Pioglitazone-Mediated Peroxisome Proliferator-Activated Receptor γ Activation Aggravates Murine Immune-Mediated Hepatitis

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
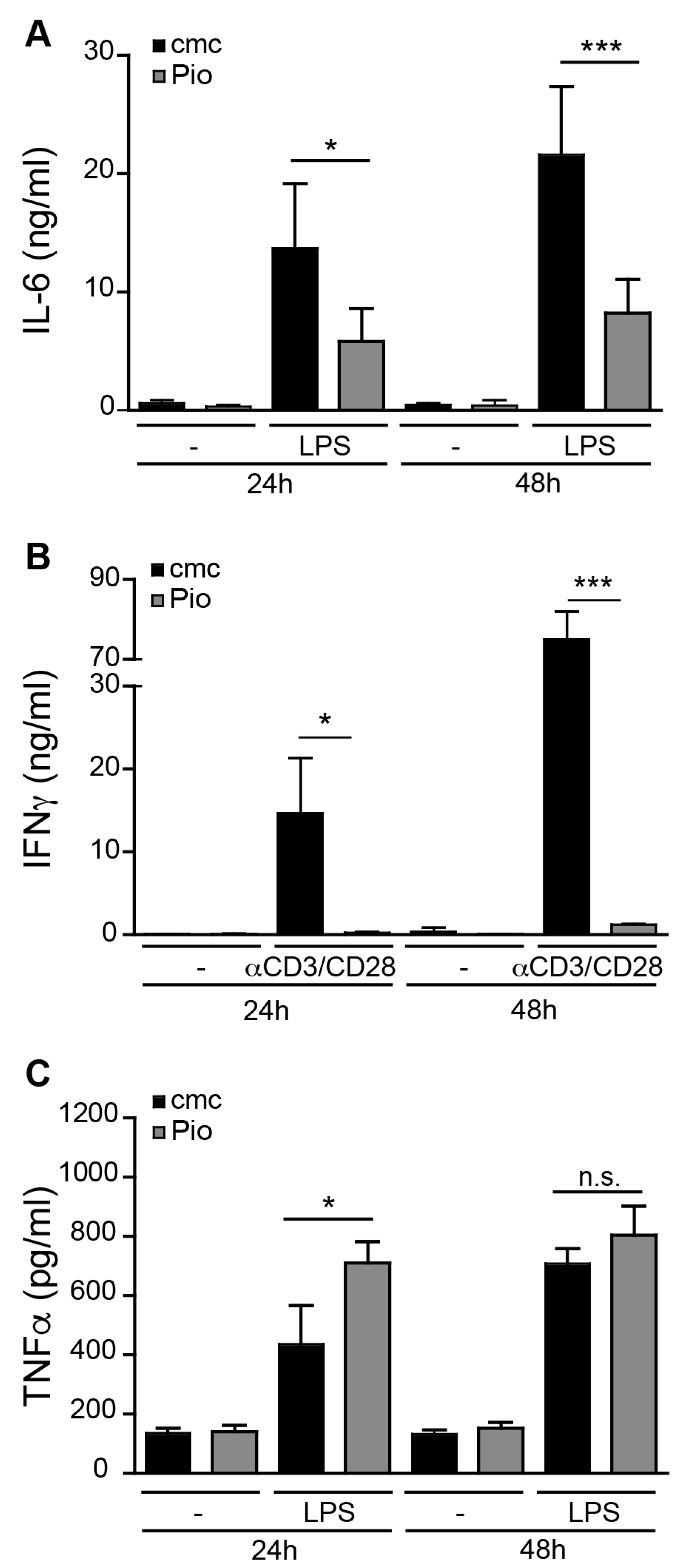
2.1. PPARγ Activation Regulates Pro-Inflammatory Activity in liver sinusoidal endothelial cells (LSEC) and T Cells but not Kupffer Cells
2.2. PPARγ Activation in LSEC Does Not Influence T Cell Modulatory Capacity
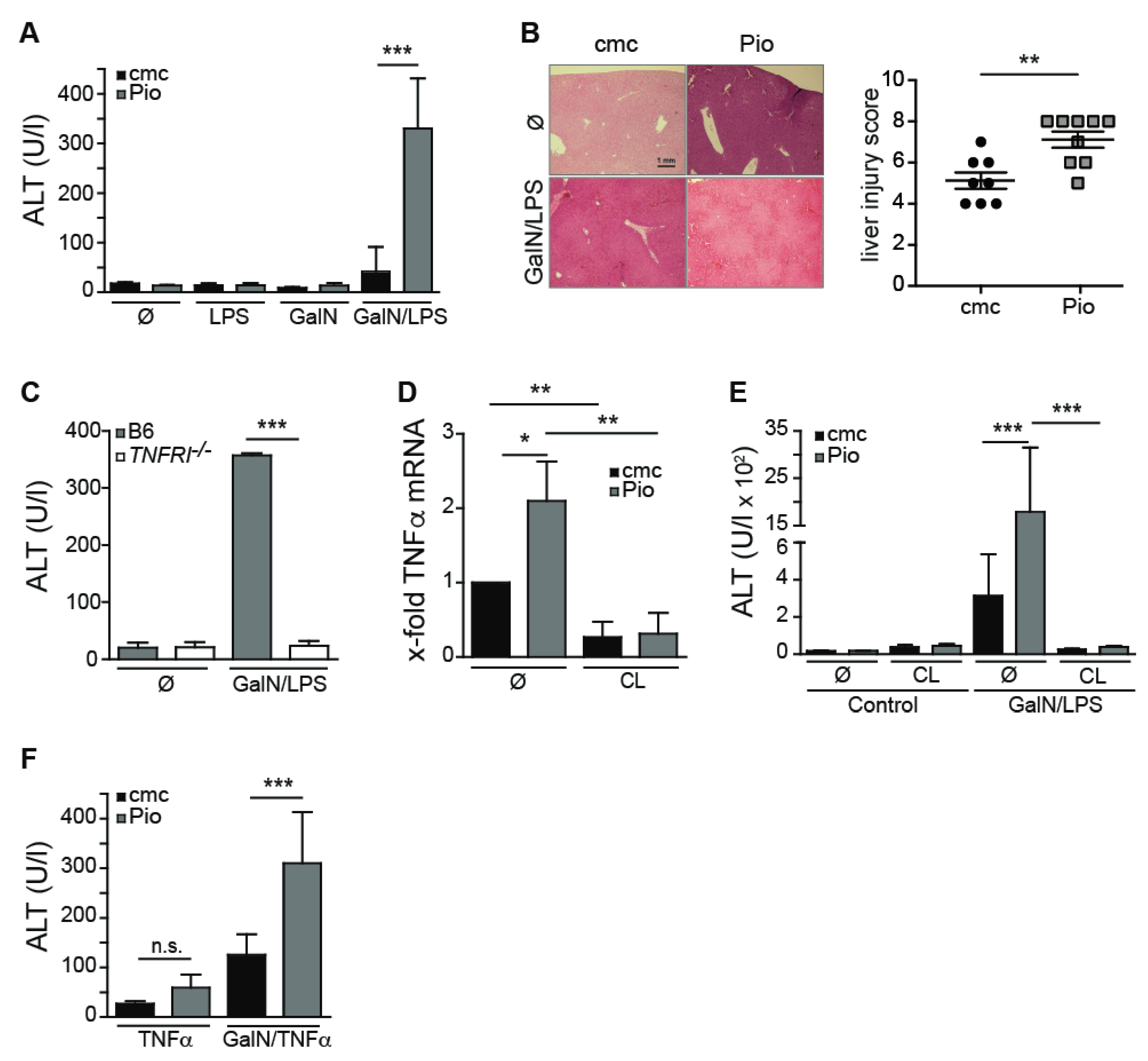
2.3. Pioglitazone-Mediated PPARγ Activation Aggravates GalN/LPS-Induced Hepatitis
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Reagents
4.3. Cell Isolation
4.4. T Cell Assays
4.5. Experimental Hepatitis (GalN/LPS, GalN/TNFα)
4.6. Histology
4.7. Depletion of Macrophages by Clodronate Liposomes
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Nowell, M.; Chima, R.; Zingarelli, B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun. 2014, 20, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Széles, L.; Töröcsik, D.; Nagy, L. PPARgamma in immunity and inflammation: Cell types and diseases. Biochim. Biophys. Acta 2007, 1771, 1014–1030. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.V.; Kei, A.; Elisaf, M.S. The current role of thiazolidinediones in diabetes management. Arch. Toxicol. 2016, 90, 1861–1881. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Dani, I.; Edenhofer, F.; Nolden, L.; Evert, B.; Paul, B.; Kolanus, W.; Klockgether, T.; Knolle, P.; Diehl, L. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J. Immunol. 2007, 178, 2122–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Yan, H.; Li, S.; Nie, W.; Fan, F.; Zhu, J. PPAR-γ agonist pioglitazone regulates dendritic cells immunogenicity mediated by DC-SIGN via the MAPK and NF-κB pathways. Int Immunopharmacol. 2016, 41, 24–34. [Google Scholar] [CrossRef]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Novak, N.; Beyer, M.; Mayer, G.; et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef]
- Guri, A.J.; Mohapatra, S.K.; Horne, W.T.; Hontecillas, R.; Bassaganya-Riera, J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 2010, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Dahten, A.; Koch, C.; Ernst, D.; Schnöller, C.; Hartmann, S.; Worm, M. Systemic PPARgamma ligation inhibits allergic immune response in the skin. J. Investig. Dermatol. 2008, 128, 2211–2218. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.-J.; Ye, H.; Wang, F.; Wang, J.-L.; Xie, M.-L. Apigenin inhibits d-galactosamine/LPS-induced liver injury through upregulation of hepatic Nrf-2 and PPARγ expressions in mice. Biochem Biophys Res Comm. 2017, 493, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Gerken, G.; Loser, E.; Dienes, H.P.; Gantner, F.; Tiegs, G.; Meyer zum Buschenfelde, K.H.; Lohse, A.W. Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 1996, 24, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Schumann, J.; Wolf, D.; Pahl, A.; Brune, K.; Papadopoulos, T.; van Rooijen, N.; Tiegs, G. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 2000, 157, 1671–1683. [Google Scholar] [CrossRef] [Green Version]
- Kern, M.; Popov, A.; Scholz, K.; Schumak, B.; Djandji, D.; Limmer, A.; Eggle, D.; Sacher, T.; Zawatzky, R.; Holtappels, R.; et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 2010, 138, 336–346. [Google Scholar] [CrossRef]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2008, 47, 296–305. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Schanz, O.; Wohlleber, D.; Abdullah, Z.; Debey-Pascher, S.; Staratschek-Jox, A.; Höchst, B.; Hegenbarth, S.; Grell, J.; Limmer, A.; et al. Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep. 2013, 3, 779–795. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.; Hucke, S.; Thimm, D.; Classen, S.; Gaarz, A.; Schultze, J.; Edenhofer, F.; Kurts, C.; Klockgether, T.; Limmer, A.; et al. Increased antigen cross-presentation but impaired cross-priming after activation of peroxisome proliferator-activated receptor gamma is mediated by up-regulation of B7H1. J. Immunol. 2009, 183, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Galanos, C.; Freudenberg, M.A.; Reutter, W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc. Natl. Acad. Sci. USA 1979, 76, 5939–5943. [Google Scholar] [CrossRef] [Green Version]
- Freudenberg, M.A.; Galanos, C. Tumor necrosis factor alpha mediates lethal activity of killed gram-negative and gram-positive bacteria in D-galactosamine-treated mice. Infect. Immun. 1991, 59, 2110–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlleber, D.; Kashkar, H.; Gärtner, K.; Frings, M.K.; Odenthal, M.; Hegenbarth, S.; Börner, C.; Arnold, B.; Hämmerling, G.; Nieswandt, B.; et al. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep. 2012, 2, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchimura, K.; Nakamuta, M.; Enjoji, M.; Irie, T.; Sugimoto, R.; Muta, T.; Iwamoto, H.; Nawata, H. Activation of retinoic X receptor and peroxisome proliferator-activated receptor-gamma inhibits nitric oxide and tumor necrosis factor-alpha production in rat Kupffer cells. Hepatology 2001, 33, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Loser, E.; Protzer, U.; Duchmann, R.; Schmitt, E.; zum Büschenfelde, K.H.; Rose-John, S.; Gerken, G. Regulation of endotoxin-induced IL-6 production in liver sinusoidal endothelial cells and Kupffer cells by IL-10. Clin. Exp. Immunol. 1997, 107, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, M.A.; Galanos, C. Induction of tolerance to lipopolysaccharide (LPS)-D-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun. 1988, 56, 1352–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schäfers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-γ Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 803–816. [Google Scholar] [CrossRef]
- Santucci, L.; Fiorucci, S.; Chiorean, M.; Brunori, P.M.; di Matteo, F.M.; Sidoni, A.; Mifliorati, G.; Morelli, A. Interleukin 10 reduces lethality and hepatic injury induced by lipopolysaccharide in galactosamine-sensitized mice. YGAST 1996, 111, 736–744. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Isley, W.L.; Oki, J.C.; Ramrakhiani, S.; Quiason, S.G.; Phillips, N.J.; Brunt, E.M. Troglitazone-induced hepatic failure leading to liver transplantation. A case report. Ann. Intern. Med. 1998, 129, 38–41. [Google Scholar] [CrossRef]
- Watkins, P.B.; Whitcomb, R.W. Hepatic dysfunction associated with troglitazone. N. Engl. J. Med. 1998, 338, 916–917. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T. Drug-induced idiosyncratic hepatotoxicity: Prevention strategy developed after the troglitazone case. Drug Metab. Pharmacokinet. 2011, 26, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Kwan, T.; Yu, C.; Chen, F.; Freedman, B.; Schafer, J.M.; Lee, E.J.; Jameson, J.L.; Jordan, V.C.; Cryns, V.L. Peroxisome proliferator-activated receptor gamma agonists promote TRAIL-induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J. Biol. Chem. 2005, 280, 6742–6751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolman, K.G.; Chandramouli, J. Hepatotoxicity of the thiazolidinediones. Clin. Liver Dis. 2003, 7, 369–379. [Google Scholar] [CrossRef]
- May, L.; Lefkowitsch, J.; Kram, M.; Rubin, D. Mixed hepatocellular-cholestatic liver injury after pioglitazone therapy. Ann. Intern. Med. 2002, 136, 449–452. [Google Scholar] [CrossRef]
- Pinto, A.; Cummings, O.; Chalasani, N. Severe but reversible cholestatic liver injury after pioglitazone therapy. Ann. Intern. Med. 2002, 137, 857. [Google Scholar] [CrossRef]
- Erhardt, A.; Biburger, M.; Papadopoulos, T.; Tiegs, G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 2007, 45, 475–485. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulte, R.; Wohlleber, D.; Unrau, L.; Geers, B.; Metzger, C.; Erhardt, A.; Tiegs, G.; van Rooijen, N.; Heukamp, L.C.; Klotz, L.; et al. Pioglitazone-Mediated Peroxisome Proliferator-Activated Receptor γ Activation Aggravates Murine Immune-Mediated Hepatitis. Int. J. Mol. Sci. 2020, 21, 2523. https://doi.org/10.3390/ijms21072523
Schulte R, Wohlleber D, Unrau L, Geers B, Metzger C, Erhardt A, Tiegs G, van Rooijen N, Heukamp LC, Klotz L, et al. Pioglitazone-Mediated Peroxisome Proliferator-Activated Receptor γ Activation Aggravates Murine Immune-Mediated Hepatitis. International Journal of Molecular Sciences. 2020; 21(7):2523. https://doi.org/10.3390/ijms21072523
Chicago/Turabian StyleSchulte, Rike, Dirk Wohlleber, Ludmilla Unrau, Bernd Geers, Christina Metzger, Annette Erhardt, Gisa Tiegs, Nico van Rooijen, Lukas C. Heukamp, Luisa Klotz, and et al. 2020. "Pioglitazone-Mediated Peroxisome Proliferator-Activated Receptor γ Activation Aggravates Murine Immune-Mediated Hepatitis" International Journal of Molecular Sciences 21, no. 7: 2523. https://doi.org/10.3390/ijms21072523