Pathogenesis of Mucopolysaccharidoses, an Update

{kind=link}
{kind=link}
Abstract
:1. Introduction
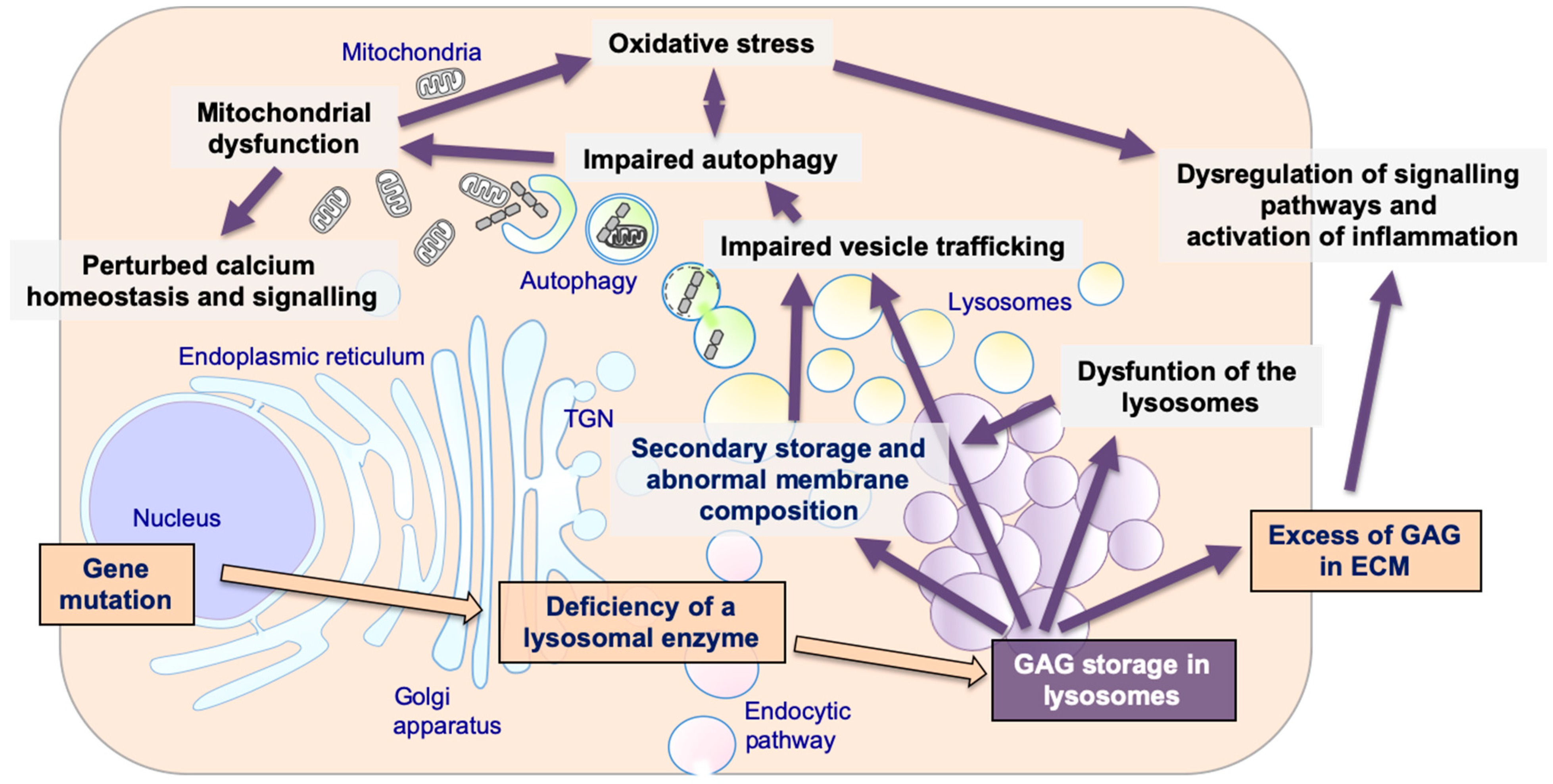
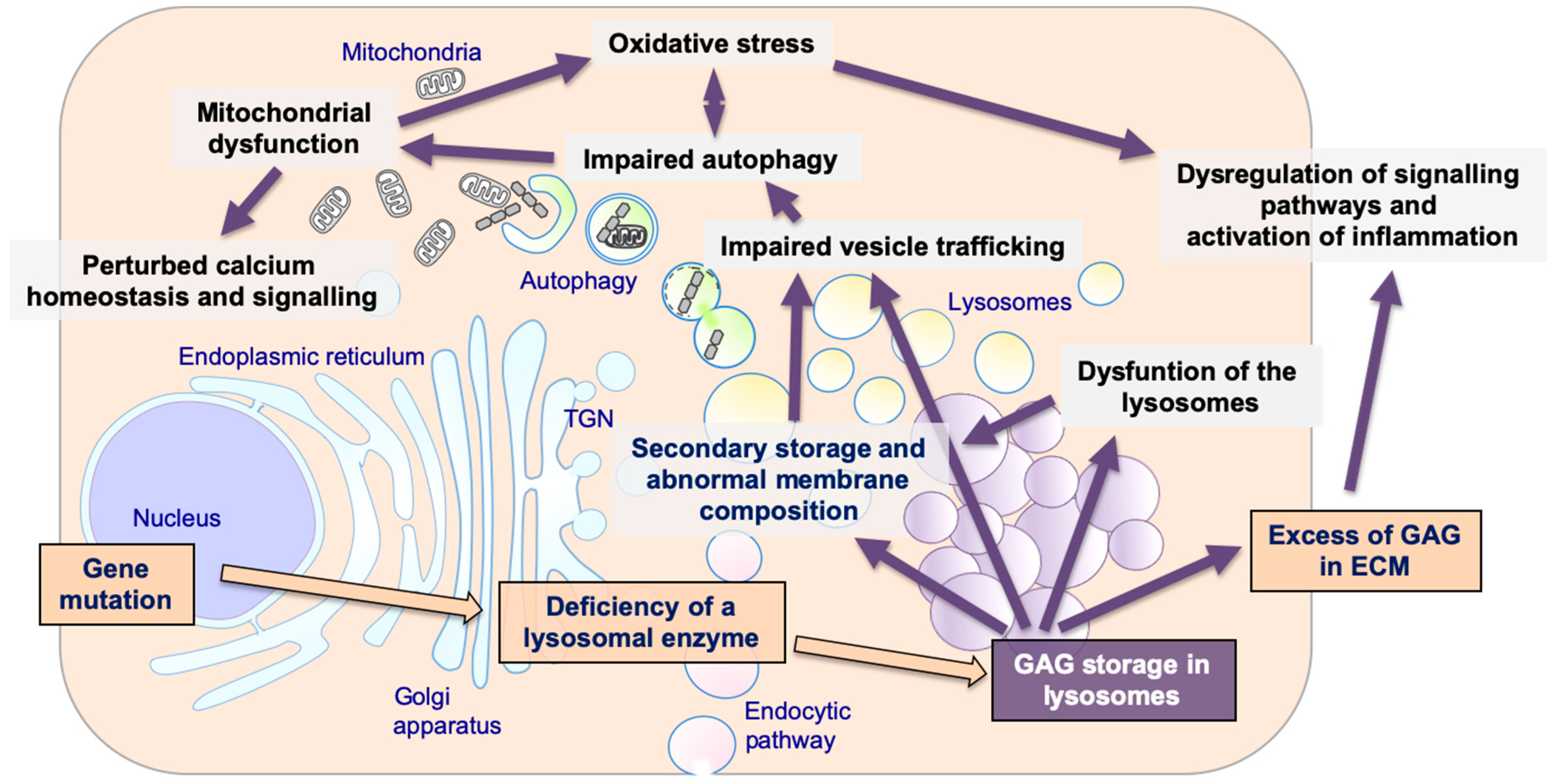
2. Mucopolysaccharidoses (MPSs), Glycosaminoglycans (GAGs), and Lysosome Biology
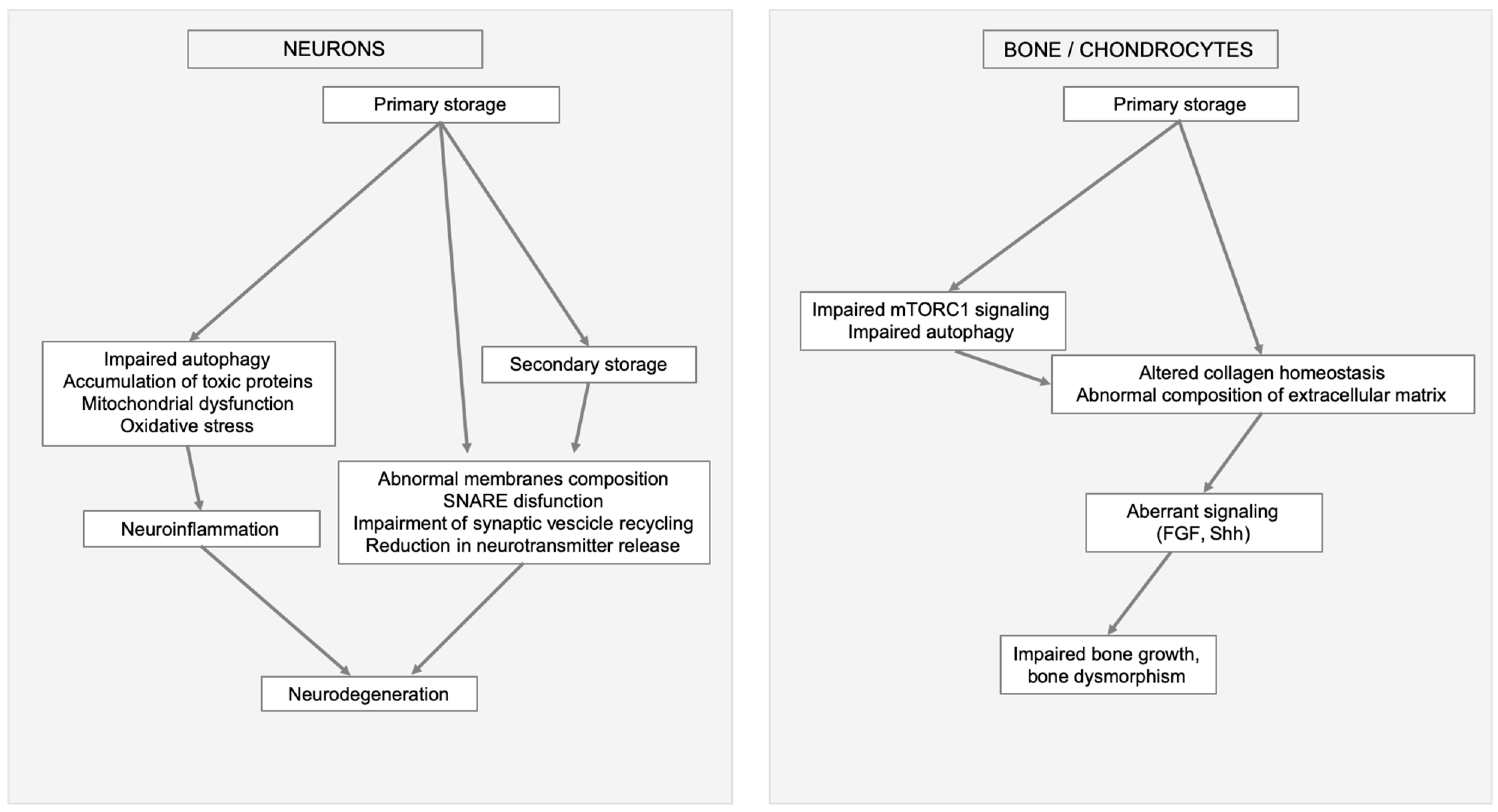
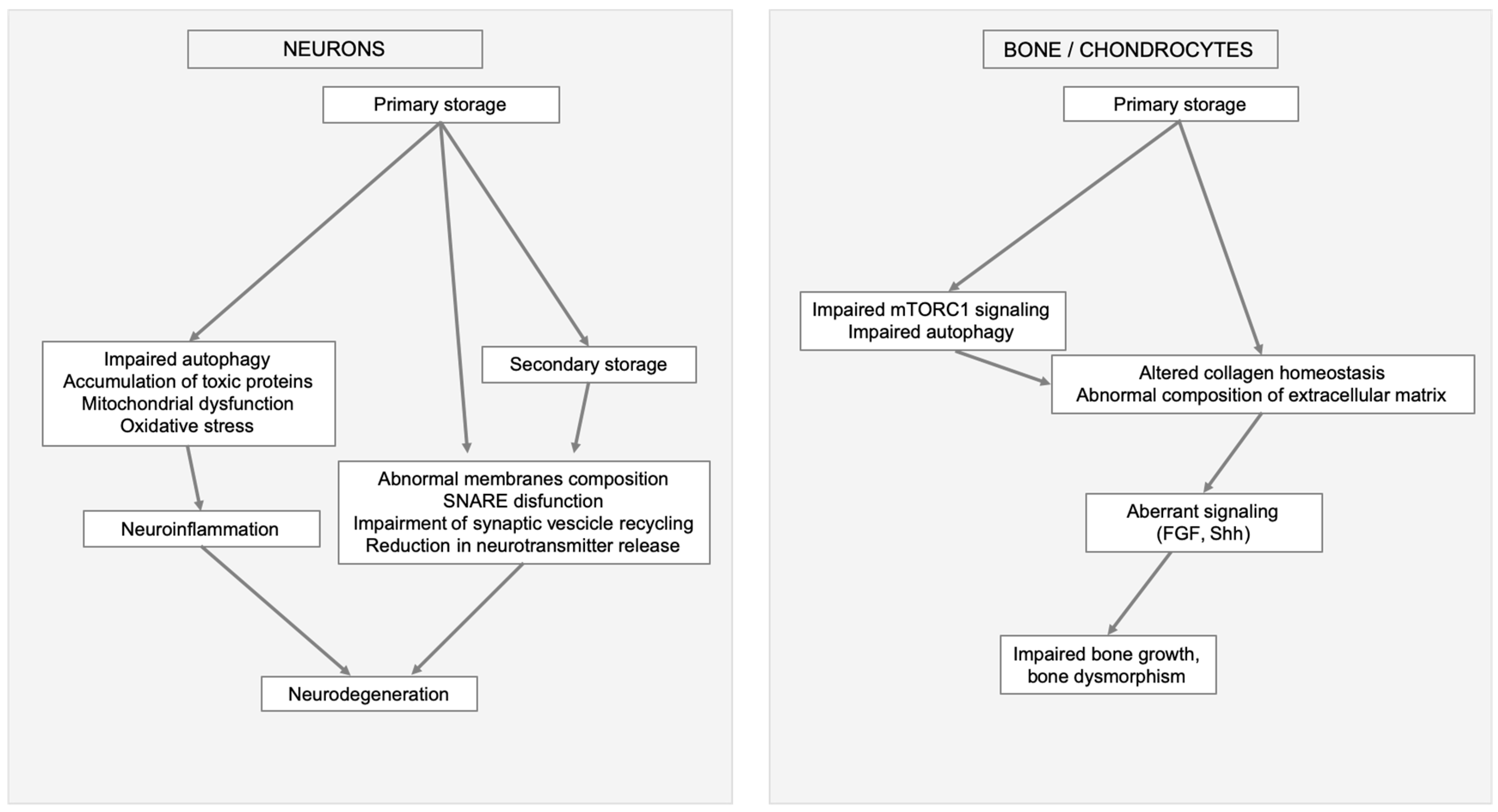
3. Lysosomal Storage and Secondary Disruption of Cellular Pathways
3.1. Secondary Storage
3.2. Abnormal Composition of Membranes and Aberrant Intracellular Trafficking
3.3. Abnormal Autophagy
3.4. Mitochondrial Dysfunction
3.5. Oxidative Stress
3.6. Alteration of Signaling Pathways
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MPS | Mucopolysaccharidosis |
| GAGs | Glycosaminoglycans |
| LSD | Lysosomal Storage Disease |
| ALP | Autophagic-lysosomal pathway |
| TFEB | Transcription factor EB |
| mTOR | Mammalian receptor of rapamycin |
| SNARE | N-ethylmaleimide-sensitive factor attachment protein receptors |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| ROS | Reactive oxygen species |
| FGF | Fibroblast growth factor |
| TLR | Toll-like receptor |
| LPS | Lipopolysaccharide |
| TNF-α | Tumor necrosis factor-α |
| IL | Interleukin |
| CCL3 | Chemochine ligand-3 |
| MyD88 | Myeloid differentiation primary response 88 |
| MIF | Macrophage migration inhibitory factor |
| BMP-4 | Bone morphogenetic protein-4 |
| GPNMB | Transmembrane glycoprotein NMB |
| SHH | Sonic hedgehog |
References
- Hunter, C. A Rare Disease in Two Brothers. Proc. R. Soc. Med. 1917, 10, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurler, G. Über einen Typ multipler Abartungen, vorwiegend am Skelettsystem. Zeitschrift für Kinderheilkunde 1920, 24, 220–234. [Google Scholar] [CrossRef]
- Morquio, L. Sur une forme de dystrophie ossuese familiale. Arch. Med. Enfants 1929, 32, 129–140. [Google Scholar]
- Sanfilippo, S.J.; Podosin, R.; Langer, L.O., Jr.; Good, R.A. Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J. Pediatrics 1963, 63, 837–838. [Google Scholar] [CrossRef]
- Maroteaux, P.; Leveque, B.; Marie, J.; Lamy, M. A New Dysostosis with Urinary Elimination of Chondroitin Sulfate B. Presse Med. 1963, 71, 1849–1852. [Google Scholar]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Hers, H.G. Inborn Lysosomal Diseases. Gastroenterology 1965, 48, 625–633. [Google Scholar] [CrossRef]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef] [Green Version]
- Ernst, S.; Langer, R.; Cooney, C.L.; Sasisekharan, R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387–444. [Google Scholar] [CrossRef]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Fraldi, A. Pathogenesis of Mucopolysaccharidoses: Dysfunction of Lysosome; Nova Publisher: Hauppauge, NY, USA, 2018; Volume 2. [Google Scholar]
- Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M.; Banecka-Majkutewicz, Z.; Banecki, B.; Wegrzyn, A.; Wegrzyn, G. Factors and processes modulating phenotypes in neuronopathic lysosomal storage diseases. Metab. Brain Dis. 2014, 29, 1–8. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, R.D.; Johnstone, A.C.; Norman, E.J.; Hopwood, J.J.; Walkley, S.U. Pathology of mucopolysaccharidosis IIIA in Huntaway dogs. Vet. Pathol. 2007, 44, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V. Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis. 2015, 3, e1049793. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef] [Green Version]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U.; Thrall, M.A.; Haskins, M.E.; Mitchell, T.W.; Wenger, D.A.; Brown, D.E.; Dial, S.; Seim, H. Abnormal neuronal metabolism and storage in mucopolysaccharidosis type VI (Maroteaux-Lamy) disease. Neuropathol. Appl. Neurobiol. 2005, 31, 536–544. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Fraldi, A.; Klein, A.D.; Medina, D.L.; Settembre, C. Brain Disorders Due to Lysosomal Dysfunction. Annu. Rev. Neurosci. 2016, 39, 277–295. [Google Scholar] [CrossRef]
- Shachar, T.; Lo Bianco, C.; Recchia, A.; Wiessner, C.; Raas-Rothschild, A.; Futerman, A.H. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov. Disord. 2011, 26, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Autophagy in astrocytes: A novel culprit in lysosomal storage disorders. Autophagy 2012, 8, 1871–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambri, I.; D’Alessio, R.; Ezhova, Y.; Giuliano, T.; Sorrentino, N.C.; Cacace, V.; De Risi, M.; Cataldi, M.; Annunziato, L.; De Leonibus, E.; et al. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 2017, 9, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef]
- Monaco, A.; Maffia, V.; Sorrentino, N.C.; Sambri, I.; Ezhova, Y.; Giuliano, T.; Cacace, V.; Nusco, E.; De Risi, M.; De Leonibus, E.; et al. The Amyloid Inhibitor CLR01 Relieves Autophagy and Ameliorates Neuropathology in a Severe Lysosomal Storage Disease. Mol. Ther. 2020, in press. [Google Scholar] [CrossRef]
- Simons, K.; Gruenberg, J. Jamming the endosomal system: Lipid rafts and lysosomal storage diseases. Trends Cell Biol. 2000, 10, 459–462. [Google Scholar] [CrossRef]
- Sobo, K.; Le Blanc, I.; Luyet, P.P.; Fivaz, M.; Ferguson, C.; Parton, R.G.; Gruenberg, J.; van der Goot, F.G. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS ONE 2007, 2, e851. [Google Scholar] [CrossRef]
- Cosma, M.P.; Pepe, S.; Annunziata, I.; Newbold, R.F.; Grompe, M.; Parenti, G.; Ballabio, A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 2003, 113, 445–456. [Google Scholar] [CrossRef]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; von Figura, K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, D.J.; Winter, M.A.; Hemsley, K.M.; Mackenzie, K.D.; Teo, E.H.; Hopwood, J.J.; Brooks, D.A.; Parkinson-Lawrence, E.J. Exocytosis is impaired in mucopolysaccharidosis IIIA mouse chromaffin cells. Neuroscience 2012, 227, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bonini, N.M.; Giasson, B.I. Snaring the function of alpha-synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Rubinsztein, D.C.; Ballabio, A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008, 4, 113–114. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Webber, D.L.; Choo, A.; Hewson, L.J.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Richards, R.I.; Hemsley, K.M.; O’Keefe, L.V. Neuronal-specific impairment of heparan sulfate degradation in Drosophila reveals pathogenic mechanisms for Mucopolysaccharidosis type IIIA. Exp. Neurol. 2018, 303, 38–47. [Google Scholar] [CrossRef]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V. Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy 2016, 12, 1059–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomeo, R.; Cinque, L.; De Leonibus, C.; Forrester, A.; Salzano, A.C.; Monfregola, J.; De Gennaro, E.; Nusco, E.; Azario, I.; Lanzara, C.; et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Investig. 2017, 127, 3717–3729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A affects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 2017, 26, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Layfield, R.; Lotz, M.; Settembre, C.; Whitehouse, C. Boning up on autophagy: The role of autophagy in skeletal biology. Autophagy 2014, 10, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Cinque, L.; Bartolomeo, R.; Di Malta, C.; De Leonibus, C.; Forrester, A. Defective collagen proteostasis and matrix formation in the pathogenesis of lysosomal storage disorders. Matrix Biol. 2018, 71–72, 283–293. [Google Scholar] [CrossRef]
- Wang, R.; Wang, G. Autophagy in Mitochondrial Quality Control. Adv. Exp. Med. Biol. 2019, 1206, 421–434. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Saffari, A.; Kolker, S.; Hoffmann, G.F.; Ebrahimi-Fakhari, D. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, I.; Sano, R.; d’Azzo, A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018, 9, 328. [Google Scholar] [CrossRef]
- Ryazantsev, S.; Yu, W.H.; Zhao, H.Z.; Neufeld, E.F.; Ohmi, K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Ariga, M.; Sato, Y.; Fujiwara, M.; Fukasawa, N.; Fukuda, T.; Takahashi, H.; Ikegami, M.; Kosuga, M.; Okuyama, T.; et al. P-Tau and Subunit c Mitochondrial ATP Synthase Accumulation in the Central Nervous System of a Woman with Hurler-Scheie Syndrome Treated with Enzyme Replacement Therapy for 12 Years. JIMD Rep. 2018, 41, 101–107. [Google Scholar] [CrossRef]
- Rego, A.C.; Oliveira, C.R. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 2003, 28, 1563–1574. [Google Scholar] [CrossRef] [Green Version]
- Villani, G.R.; Di Domenico, C.; Musella, A.; Cecere, F.; Di Napoli, D.; Di Natale, P. Mucopolysaccharidosis IIIB: Oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res. 2009, 1279, 99–108. [Google Scholar] [CrossRef]
- Villani, G.R.; Chierchia, A.; Di Napoli, D.; Di Natale, P. Unfolded protein response is not activated in the mucopolysaccharidoses but protein disulfide isomerase 5 is deregulated. J. Inherit. Metab. Dis. 2012, 35, 479–493. [Google Scholar] [CrossRef]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Therond, P.; Scherman, D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: The positive effect of long-term aspirin treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef]
- Pereira, V.G.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef]
- Filippon, L.; Vanzin, C.S.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; Peralba Mdo, C.; Schwartz, I.V.; Giugliani, R.; Vargas, C.R. Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Trudel, S.; Trecherel, E.; Gomila, C.; Peltier, M.; Aubignat, M.; Gubler, B.; Morliere, P.; Heard, J.M.; Ausseil, J. Oxidative stress is independent of inflammation in the neurodegenerative Sanfilippo syndrome type B. J. Neurosci. Res. 2015, 93, 424–432. [Google Scholar] [CrossRef]
- von Zastrow, M.; Sorkin, A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007, 19, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2018, 141, 99–116. [Google Scholar] [CrossRef] [Green Version]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef]
- Parente, M.K.; Rozen, R.; Cearley, C.N.; Wolfe, J.H. Dysregulation of gene expression in a lysosomal storage disease varies between brain regions implicating unexpected mechanisms of neuropathology. PLoS ONE 2012, 7, e32419. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Schuchman, E.H.; Ge, Y.; Lai, A.; Borisov, Y.; Faillace, M.; Eliyahu, E.; He, X.; Iatridis, J.; Vlassara, H.; Striker, G.; et al. Pentosan polysulfate: A novel therapy for the mucopolysaccharidoses. PLoS ONE 2013, 8, e54459. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Tomatsu, S.; Sikora, T.; Kubaski, F.; Frohbergh, M.; Guevara, J.M.; Wang, R.Y.; Vera, M.; Kang, J.L.; Smith, L.J.; et al. Pentosan Polysulfate: Oral Versus Subcutaneous Injection in Mucopolysaccharidosis Type I Dogs. PLoS ONE 2016, 11, e0153136. [Google Scholar] [CrossRef] [Green Version]
- Orii, K.; Lim, A.; Tomatsu, S.; Stapleton, M.; Suzuki, Y.; Simonaro, C.M.; Schuchman, E.H.; Fukao, T.; Matsumoto, T. Safety Study of Sodium Pentosan Polysulfate for Adult Patients with Mucopolysaccharidosis Type II. Diagnostics 2019, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Yasuda, E.; Patel, P.; Ruhnke, K.; Shimada, T.; Mackenzie, W.G.; Mason, R.; Thacker, M.M.; Theroux, M.; Montano, A.M.; et al. Morquio A syndrome: Diagnosis and current and future therapies. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 141–151. [Google Scholar]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef] [Green Version]
- Kingma, S.D.K.; Wagemans, T.; L, I.J.; Bronckers, A.; van Kuppevelt, T.H.; Everts, V.; Wijburg, F.A.; van Vlies, N. Altered interaction and distribution of glycosaminoglycans and growth factors in mucopolysaccharidosis type I bone disease. Bone 2016, 88, 92–100. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. FGF signaling deregulation is associated with early developmental skeletal defects in animal models for mucopolysaccharidosis type II (MPSII). Hum. Mol. Genet. 2018, 27, 2262–2275. [Google Scholar] [CrossRef]
- Peck, S.H.; O’Donnell, P.J.; Kang, J.L.; Malhotra, N.R.; Dodge, G.R.; Pacifici, M.; Shore, E.M.; Haskins, M.E.; Smith, L.J. Delayed hypertrophic differentiation of epiphyseal chondrocytes contributes to failed secondary ossification in mucopolysaccharidosis VII dogs. Mol. Genet. Metab. 2015, 116, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Peck, S.H.; Tobias, J.W.; Shore, E.M.; Malhotra, N.R.; Haskins, M.E.; Casal, M.L.; Smith, L.J. Molecular profiling of failed endochondral ossification in mucopolysaccharidosis VII. Bone 2019, 128, 115042. [Google Scholar] [CrossRef] [PubMed]
- Heppner, J.M.; Zaucke, F.; Clarke, L.A. Extracellular matrix disruption is an early event in the pathogenesis of skeletal disease in mucopolysaccharidosis I. Mol. Genet. Metab. 2015, 114, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Urbani, A.; Salvalaio, M.; Bellesso, S.; Cieri, D.; Zancan, I.; Filocamo, M.; Bonaldo, P.; Szabo, I.; Tomanin, R.; et al. Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum. Mol. Genet. 2017, 26, 1643–1655. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca(2+) homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011, 50, 200–205. [Google Scholar] [CrossRef]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. https://doi.org/10.3390/ijms21072515
Fecarotta S, Tarallo A, Damiano C, Minopoli N, Parenti G. Pathogenesis of Mucopolysaccharidoses, an Update. International Journal of Molecular Sciences. 2020; 21(7):2515. https://doi.org/10.3390/ijms21072515
Chicago/Turabian StyleFecarotta, Simona, Antonietta Tarallo, Carla Damiano, Nadia Minopoli, and Giancarlo Parenti. 2020. "Pathogenesis of Mucopolysaccharidoses, an Update" International Journal of Molecular Sciences 21, no. 7: 2515. https://doi.org/10.3390/ijms21072515
APA StyleFecarotta, S., Tarallo, A., Damiano, C., Minopoli, N., & Parenti, G. (2020). Pathogenesis of Mucopolysaccharidoses, an Update. International Journal of Molecular Sciences, 21(7), 2515. https://doi.org/10.3390/ijms21072515