Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Colorectal Cancer and Oxidative DNA Damage
2.1. DNA Damage and Colorectal Cancer Pathogenesis
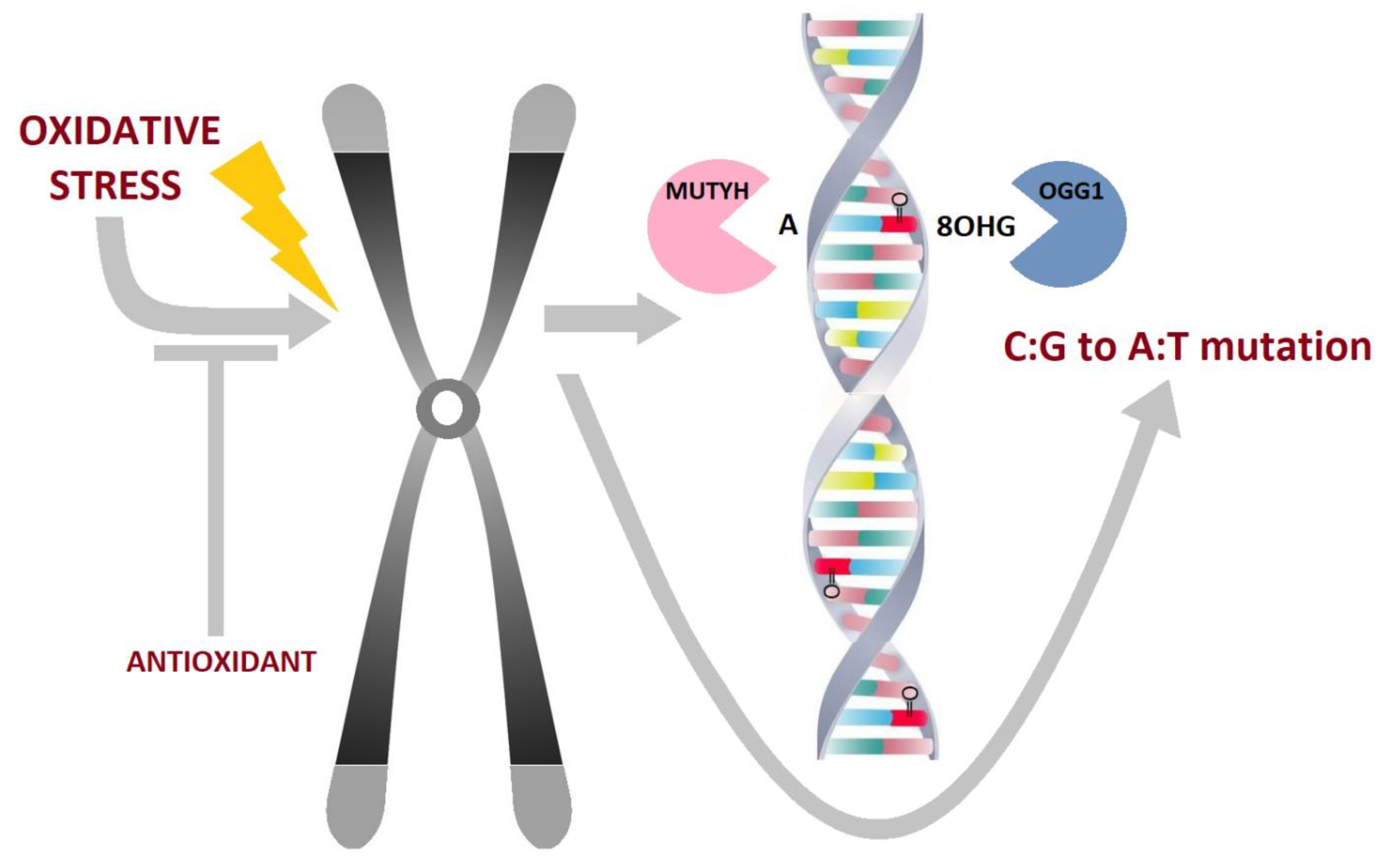
2.2. Oxidative DNA Damage, Characteristics, Biologic Properties and Relevance
2.3. The Repair of Oxidative DNA Damage
2.4. Oxidative DNA Damage, its Repair and Implications in Colorectal Carcinogenesis
2.4.1. Hereditary Syndromes with Defects in Glycosylases Predisposing Colorectal Cancer
2.4.2. Sporadic Colorectal Cancer
2.4.3. Base Excision Repair Capacity in Sporadic Colorectal Cancer
2.4.4. Sporadic Colorectal Cancer and Gene Variants in Base Excision Repair
2.5. Colorectal Cancer, Oxidative Damage and Intestinal Microenvironment
Oxidative Damage, Intestinal Microenvironment and CRC Prevention
3. Possible Utilization of Oxidative DNA Damage in Colorectal Cancer Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5FU | 5-fluorouracil |
| 8-oxo-dG | 8-oxo-7,8-dihydro-2´deoxyguanosine |
| BER | base excision repair |
| CIMP | CpG island methylator phenotype |
| CIN | chromosomal instability |
| CRC | colorectal cancer |
| DDR | DNA damage response |
| dNTPs | deoxynucleotide triphosphates |
| DRC | DNA excision repair capacity |
| EFS | event-free survival |
| FAPY | 2,6-diamino-4-hydroxy-5-formamidopyrimidine |
| HR | homologous recombination repair |
| LigI | human DNA ligase I |
| LigIII | human DNA ligase III |
| LOH | loss of heterozygosity |
| hOOG1 | human 8-oxo-dG DNA N-glycosylase 1 |
| MAP | MUTYH-associated polyposis |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| MSS | microsatellite stable |
| MTH1 | human mutT homolog 1 |
| MUTYH, MYH | mutY DNA glycosylase |
| NATS | NTHL1-associated tumor syndrome |
| NER | nucleotide excision repair |
| OS | overall survival |
| ROS | reactive oxygen species |
| SNP | single nucleotide polymorphism |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, H.; Chen, C. The colorectal cancer epidemic: Challenges and opportunities for primary, secondary and tertiary prevention. Br. J. Cancer 2018, 119, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Medina Pabon, M.A.; Babiker, H.M. A Review Of Hereditary Colorectal Cancers. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2019. [Google Scholar]
- Pawlik, T.M.; Raut, C.P.; Rodriguez-Bigas, M.A. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis. Markers 2004, 20, 199–206. [Google Scholar] [CrossRef]
- Yamagishi, H.; Kuroda, H.; Imai, Y.; Hiraishi, H. Molecular pathogenesis of sporadic colorectal cancers. Chin. J. Cancer 2016, 35, 4. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.R.; Azqueta, A.; Langie, S.A.S. Effects of micronutrients on DNA repair. Eur. J. Nutr. 2012, 51, 261–279. [Google Scholar] [CrossRef]
- Kompella, P.; Vasquez, K.M. Obesity and cancer: A mechanistic overview of metabolic changes in obesity that impact genetic instability. Mol. Carcinog. 2019, 58, 1531–1550. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Jenab, M.; Gunter, M.J. Adiposity and gastrointestinal cancers: Epidemiology, mechanisms and future directions. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 659–670. [Google Scholar] [CrossRef]
- Chakraborty, A.; Ferk, F.; Simić, T.; Brantner, A.; Dusinská, M.; Kundi, M.; Hoelzl, C.; Nersesyan, A.; Knasmüller, S. DNA-protective effects of sumach (Rhus coriaria L.), a common spice: Results of human and animal studies. Mutat. Res. 2009, 661, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Lorenzo, Y.; Karlsen, A.; Carlsen, M.H.; Novosadova, V.; Blomhoff, R.; Vodicka, P.; Collins, A.R. Both genetic and dietary factors underlie individual differences in DNA damage levels and DNA repair capacity. DNA Repair 2014, 16, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Méplan, C.; Hesketh, J. Selenium and cancer: A story that should not be forgotten-insights from genomics. Cancer Treat. Res. 2014, 159, 145–166. [Google Scholar] [CrossRef]
- Simonelli, V.; Mazzei, F.; D’Errico, M.; Dogliotti, E. Gene susceptibility to oxidative damage: From single nucleotide polymorphisms to function. Mutat. Res. 2012, 731, 1–13. [Google Scholar] [CrossRef]
- Tudek, B.; Speina, E. Oxidatively damaged DNA and its repair in colon carcinogenesis. Mutat. Res. 2012, 736, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ding, P.; Xie, C.; Ye, C.; Ye, M.; Pan, C.; Cao, X.; Zhang, S.; Zheng, S. Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget 2017, 8, 75767–75777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e1173. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Hernández, G.; Ramírez, M.J.; Minguillón, J.; Quiles, P.; Ruiz de Garibay, G.; Aza-Carmona, M.; Bogliolo, M.; Pujol, R.; Prados-Carvajal, R.; Fernández, J.; et al. Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Nat. Commun. 2018, 9, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niskakoski, A.; Pasanen, A.; Lassus, H.; Renkonen-Sinisalo, L.; Kaur, S.; Mecklin, J.-P.; Bützow, R.; Peltomäki, P. Molecular changes preceding endometrial and ovarian cancer: A study of consecutive endometrial specimens from Lynch syndrome surveillance. Mod. Pathol. 2018, 31, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; HA, A.L.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Manini, P.; De Palma, G.; Andreoli, R.; Marczynski, B.; Hanova, M.; Mozzoni, P.; Naccarati, A.; Vodickova, L.; Hlavac, P.; Mutti, A.; et al. Biomarkers of nucleic acid oxidation, polymorphism in, and expression of, hOGG1 gene in styrene-exposed workers. Toxicol. Lett. 2009, 190, 41–47. [Google Scholar] [CrossRef]
- Naccarati, A.; Pardini, B.; Hemminki, K.; Vodicka, P. Sporadic colorectal cancer and individual susceptibility: A review of the association studies investigating the role of DNA repair genetic polymorphisms. Mutat. Res. 2007, 635, 118–145. [Google Scholar] [CrossRef]
- Vodicka, P.; Kumar, R.; Stetina, R.; Sanyal, S.; Soucek, P.; Haufroid, V.; Dusinska, M.; Kuricova, M.; Zamecnikova, M.; Musak, L.; et al. Genetic polymorphisms in DNA repair genes and possible links with DNA repair rates, chromosomal aberrations and single-strand breaks in DNA. Carcinogenesis 2004, 25, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Ohno, M.; Sakumi, K.; Fukumura, R.; Furuichi, M.; Iwasaki, Y.; Hokama, M.; Ikemura, T.; Tsuzuki, T.; Gondo, Y.; Nakabeppu, Y. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014, 4, 4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, S.S. DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagenesis 2013, 54, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Vodenkova, S.; Opattova, A.; Vodickova, L. DNA damage and repair measured by comet assay in cancer patients. Mutat. Res. 2019, 843, 95–110. [Google Scholar] [CrossRef]
- Radom, M.; Machnicka, M.A.; Krwawicz, J.; Bujnicki, J.M.; Formanowicz, P. Petri net-based model of the human DNA base excision repair pathway. PLoS ONE 2019, 14, e0217913. [Google Scholar] [CrossRef] [Green Version]
- Köger, N.; Brieger, A.; Hinrichsen, I.M.; Zeuzem, S.; Plotz, G. Analysis of MUTYH alternative transcript expression, promoter function, and the effect of human genetic variants. Hum. Mutat. 2019, 40, 472–482. [Google Scholar] [CrossRef]
- Wang, R.; Hao, W.; Pan, L.; Boldogh, I.; Ba, X. The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 2018, 75, 3741–3750. [Google Scholar] [CrossRef] [Green Version]
- Leitner-Dagan, Y.; Sevilya, Z.; Pinchev, M.; Kramer, R.; Elinger, D.; Roisman, L.C.; Rennert, H.S.; Schechtman, E.; Freedman, L.; Rennert, G.; et al. N-methylpurine DNA glycosylase and OGG1 DNA repair activities: Opposite associations with lung cancer risk. J. Natl. Cancer Inst. 2012, 104, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Sjolund, A.B.; Senejani, A.G.; Sweasy, J.B. MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 2013, 743–744, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaria, P.; Svilar, D.; Brown, A.R.; Wang, X.-h.; Sobol, R.W.; Wyatt, M.D. SMUG1 but not UNG DNA glycosylase contributes to the cellular response to recovery from 5-fluorouracil induced replication stress. Mutat. Res. 2013, 743–744, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexeeva, M.; Moen, M.N.; Grøsvik, K.; Tesfahun, A.N.; Xu, X.M.; Muruzábal-Lecumberri, I.; Olsen, K.M.; Rasmussen, A.; Ruoff, P.; Kirpekar, F.; et al. Excision of uracil from DNA by hSMUG1 includes strand incision and processing. Nucleic Acids Res. 2019, 47, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Goto, M.; Tao, H.; Yoshimura, K.; Nakamura, S.; Misawa, K.; Sugimura, H. Defective repair capacity of variant proteins of the DNA glycosylase NTHL1 for 5-hydroxyuracil, an oxidation product of cytosine. Free Radic. Biol. Med. 2019, 131, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Slyvka, A.; Mierzejewska, K.; Bochtler, M. Nei-like 1 (NEIL1) excises 5-carboxylcytosine directly and stimulates TDG-mediated 5-formyl and 5-carboxylcytosine excision. Sci. Rep. 2017, 7, 9001. [Google Scholar] [CrossRef] [Green Version]
- Sarker, A.H.; Chatterjee, A.; Williams, M.; Lin, S.; Havel, C.; Jacob, P., 3rd; Boldogh, I.; Hazra, T.K.; Talbot, P.; Hang, B. NEIL2 protects against oxidative DNA damage induced by sidestream smoke in human cells. PLoS ONE 2014, 9, e90261. [Google Scholar] [CrossRef]
- Han, D.; Schomacher, L.; Schüle, K.M.; Mallick, M.; Musheev, M.U.; Karaulanov, E.; Krebs, L.; von Seggern, A.; Niehrs, C. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife 2019, 8, e49044. [Google Scholar] [CrossRef]
- Minko, I.G.; Christov, P.P.; Li, L.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Processing of N(5)-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair 2019, 73, 49–54. [Google Scholar] [CrossRef]
- Massaad, M.J.; Zhou, J.; Tsuchimoto, D.; Chou, J.; Jabara, H.; Janssen, E.; Glauzy, S.; Olson, B.G.; Morbach, H.; Ohsumi, T.K.; et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J. Clin. Investig. 2016, 126, 4219–4236. [Google Scholar] [CrossRef]
- Da, L.-T.; Shi, Y.; Ning, G.; Yu, J. Dynamics of the excised base release in thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 2018, 46, 568–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.; Liu, L.; Yang, Q.-L.; Wang, Y.; Xu, P.; Zhang, L.; Liu, S.; Dai, Q.; Ji, Q.; Xu, G.-L.; et al. Thymine DNA glycosylase recognizes the geometry alteration of minor grooves induced by 5-formylcytosine and 5-carboxylcytosine. Chem. Sci. 2019, 10, 7407–7417. [Google Scholar] [CrossRef] [Green Version]
- Weiser, B.P.; Rodriguez, G.; Cole, P.A.; Stivers, J.T. N-terminal domain of human uracil DNA glycosylase (hUNG2) promotes targeting to uracil sites adjacent to ssDNA-dsDNA junctions. Nucleic Acids Res. 2018, 46, 7169–7178. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Grist, S.; Gardner, J.; Suthers, G.; Wilson, T.M.; Lu, A.L. Functional characterization of human MutY homolog (hMYH) missense mutation (R231L) that is linked with hMYH-associated polyposis. Cancer Lett. 2007, 250, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, S.A.; Morreau, H.; de Miranda, N.F.C.C.; van Wezel, T. The missing heritability of familial colorectal cancer. Mutagenesis 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Kow, Y.W.; Mitra, S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis 2003, 24, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Sakumi, K.; Furuichi, M.; Tsuzuki, T.; Kakuma, T.; Kawabata, S.; Maki, H.; Sekiguchi, M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 1993, 268, 23524–23530. [Google Scholar]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Takao, M.; Kanno, S.-I.; Kobayashi, K.; Zhang, Q.-M.; Yonei, S.; van der Horst, G.T.J.; Yasui, A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J. Biol. Chem. 2002, 277, 42205–42213. [Google Scholar] [CrossRef] [Green Version]
- Galick, H.A.; Kathe, S.; Liu, M.; Robey-Bond, S.; Kidane, D.; Wallace, S.S.; Sweasy, J.B. Germ-line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proc. Natl. Acad. Sci. USA 2013, 110, 14314–14319. [Google Scholar] [CrossRef] [Green Version]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Rai, P.; Sobol, R.W. Mechanisms of MTH1 inhibition-induced DNA strand breaks: The slippery slope from the oxidized nucleotide pool to genotoxic damage. DNA Repair 2019, 77, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Urrabaz, R.; Nguyen, M.; Marty, J.; Stringer, S.; Cruz, E.; Medina-Gundrum, L.; Weitman, S. Elevated expression of DNA ligase I in human cancers. Clin. Cancer Res. 2001, 7, 4143–4148. [Google Scholar] [PubMed]
- Saquib, M.; Ansari, M.I.; Johnson, C.R.; Khatoon, S.; Kamil Hussain, M.; Coop, A. Recent advances in the targeting of human DNA ligase I as a potential new strategy for cancer treatment. Eur. J. Med. Chem. 2019, 182, 111657. [Google Scholar] [CrossRef]
- McNally, J.R.; O’Brien, P.J. Kinetic analyses of single-stranded break repair by human DNA ligase III isoforms reveal biochemical differences from DNA ligase I. J. Biol. Chem. 2017, 292, 15870–15879. [Google Scholar] [CrossRef] [Green Version]
- Adachi, S.; Takemoto, K.; Hirosue, T.; Hosogai, Y. Spontaneous and 2-nitropropane induced levels of 8-hydroxy-2′-deoxyguanosine in liver DNA of rats fed iron-deficient or manganese- and copper-deficient diets. Carcinogenesis 1993, 14, 265–268. [Google Scholar] [CrossRef]
- Kasai, H.; Crain, P.F.; Kuchino, Y.; Nishimura, S.; Ootsuyama, A.; Tanooka, H. Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 1986, 7, 1849–1851. [Google Scholar] [CrossRef]
- Wood, M.L.; Dizdaroglu, M.; Gajewski, E.; Essigmann, J.M. Mechanistic studies of ionizing radiation and oxidative mutagenesis: Genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry 1990, 29, 7024–7032. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Gackowski, D.; Foksinski, M.; Rozalski, R.; Roszkowski, K.; Jaruga, P. Oxidative DNA damage: Assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome. Free Radic. Biol. Med. 2002, 33, 192–200. [Google Scholar] [CrossRef]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; De Luca, G.; Degan, P.; Parlanti, E.; Dogliotti, E.; Barnes, D.E.; Lindahl, T.; Yang, H.; Miller, J.H.; Bignami, M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004, 64, 4411–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assié, G.; André, T.; Hélias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.-F.; et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weren, R.D.A.; Ligtenberg, M.J.; Geurts van Kessel, A.; De Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, E.F.R.; Ribeiro, M.L.; Magro, D.O.; Carvalho, J.; Kanno, D.T.; Martinez, C.A.R.; Coy, C.S.R. tissue expresion of the genes mutyh and ogg1 in patients with sporadic colorectal cancer. Arq. Bras. Cir. Dig. 2017, 30, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Vodenkova, S.; Jiraskova, K.; Urbanova, M.; Kroupa, M.; Slyskova, J.; Schneiderova, M.; Levy, M.; Buchler, T.; Liska, V.; Vodickova, L.; et al. Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients. DNA Repair 2018, 72, 77–85. [Google Scholar] [CrossRef]
- Koketsu, S.; Watanabe, T.; Nagawa, H. Expression of DNA repair protein: MYH, NTH1, and MTH1 in colorectal cancer. Hepatogastroenterology 2004, 51, 638–642. [Google Scholar]
- Zhang, X.; Song, W.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Sun, Q. Expression and function of MutT homolog 1 in distinct subtypes of breast cancer. Oncol. Lett. 2017, 13, 2161–2168. [Google Scholar] [CrossRef] [Green Version]
- Furlan, D.; Trapani, D.; Berrino, E.; Debernardi, C.; Panero, M.; Libera, L.; Sahnane, N.; Riva, C.; Tibiletti, M.G.; Sessa, F.; et al. Oxidative DNA damage induces hypomethylation in a compromised base excision repair colorectal tumourigenesis. Br. J. Cancer 2017, 116, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Farkas, S.A.; Vymetalkova, V.; Vodickova, L.; Vodicka, P.; Nilsson, T.K. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/beta-catenin signaling pathway genes. Epigenomics 2014, 6, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybylowska, K.; Kabzinski, J.; Sygut, A.; Dziki, L.; Dziki, A.; Majsterek, I. An association selected polymorphisms of XRCC1, OGG1 and MUTYH gene and the level of efficiency oxidative DNA damage repair with a risk of colorectal cancer. Mutat. Res. 2013, 745–746, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Korenkova, V.; Collins, A.R.; Prochazka, P.; Vodickova, L.; Svec, J.; Lipska, L.; Levy, M.; Schneiderova, M.; Liska, V.; et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin. Cancer Res. 2012, 18, 5878–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, L.K.; Moreno, N.C.; Rocha, C.R.R.; Munford, V.; Santos, V.; Soltys, D.T.; Garcia, C.C.M.; Sarasin, A.; Menck, C.F.M. XPD/ERCC2 mutations interfere in cellular responses to oxidative stress. Mutagenesis 2019, 34, 341–354. [Google Scholar] [CrossRef]
- Melis, J.P.M.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-H.; Kang, T.-H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Shi, T.-Y.; Zhu, M.-L.; Wang, M.-Y.; Li, Q.-X.; Wei, Q.-Y. Associations of Lys939Gln and Ala499Val polymorphisms of the XPC gene with cancer susceptibility: A meta-analysis. Int. J. Cancer 2013, 133, 1765–1775. [Google Scholar] [CrossRef]
- Slyskova, J.; Naccarati, A.; Pardini, B.; Polakova, V.; Vodickova, L.; Smerhovsky, Z.; Levy, M.; Lipska, L.; Liska, V.; Vodicka, P. Differences in nucleotide excision repair capacity between newly diagnosed colorectal cancer patients and healthy controls. Mutagenesis 2012, 27, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Stetina, R.; Polakova, V.; Tulupova, E.; Naccarati, A.; Vodickova, L.; Kumar, R.; Hanova, M.; Pardini, B.; Slyskova, J.; et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis 2007, 28, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Hsieh, L.-L.; Tang, R.; Santella, R.M.; Chang-Chieh, C.R.; Yeh, C.-C. Association between polymorphisms of APE1 and OGG1 and risk of colorectal cancer in Taiwan. World J. Gastroenterol. 2016, 22, 3372–3380. [Google Scholar] [CrossRef] [Green Version]
- Pardini, B.; Naccarati, A.; Novotny, J.; Smerhovsky, Z.; Vodickova, L.; Polakova, V.; Hanova, M.; Slyskova, J.; Tulupova, E.; Kumar, R.; et al. DNA repair genetic polymorphisms and risk of colorectal cancer in the Czech Republic. Mutat. Res. 2008, 638, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.-L.; Han, F.-F.; Wang, H.-Y.; Wang, L. Meta-analysis of the association between hOGG1 Ser326Cys polymorphism and risk of colorectal cancer based on case--control studies. J. Cancer Res. Clin. Oncol. 2012, 138, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Kinnersley, B.; Buch, S.; Castellví-Bel, S.; Farrington, S.M.; Forsti, A.; Hampe, J.; Hemminki, K.; Hofstra, R.M.W.; Northwood, E.; Palles, C.; et al. Re: Role of the oxidative DNA damage repair gene OGG1 in colorectal tumorigenesis. J. Natl. Cancer Inst. 2014, 106, dju086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; He, B.-S.; Pan, Y.-Q.; Xu, Y.-Q.; Wang, S.-K. Association of OGG1 Ser326Cys polymorphism with colorectal cancer risk: A meta-analysis. Int. J. Colorectal Dis. 2011, 26, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Rosa, F.; Barone, E.; Di Gaetano, C.; Slyskova, J.; Novotny, J.; Levy, M.; Garritano, S.; Vodickova, L.; Buchler, T.; et al. Variation within 3′-UTRs of base excision repair genes and response to therapy in colorectal cancer patients: A potential modulation of microRNAs binding. Clin. Cancer Res. 2013, 19, 6044–6056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiraskova, K.; Hughes, D.J.; Brezina, S.; Gumpenberger, T.; Veskrnova, V.; Buchler, T.; Schneiderova, M.; Levy, M.; Liska, V.; Vodenkova, S.; et al. Functional Polymorphisms in DNA Repair Genes Are Associated with Sporadic Colorectal Cancer Susceptibility and Clinical Outcome. Int. J. Mol. Sci. 2018, 20, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Lorenzo Bermejo, J.; Chang-Claude, J.; Hoffmeister, M.; Fernández-Rozadilla, C.; Carracedo, A.; Castells, A.; Castellví-Bel, S.; The EPICOLON Consortium Members of the EPICOLON Consortium; Naccarati, A.; et al. Meta-analysis of mismatch repair polymorphisms within the cogent consortium for colorectal cancer susceptibility. PLoS ONE 2013, 8, e72091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tlaskalová-Hogenová, H.; Stěpánková, R.; Kozáková, H.; Hudcovic, T.; Vannucci, L.; Tučková, L.; Rossmann, P.; Hrnčíř, T.; Kverka, M.; Zákostelská, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell. Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Park, H.E.; Kim, J.H.; Cho, N.-Y.; Lee, H.S.; Kang, G.H. Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows Arch. 2017, 471, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimesova, K.; Jiraskova Zakostelska, Z.; Tlaskalova-Hogenova, H. Oral Bacterial and Fungal Microbiome Impacts Colorectal Carcinogenesis. Front. Microbiol. 2018, 9, 774. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Hsu, C.W.; Sowers, M.L.; Hsu, W.; Eyzaguirre, E.; Qiu, S.; Chao, C.; Mouton, C.P.; Fofanov, Y.; Singh, P.; Sowers, L.C. How does inflammation drive mutagenesis in colorectal cancer? Trends Cancer Res. 2017, 12, 111–132. [Google Scholar]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- De Barrios, O.; Sanchez-Moral, L.; Cortés, M.; Ninfali, C.; Profitós-Pelejà, N.; Martínez-Campanario, M.C.; Siles, L.; Del Campo, R.; Fernández-Aceñero, M.J.; Darling, D.S.; et al. ZEB1 promotes inflammation and progression towards inflammation-driven carcinoma through repression of the DNA repair glycosylase MPG in epithelial cells. Gut 2019, 68, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.T.; Chiu, Y.T.; Wei, P.Y.; Chiang, C.W.; Fang, H.L.; Wei, S.C. Microbiota and gastrointestinal cancer. J. Formos. Med. Assoc. 2019, 118 (Suppl. 1), S32–S41. [Google Scholar] [CrossRef]
- Møller, P.; Loft, S. Dietary antioxidants and beneficial effect on oxidatively damaged DNA. Free Radic. Biol. Med. 2006, 41, 388–415. [Google Scholar] [CrossRef] [PubMed]
- Hoelzl, C.; Knasmüller, S.; Misík, M.; Collins, A.; Dusinská, M.; Nersesyan, A. Use of single cell gel electrophoresis assays for the detection of DNA-protective effects of dietary factors in humans: Recent results and trends. Mutat. Res. 2009, 681, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Shoji, M.; Hoshigami, H.; Watanabe, K.; Watanabe, K.; Takatsuzu, T.; Yasuda, S.; Igoshi, K.; Kinoshita, H. Antioxidant capacity of soymilk yogurt and exopolysaccharides produced by lactic acid bacteria. Biosci. Microbiota Food Health 2019, 38, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Rejhová, A.; Opattová, A.; Čumová, A.; Slíva, D.; Vodička, P. Natural compounds and combination therapy in colorectal cancer treatment. Eur. J. Med. Chem. 2018, 144, 582–594. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; de Vos, W.M. Effect of diet on the intestinal microbiota and its activity. Curr. Opin. Gastroenterol. 2014, 30, 189–195. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.-J.; Tveit, K.-M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Colorectal. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyskova, J.; Cordero, F.; Pardini, B.; Korenkova, V.; Vymetalkova, V.; Bielik, L.; Vodickova, L.; Pitule, P.; Liska, V.; Matejka, V.M.; et al. Post-treatment recovery of suboptimal DNA repair capacity and gene expression levels in colorectal cancer patients. Mol. Carcinog. 2015, 54, 769–778. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Wilson, D.M., 3rd. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci. 2009, 66, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Chen, W.; Lian, W.; Yuan, Y.; Li, M. The synergistic effects of oxaliplatin and piperlongumine on colorectal cancer are mediated by oxidative stress. Cell Death Dis. 2019, 10, 600. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef]
- Opattova, A.; Horak, J.; Vodenkova, S.; Kostovcikova, K.; Cumova, A.; Macinga, P.; Galanova, N.; Rejhova, A.; Vodickova, L.; Kozics, K.; et al. Ganoderma Lucidum induces oxidative DNA damage and enhances the effect of 5-Fluorouracil in colorectal cancer in vitro and in vivo. Mutat. Res. 2019, 845, 403065. [Google Scholar] [CrossRef]
- Liu, W.; Gu, J.; Qi, J.; Zeng, X.-N.; Ji, J.; Chen, Z.-Z.; Sun, X.-L. Lentinan exerts synergistic apoptotic effects with paclitaxel in A549 cells via activating ROS-TXNIP-NLRP3 inflammasome. J. Cell Mol. Med. 2015, 19, 1949–1955. [Google Scholar] [CrossRef]
- Morano, F.; Corallo, S.; Niger, M.; Barault, L.; Milione, M.; Berenato, R.; Moretto, R.; Randon, G.; Antista, M.; Belfiore, A.; et al. Temozolomide and irinotecan (TEMIRI regimen) as salvage treatment of irinotecan-sensitive advanced colorectal cancer patients bearing MGMT methylation. Ann. Oncol. 2018, 29, 1800–1806. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.S.; Banerjee, S.; Aneja, R.; Sarkar, F.H.; Ostrov, D.A.; Narayan, S. DNA polymerase β as a novel target for chemotherapeutic intervention of colorectal cancer. PLoS ONE 2011, 6, e16691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishita, T.; Okamoto, T.; Akamine, T.; Takamori, S.; Takada, K.; Katsura, M.; Toyokawa, G.; Shoji, F.; Shimokawa, M.; Oda, Y.; et al. Association of MTH1 expression with the tumor malignant potential and poor prognosis in patients with resected lung cancer. Lung Cancer 2017, 109, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.; Saeki, H.; Nakashima, Y.; Iimori, M.; Kitao, H.; Oki, E.; Oda, Y.; Nakabeppu, Y.; Kakeji, Y.; Maehara, Y. Prognostic impact of MutT homolog-1 expression on esophageal squamous cell carcinoma. Cancer Med. 2017, 6, 258–266. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, L.; Yang, J.; Qiao, H.; Li, L.; Guo, Q.; Ma, J.; Zhao, L.; Wang, J.; Jiang, G.; et al. Potent and specific MTH1 inhibitors targeting gastric cancer. Cell Death Dis. 2019, 10, 434. [Google Scholar] [CrossRef] [Green Version]
- Abbas, H.H.K.; Alhamoudi, K.M.H.; Evans, M.D.; Jones, G.D.D.; Foster, S.S. MTH1 deficiency selectively increases non-cytotoxic oxidative DNA damage in lung cancer cells: More bad news than good? BMC Cancer 2018, 18, 423. [Google Scholar] [CrossRef] [Green Version]
- Frattini, M.; Balestra, D.; Suardi, S.; Oggionni, M.; Alberici, P.; Radice, P.; Costa, A.; Daidone, M.G.; Leo, E.; Pilotti, S.; et al. Different genetic features associated with colon and rectal carcinogenesis. Clin. Cancer Res. 2004, 10, 4015–4021. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Malietzis, G.; Askari, A.; Bernardo, D.; Al-Hassi, H.O.; Clark, S.K. Is right-sided colon cancer different to left-sided colorectal cancer?-a systematic review. Eur. J. Surg Oncol. 2015, 41, 300–308. [Google Scholar] [CrossRef]
- Punt, C.J.A.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Hoogerbrugge, N. NTHL1 defines novel cancer syndrome. Oncotarget 2015, 6, 34069–34070. [Google Scholar] [CrossRef] [Green Version]
- Rolseth, V.; Luna, L.; Olsen, A.K.; Suganthan, R.; Scheffler, K.; Neurauter, C.G.; Esbensen, Y.; Kuśnierczyk, A.; Hildrestrand, G.A.; Graupner, A.; et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci. Rep. 2017, 7, 4384. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, G.J.; Huynh, M.; Rai, P. MTH1 as a Chemotherapeutic Target: The Elephant in the Room. Cancers 2017, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Corrado, A.; Paolicchi, E.; Cugliari, G.; Berndt, S.I.; Bezieau, S.; Bien, S.A.; Brenner, H.; Caan, B.J.; Campbell, P.T.; et al. DNA repair and cancer in colon and rectum: Novel players in genetic susceptibility. Int. J. Cancer 2020, 146, 363–372. [Google Scholar] [CrossRef]
- Forsti, A.; Frank, C.; Smolkova, B.; Kazimirova, A.; Barancokova, M.; Vymetalkova, V.; Kroupa, M.; Naccarati, A.; Vodickova, L.; Buchancova, J.; et al. Genetic variation in the major mitotic checkpoint genes associated with chromosomal aberrations in healthy humans. Cancer Lett. 2016, 380, 442–446. [Google Scholar] [CrossRef]
- Vodicka, P.; Musak, L.; Frank, C.; Kazimirova, A.; Vymetalkova, V.; Barancokova, M.; Smolkova, B.; Dzupinkova, Z.; Jiraskova, K.; Vodenkova, S.; et al. Interactions of DNA repair gene variants modulate chromosomal aberrations in healthy subjects. Carcinogenesis 2015, 36, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Vodenkova, S.; Buchler, T.; Vodickova, L. DNA repair capacity and response to treatment of colon cancer. Pharmacogenomics 2019, 20, 1225–1233. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Richard, C.; Chevrier, S.; Végran, F.; Boidot, R. Efficiency of olaparib in colorectal cancer patients with an alteration of the homologous repair protein. World J. Gastroenterol. 2016, 22, 10680–10686. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Howes, T.R.L.; Wiest, N.E. DNA ligases as therapeutic targets. Transl. Cancer Res. 2013, 2, 1219. [Google Scholar] [PubMed]


{kind=link}
| Glycosylase Name | Gene | Enzyme Commission Number | Biologic Function | Reference |
|---|---|---|---|---|
| Adenine DNA glycosylase | MUTYH | 3.2.2.31 | MUTYH is a monofunctional DNA glycosylase which, after the replication, removes adenines mispaired with 8-oxo-dG. | Koger et al., 2019 [40] |
| N-glycosylase/DNA lyase | OGG1 | 4.2.99.18 | OGG1 acts in cooperation with MUTYH. It is a major glycosylase for the removal of 8-oxo-dG. It possesses also an intrinsic AP lyase activity at abasic sites. | Wang et al., 2018 [41] |
| DNA-3-methyladenine glycosylase | MPG | 3.2.2.21 | MPG removes a variety of alkylated (3-methyladenine, 7-methylguanine) and deaminated (hypoxanthine) purines. It also recognizes and removes secondary oxidative lesions such as 1,N6-ethenoadenine. | Leitner-Dagan et al., 2012 [42] |
| Methyl-CpG-binding domain protein 4 | MBD4 | 3.2.2.- | MBD4 preferentially binds to CpG sites and guards DNA against deamination of cytosine to uracil or 5-methylcytosine to thymine. | Sjolund et al., 2013 [43] |
| Single-strand selective monofunctional uracil DNA glycosylase | SMUG1 | 3.2.2.- | SMUG1 belongs to the uracil DNA glycosylase superfamily. It is a back-up uracil DNA glycosylase removing a wide variety of oxidized pyrimidines such as 5-hydroxyuracil, 5-hydroxymethyluracil, 5-formyluracil and 5-carboxyuracil In addition to that, SMUG1 has also an activity towards 5-fluorouracil, a commonly used chemotherapeutic agent to treat CRC. | Nagaria et al., 2013 [44], Alexeeva et al., 2019 [45] |
| Endonuclease III-like protein 1 | NTH1 | 4.2.99.18 | NTH1 cleaves a broad range of lesions such as thymine glycol, 5-hydroxyuracil, 5-formyluracil, 5-hydroxycytosine, 5-hydroxy-6-hydrothymine, 5,6-dihydroxycytosine, 5,6-dihydrouracil and formamidopyrimidine. | Shinmura et al., 2019 [46] |
| Endonuclease VIII-like 1 | NEIL1 | 4.2.99.18 | NEIL1 acts at the replication fork and it is implicated in direct removal of the 5-carboxylcytosine. Further, it stimulates TDG-mediated excision of 5-formylcytosine and 5-carboxylcytosine. | Slyvka et al., 2017 [47] |
| Endonuclease VIII-like 2 | NEIL2 | 4.2.99.18 | NEIL2 takes part in the transcription-coupled BER. It excises 8-oxoguanine, thymine glycol, formamidopyrimidine lesions and oxidative products of cytosine, particularly 5-hydroxyuracil and 5-hydroxycytosine. | Sarker et al., 2014 [48], Han et al., 2019 [49], Minko et al., 2019 [50] |
| Endonuclease VIII-like 3 | NEIL3 | 4.2.99.18 | NEIL3 acts preferentially on ssDNA. It removes spiroiminodihydantoin and guanidinohydantoin, further oxidation products of 8-oxo-7,8-dihydroguanine. It is also implicated in the repair of formamidopyrimidine DNA adducts. | Massaad et al., 2016 [51], Minko et al., 2019 [50] |
| G/T mismatch-specific thymine DNA glycosylase | TDG | 3.2.2.29 | TDG recognizes U-G or T-G mismatches caused by the deamination of the cytosine or 5-methylcytosine. Therefore, it prevents the formation of a C→T mutation. Further, it excises oxidized products of the 5-methylcytosine and 5-hydroxymethylcytosine, such as the 5-formylcytosine and 5-carboxycytosine. | Da et al. 2018 [52], Fu et al., 2019 [53] |
| Uracil-DNA glycosylase | UNG | 3.2.2.27 | UNG hydrolyzes uracil from both ss and dsDNA, leaving an apyrimidinic site. Such lesions can arise due to deamination of cytosine or due to misincorporation of dUMPs during replication or repair. | Weiser et al., 2018 [54] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. https://doi.org/10.3390/ijms21072473
Vodicka P, Urbanova M, Makovicky P, Tomasova K, Kroupa M, Stetina R, Opattova A, Kostovcikova K, Siskova A, Schneiderova M, et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. International Journal of Molecular Sciences. 2020; 21(7):2473. https://doi.org/10.3390/ijms21072473
Chicago/Turabian StyleVodicka, Pavel, Marketa Urbanova, Pavol Makovicky, Kristyna Tomasova, Michal Kroupa, Rudolf Stetina, Alena Opattova, Klara Kostovcikova, Anna Siskova, Michaela Schneiderova, and et al. 2020. "Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients" International Journal of Molecular Sciences 21, no. 7: 2473. https://doi.org/10.3390/ijms21072473
APA StyleVodicka, P., Urbanova, M., Makovicky, P., Tomasova, K., Kroupa, M., Stetina, R., Opattova, A., Kostovcikova, K., Siskova, A., Schneiderova, M., Vymetalkova, V., & Vodickova, L. (2020). Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. International Journal of Molecular Sciences, 21(7), 2473. https://doi.org/10.3390/ijms21072473