed that increased levels of IL-1β, derived from the microenvironment of malignant cells, activate inflammation that promotes invasiveness [
2].
Previous work by our group has shown that binding of the inflammatory cytokine IL-1β to its receptor IL-1RI, present in non-invasive MCF-7 breast cancer cells, triggered the initiation of epithelial–mesenchymal transition (EMT) by activation of the signaling pathway IL-1β/IL-1R/β-catenin [
3,
4,
5]. The transition was initiated by striking modifications of the intercellular junctions and the actin cytoskeleton of the epithelial cells. The cells detached from each other acquiring a mesenchymal morphology and increased migration and invasiveness [
3]. Disorganization of cell–cell contacts led to internalization of cell junction proteins among those β-catenins which were translocated to the nucleus. The β-catenin acted as a transcriptional coactivator, modulating the expression of genes and proteins downstream of the IL-1β-activated signaling pathway to complete the EMT and the acquisition of an aggressive phenotype of the so-called 6D cells. These data supported the proposal by us and other authors of a relationship between cancer development and an inflammatory microenvironment [
6,
7].
For several years cannabidiol, CBD, a constituent of
Cannabis sativa without psychotropic effects, has been empirically used as an anti-inflammatory drug and modulator of cancer progression. Recent studies highlighted that CBD is toxic at different concentrations in diverse cells, making the results obtained in cell models and the clinic difficult to interpret and, therefore, for defining the proper dose for patients [
8]. On the other hand, in vitro studies have shown that activation of the cannabinoid receptors modulates different steps of tumorigenesis in several types of cancer [
9,
10]. It is known that CBD downregulates metastasis and replication in highly invasive cells by inhibiting expression of the
ID-1 gene [
11]. Cannabidiol has also been proposed as an inducer of apoptosis and autophagy, two mechanisms involved in decrease of cancer cell growth [
12]. These reports have suggested that CBD has a potential role in the treatment of tumors and chronic inflammatory diseases. Therefore, a better understanding of the cellular and molecular mechanisms underlying CBD activities is imperative for its safe administration in patients, particularly when treatment is prolonged [
8,
13].
Our present work was directed to explore if the anti-inflammatory activity of CBD could hinder and reverse the IL-1β-induced EMT, leading to malignancy. We used our breast cancer invasive 6D cells model [
4,
5]. It was found that 6D cells have high levels of CBD receptor CB1. CBD bound to CB1 is internalized and released in the cytoplasm. At this point, inactivation of AKT by CBD results in the inhibition of β-catenin nuclear translocation and downregulation of genes and proteins identified as markers of malignancy in the activated EMT. The inactivation of AKT by CBD increased β-catenin and E-cadherin expression, and their relocation at the cell contacts to form adherens junctions and recover an epithelial phenotype.
3. Discussion
In vitro and
in vivo studies have shown that CBD functions differ in every type of cancer and depends on the expression of cannabinoid receptors linked to a variety of signaling pathways [
13]. Aggressive breast cancer cell lines, such as MDA-MB-231, colon CaCo2 cells, and lung tissue A549, have been reported to have great sensitivity to CBD, while non-invasive cells are not affected [
12,
21]. The present work is focused on CBD blockage and reversion of cellular and molecular mechanisms induced by IL-1β that transformed non-invasive cells to an invasive phenotype. In the invasive 6D cells a significant increase of receptor CB1 expression was found, while it was expressed in low levels in the non-invasive control MCF-7 cells. This condition could explain the higher sensitivity of 6D cells to the treatment with CBD as shown in
Figure 1. The specificity of binding of CBD to CB1, was determined using the agonist AM251. Previous treatment of 6D cells with the agonist and then CBD increased cell viability, indicating the dependency of viability on the levels of the receptor. Analysis of CB1mRNA expression showed that CBD had no effect on the transcription of gene
CNR1 but greatly decreased the levels of the receptor, cell migration, and resistance to cisplatin. The negative regulation of CB1 levels by CBD could occur by internalization of the CB1/CBD complex and the recycling of CB1 to different cellular pathways. In the central nervous system, the CB1/CBD complex has been reported to be internalized and degraded and also that de novo synthesized receptor is directed to the cell surface [
22,
23]. The CBD-treated cells lost almost 50% of their CB1 receptors and important features of malignancy; therefore, it would be expected to be less aggressive cells.
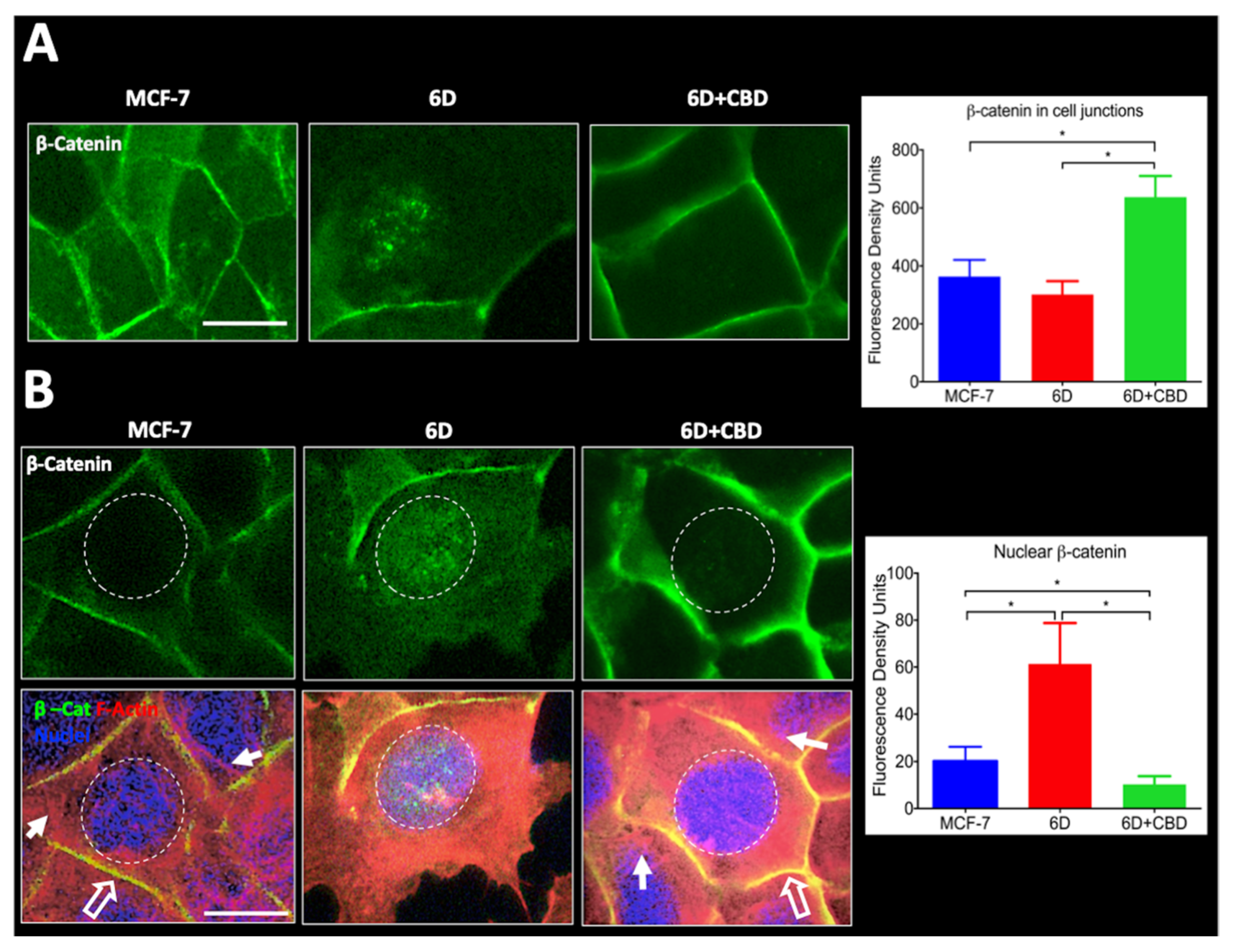
For migration to occur, loss of cell contacts and the disorganization of the actin cytoskeleton that maintain epithelial architecture have to take place. We found that CBD upregulated the levels of E-cadherin and β-catenin mRNAs, and the proteins reestablished their localization at the adherens junctions. These processes favored the recovery of cell contacts and the epithelial morphology therefore blocking cell migration.
Furthermore, we report here that CBD is an inhibitor of β-catenin nuclear translocation. We have previously shown that the endocannabinoid anandamide blocks β-catenin entry into the nucleus induced by IL-1β during the EMT. Degradation of cytoplasmic β-catenin inhibited the expression of malignancy-related genes [
5]. In both cases, the two cannabinoids blocked the nuclear translocation. However, anandamide-induced degradation of cytoplasmic β-catenin, while CBD relocated β-catenin at the adherens junctions. In both cases the expression of malignant markers in 6D cells was greatly reduced [
5,
16,
17]. In this work, we analyzed the expression of two genes,
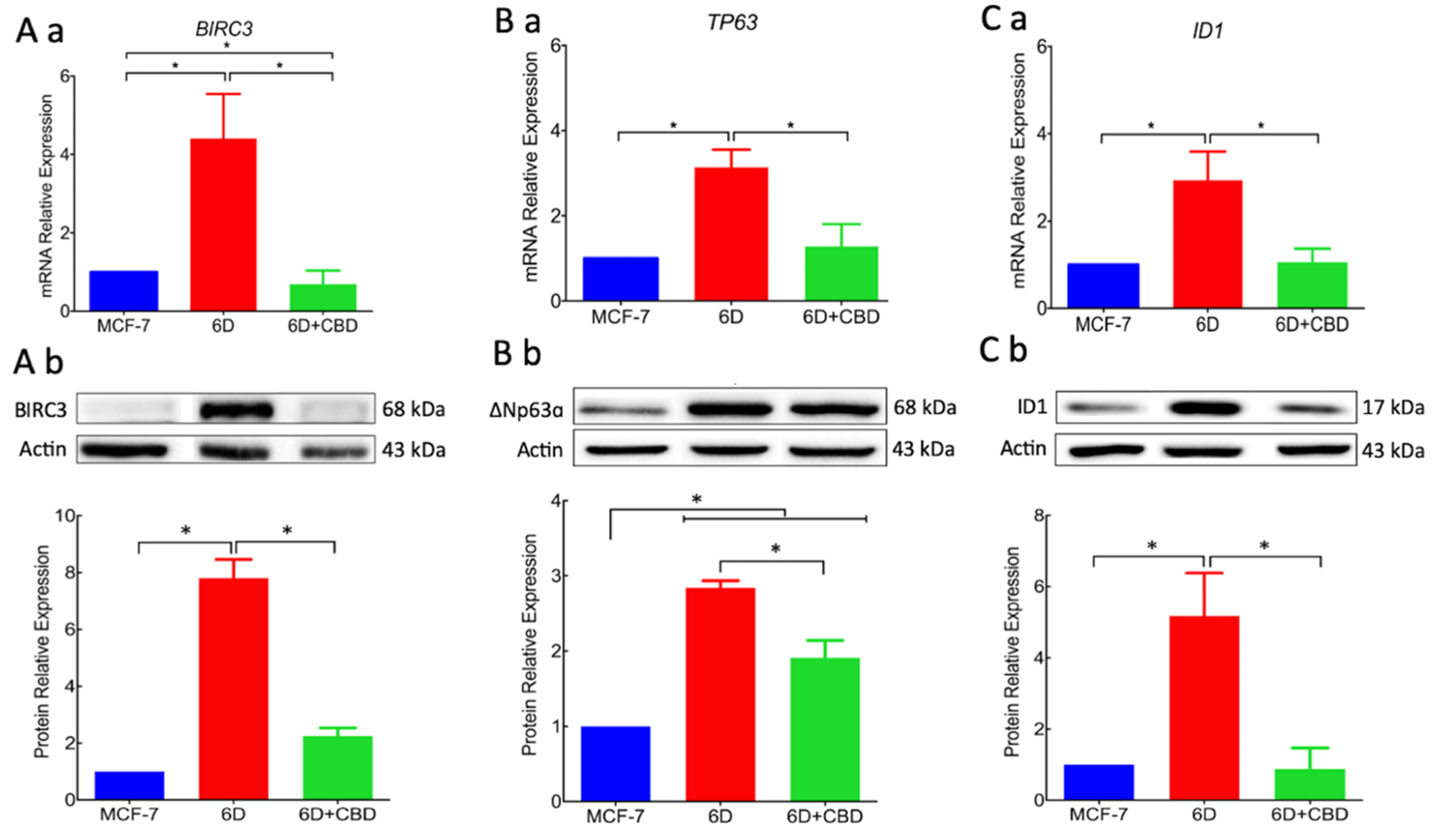
BIRC3 and
TP63, the expression of which was increased by the activation of the IL-1β/IL-1R/β-catenin pathway. Over-expression of proteins BIRC3 and ∆Np63α, two important effectors in the pathways that regulate resistance to doxorubicin and cisplatin, were significantly reduced by CBD. Downregulation of resistance to cisplatin by CBD explains the acquired sensitivity to drugs by 6D cells. All these activities of CBD support its role as an inhibitor of the IL-1β/IL-1R/β-catenin pathway and a possible inducer of mesenchymal reversion to an epithelial phenotype (drug-sensitive).
Additionally, we provide here the first evidence that the gene
ID1 and the protein ID1 are overexpressed in 6D cells stimulated with IL-1β. However, we cannot conclude that the protein expression occurs via the IL-1β/IL-1R/β-catenin pathway. ID1 protein has been reported in actively proliferating cells and has been identified in more than 20 tumorigenic cancer types [
24]. Significant reduction by CBD of ID1 expression and other protein markers of malignancy supports our hypothesis that CBD has a role in the reversion of a malignant phenotype. It has been reported that overexpression of ID1 in advanced bladder tumor cells is associated with the expression of mesenchymal markers, and when downregulated by ID-1si-RNA, the cells expressed epithelial markers [
25].
Activation of AKT is a crucial step in the IL-1β-induced EMT which regulates downstream progression of the IL-1β/IL-1R/β-catenin pathway. The present results showed that CBD, through downregulation of AKT phosphorylation, inhibited downstream activity of the IL-1β pathway and the expression of malignant markers such as migration and drug resistance. When wortmannin, a specific inhibitor of AKT phosphorylation, was added to the 6D cells, these showed reduced migration to levels similar to those in cells treated with CBD. It was very recently shown in an in silico study that CBD binds to the active site of PI3K and AKT [
26]. As a consequence of AKT inactivation by CBD, a crucial point for crosstalk in the IL-1β- and CBD-induced pathways, 6D cells could undergo reversion to an epithelial phenotype.
The acidic precursor of CBD (cannabidiolic acid, CBDA) has been able to inhibit the migration of breast cancer cells and to downregulate the proto-oncogene c-fos and the cyclooxygenase-2 (COX-2), highlighting the possibility that CBDA might act on a common pathway of inflammation and cancer mechanisms [
27]. Here, we demonstrated this association, for the first time, between CBD and the inflammatory IL-1β/IL-1R/β-catenin pathway through CB1 activation.
A diagrammatic model of our results and hypothesis about CBD induction of phenotype reversion is shown in
Figure 8.
Cell reprogramming has been thought to depend mainly on a genetic program; however, over-expression of E-cadherin has been shown to replace the presence of the gene
Oct4, thought to be a key participant in the process. A number of factors have been identified that inhibit or promote reprogramming through recognition of molecular programs involving PI3K/AKT and Wnt/β-catenin [
28]. We observed these processes in vitro when non-invasive MCF-7 breast cancer cells were treated with IL-1β, inducing an EMT program that changed the cells into an aggressive mesenchymal phenotype. Our present data showed that CBD could be the inducer of a mesenchymal–epithelial transition that would revert 6D cells to a non-invasive phenotype. This ability of CBD may be very useful in cancer therapy, as malignant cells treated with CBD will become sensitive to conventional cancer therapy.
Supplementary Materials Figure S1 shows the preliminary results obtained with an in vivo model using female nu/nu mice. When 6D cells were injected subcutaneously into the right flank of the body, tumors of approximately 8 mm in diameter developed after 40 days. The CBD, injected at that time directly into the tumor, reduced the tumor size to 50% after 72 days. These data show that CBD has the potential to act as an anti-tumorigenic drug.
4. Materials and Methods
4.1. Reagents
The RH-Oil5™ containing a concentration of 23.36 mg/mL of purified CBD was acquired from (HempMeds™, Monterrey, NL. México). A 1000 µM stock solution was prepared by dilution in DMSO. Aliquots from this stock were taken to obtain CBD concentrations to make a dose–response curve. Coconut oil (do TERRA
TM Pleasant Grove, UT, USA) 0.01 % final concentration was used as the vehicle. Wortmannin (Sigma–Aldrich, St. Louis, MO, USA) was used at 250 nM and cisplatin (CDDP, PISA™ Pharmaceutics, Guadalajara, Mexico) was used at 100 μM. Both compounds were used as previously reported in 6D cells [
3,
17]. The AM251 (Sigma–Aldrich, St Missouri, MO, USA) was used at 100 nM as CB1 receptor antagonist.
4.2. Primary Antibodies
The human antibodies used were anti-∆Np63α, anti-CB1, anti-cIAP2/BIRC3, and anti-ID1 (GeneTex, Irvine, CA, USA). Anti-AKT and anti-Phospho-AKT-Ser473 (Cell Signaling Technology, Danvers, MA, USA); anti-E-cadherin (BD; Baltimore, MD, USA); anti-β-catenin (Thermo Scientific, Waltham, MA, USA).
4.3. Cell Culture
The MCF-7 cells were obtained from (ATCC, Manasas, VA, USA). The 6D cells, a clone selected from MCF-7 non-invasive cells that was highly responsive to IL-1β stimulus and transformation to malignancy by the IL-1β-induced EMT [
4,
5], were used as the cellular model. All the cells were cultured in DMEM-F12 medium supplemented with 10% fetal bovine serum (FBS), penicillin (5000 U/mL), and streptomycin (5000 μg/mL) from Gibco BRL (Grand Island, NY, USA). Cultures were incubated at 37 °C with 5% CO
2. Before performing the experiments with CBD, the 6D cells were incubated with 20 ng/mL of human recombinant IL-1β for 48 h (to amplify their response to IL-1β stimulus) and then rinsed and cultured with regular medium (Peprotech, Rocky Hill, NJ, USA).
4.4. Cell Viability Assays in the Presence of CBD
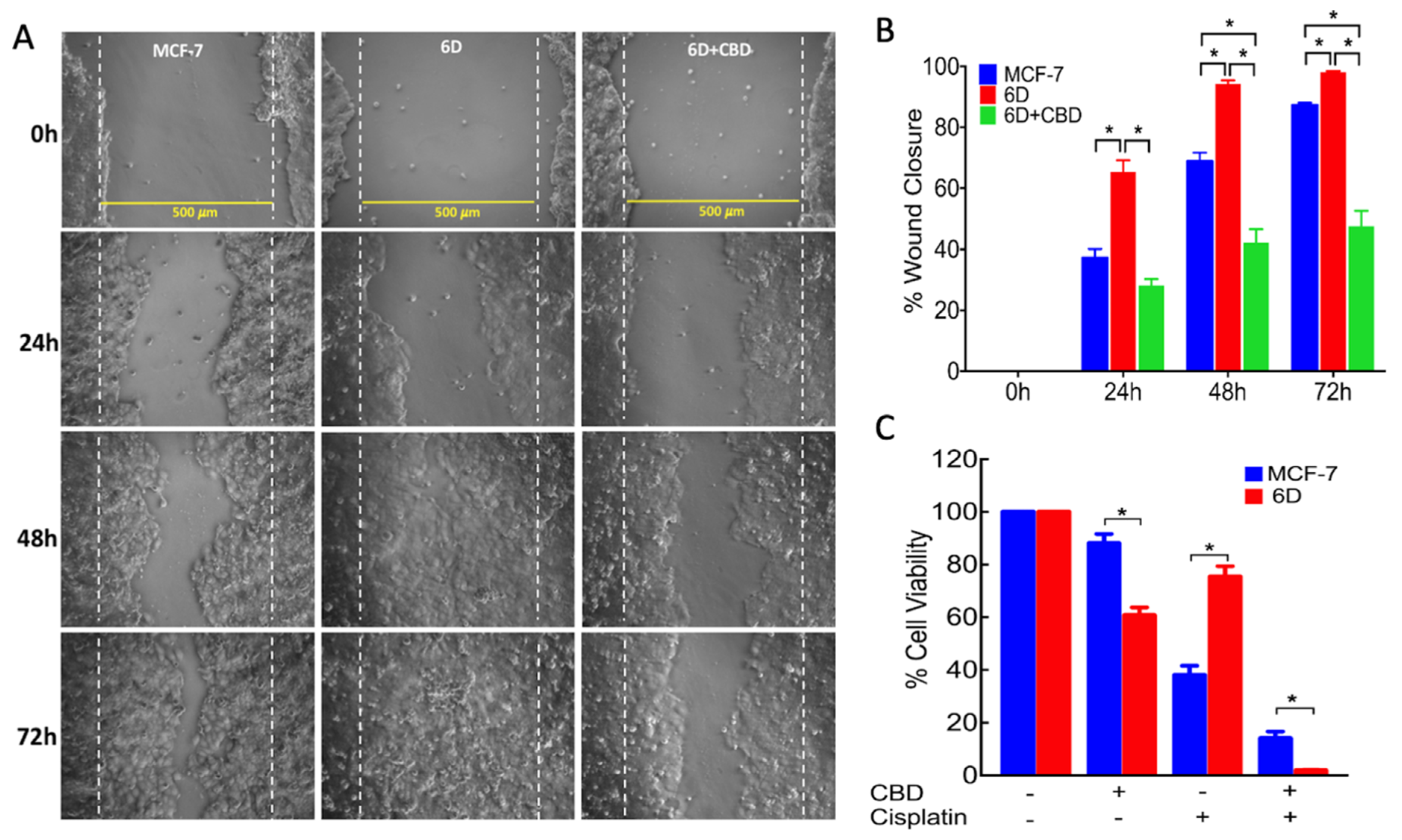
The MCF7 and 6D cells (2 × 104 cells/well) cultured in 96 well plates for 48 h, as indicated above, were switched to culture medium only supplemented with 1% FBS for 18 h and then divided in two groups: (1) cells treated with CBD at final concentrations of 5, 10, 15, and 20 µM; and (2) cells treated with coconut oil (vehicle) at a final concentration of 0.01%. After 48 h, 10 µL of WST-1 solution were added to each well (Roche Applied Science, Mannheim, Germany) and after 2 h the optical density, resulting from the reduction of tetrazolium salt in the solution by the viable cells, was measured at 450 nm in a microplate reader (Sunrise™, Tecan, Switzerland). The viability of MCF-7 cells, used as control of non-invasiveness, and that of 6D cells cultured in medium with only 1% FBS in the absence of CBD were given 100% values. To analyze viability data, three independent experiments were carried out (biological replicates) and each one was performed in triplicate (assay replication, n = 9).
4.5. CB1 Receptor Antagonist
The specific antagonist of the CB1 receptor AM251 was utilized at a final concentration of 100 nM [
9]. The AM251 was added to the cell cultures prior to addition of CBD, and then the cells were cultured for 48 h as indicated above.
4.6. Resistance to Cisplatin
Fifty thousand cells (MCF-7 or 6D) per well were seeded in 96 well culture plates and incubated for 24 h, then switched to medium with only 1% of FBS for 18 h. After this, cells were treated with different conditions: (1) cells with 100 µM cisplatin; (2) cells with 10 µM CBD; (3) cells with 10 µM CBD and 100 µM cisplatin; (4) cells only incubated in 1% FBS culture medium. All the cells were incubated for 48 h at 37 °C. To quantify cell viability for each condition the WST-1 assay, described above, was utilized.
4.7. Wound Healing Assay
The migrating ability of the cells was determined by the wound healing assay adapted from Shi and collaborators [
29]. Parental MCF-7 and 6D cells were grown in 24 well dishes to reach 90% confluence in normal culture medium. Then medium was changed to contain only 1% FBS and cells cultured for 18 h. The monolayers were wounded in a confluent zone by scratching the cells with a sterilized 10 µL pipette tip to obtain a clear area of 500 µm width. The wounded monolayers were washed with 1× PBS to eliminate debris and incubated in 1% FBS culture medium in the presence or absence of 20 ng/mL IL-1β, 10 µM CBD or 250 µM wortmannin for 1 h. After this time, cell migration was registered at 0, 24, 48, and 72 h using a phase-contrast objective in an Olympus inverted microscope. Cells protruding from the wound borders and those that had migrated into the wounded area were evaluated quantitatively by image analysis, using the Image-Pro Plus software. Measurements were carried out in three independent experiments (biological replicates) and each one was performed in quadruplicate (assay replication,
n = 12).
4.8. Gene Expression and Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from the cells using Trizol™ reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s recommendations. Integrity of total RNA components was assessed by electrophoresis in 2% agarose gels. The nucleic acid yield, quantification and purity (ratio 260/280nm) were analyzed using Nanodrop 2000, Microvolume UV-visible spectrophotometer (Thermo- Scientific, Wilmington, DE, USA). Samples were stored at −20 °C.
The cDNA synthesis was performed according to the High Capacity cDNA Reverse Transcription protocol (Applied Biosystems, CA USA) using 500 ng of total RNA. Reactions contained 2 µL of 10× RT Buffer, 0.8 µL of 25× dNTPs mix (100 mM), 2 µL 10× random primer set, 50 U Multi-Scribe Reverse Transcriptase, and nuclease-free water to make a volume of 10 µL.
Table S1 shows the primer sequences of the selected genes [
5,
30] and qRT-PCR conditions. Each primer pairs for the target gene were designed based on the sequence data obtained from GenBank. Real-time PCR was performed using Luminaris color HiGreen qPCR master mix (Thermo Fisher Scientific) in the Step One Real-Time PCR system (Thermo Fisher Scientific) according to the manufacturer’s protocol. qPCRs contained 0.3 µM of each RT-PCR primers pair, 5 µL of 2× Master mix, 1 µg cDNA and nuclease-free water to a final volume of 10 µL. Thermal cycling conditions were as following 1 cycle at 50 °C for 2 min, 1 cycle at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. Three cDNA samples from each condition were analyzed. Data were normalized with the
RPLP0 housekeeping gene. Quantification of gene expression and relative expression were calculated with Analysis of quantitative RT-qPCR data (∆Rn) using the LinRegPCR (ver. 11.0, Academic Medical Centre, University of Amsterdam, Amsterdam, The Netherlands) software.
4.9. SDS-PAGE and Western Blotting
Protein extracts were obtained from cell lysates using 1× RIPA buffer supplemented with Complete™ Protease Inhibitor Cocktail (Roche Applied Science, Mannheim, Germany). Protein concentrations were determined by the BCA method (Pierce™ BCA Protein Assay Kit). Thirty micrograms of protein were loaded per lane and separated by SDS-PAGE in 10% polyacrylamide gels, blotted onto nitrocellulose membranes and blocked with non-fat milk. The membranes were exposed to the anti-human antibodies listed in the Materials Section. The anti-actin monoclonal antibody, kindly donated by JM Hernández (CINVESTAV-IPN), was utilized to detect actin. The HRP-tagged secondary antibodies were anti-rabbit or anti-mouse (1:5000) (Jackson Immunoresearch, West Grove, PA, USA). Chemiluminescent detection was done with Immobilon™ and recorded on a ChemiDoc imaging device (Bio-Rad Laboratories, Hercules, CA, USA) for densitometric analyses with ImageLab™ software (v 6.0, Bio-Rad Laboratories, CA, USA). All proteins were identified by Western blot from three independent experiments (biological replicates, n = 3).
4.10. Immunofluorescence
For immuno-localization MCF-7 and 6D cells were grown on glass coverslips and fixed with 3.7% formaldehyde for 20 min and permeabilized with 0.1% Triton X-100 in PBS 1× for 5 min at RT. Cells were treated with PBS containing 0.1% Tween 20, 2% BSA, and rinsed with PBS before being exposed for 1 h at 37 °C to the different primary antibodies: β-Catenin (1:100 dilution) and E-Cadherin (1:100 dilution). Cells were then incubated with anti-mouse IgG or anti-Rabbit IgG conjugated with Alexa 488 at 1:100 dilution for 1 h at RT. Visualization of actin was performed staining with TRITC-phalloidin for 20 min at RT. Nuclei were stained with a 0.1% 4’,6-diamidino-2-phenylindole (DAPI) in a PBS/Triton solution for 5 min. Coverslips were mounted with VectaShield H-1000. Cells were observed in an Olympus 50× epifluorescence inverted microscope. All proteins were identified by immunofluorescence from three independent experiments (biological replicates, n = 3). Images acquired with a digital camera Olympus DP72 were analyzed with Image-pro Plus software (v. 3.0., Media Cybernetics, Rockville, MD, USA).
4.11. β-catenin Quantification by Image Analysis
Cell images captured for immuno-localization were utilized to quantify the amount of β-catenin in specific cell compartments (intercellular junctions and nuclei) using fluorescence density analysis. Before starting the analysis, the captured images were subjected to a deconvolution process to eliminate background noise (Deconvolution Image J software, Rasband, W.S., U. S. National Institutes of Health, Bethesda, MD, USA). Subsequently, 150 cells in six randomly chosen fields were selected to quantify the relative levels of the β-catenin signal associated with the intercellular junctions using a surface plot tool. The obtained data were used for statistical analysis. To measure the nuclear β-catenin, cells were stained with Alexa 488 and counterstained with TRITC-Phalloidin to set cell limits and visualize actin organization and DAPI to identify the nuclei. Considering the size and roundness of the nuclei (marked by a dotted circle) IF quantification of these areas was performed. One hundred and fifty cells were examined from three independent experiments. IF data were used for statistical determinations. The captured images of cells were processed in Image-Pro Plus Ver. 7.0, and Image J software.
4.12. Statistical Analysis
Data are presented as mean ± SD. In all cases, they represent at least three independent determinations (biological replicates) each done in triplicate. The v 6.0 of GraphPad Software (La Jolla CA, USA) was used for statistical analysis. Multiple comparisons were done using 2-way ANOVA and the Dunnett’s multiple comparisons test. p-Values ≤ 0.05 were considered significant.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







