Translational Control of Secretory Proteins in Health and Disease

Abstract
:1. Introduction
2. Synthesis and Transport of Secretory Proteins
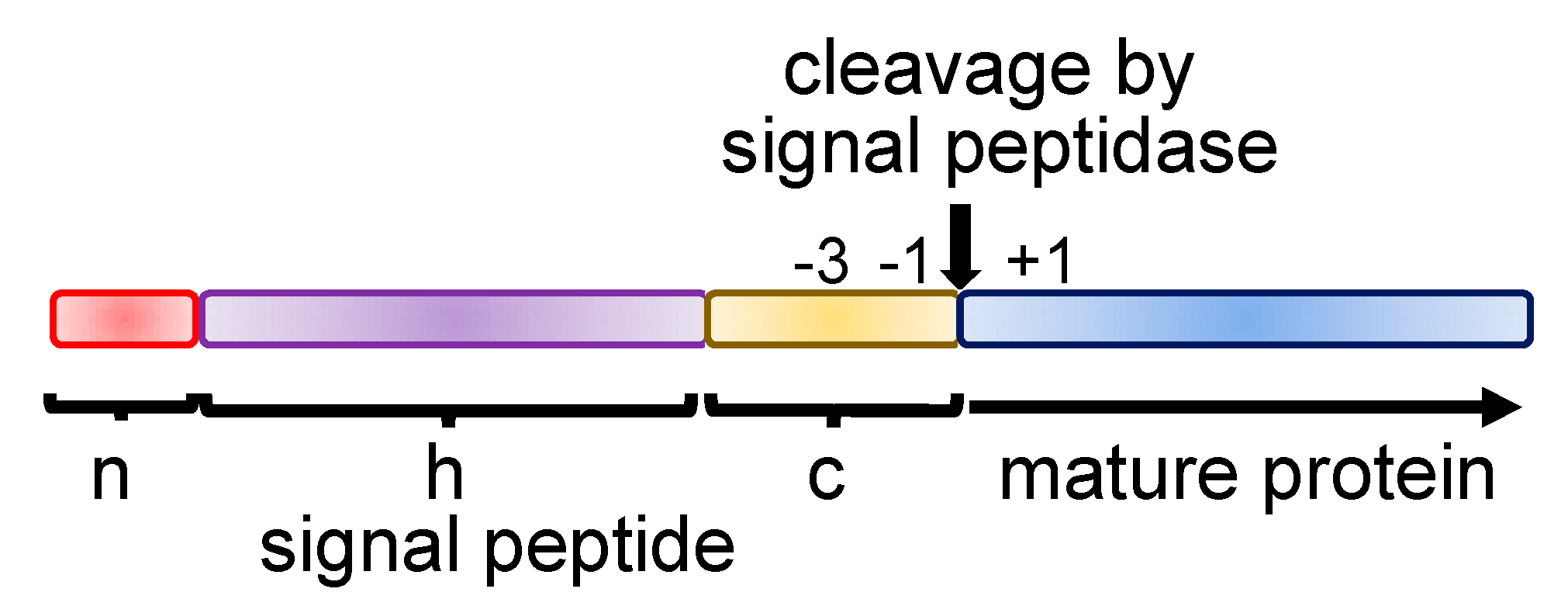
2.1. Secretory Proteins Are Synthesized as Precursors
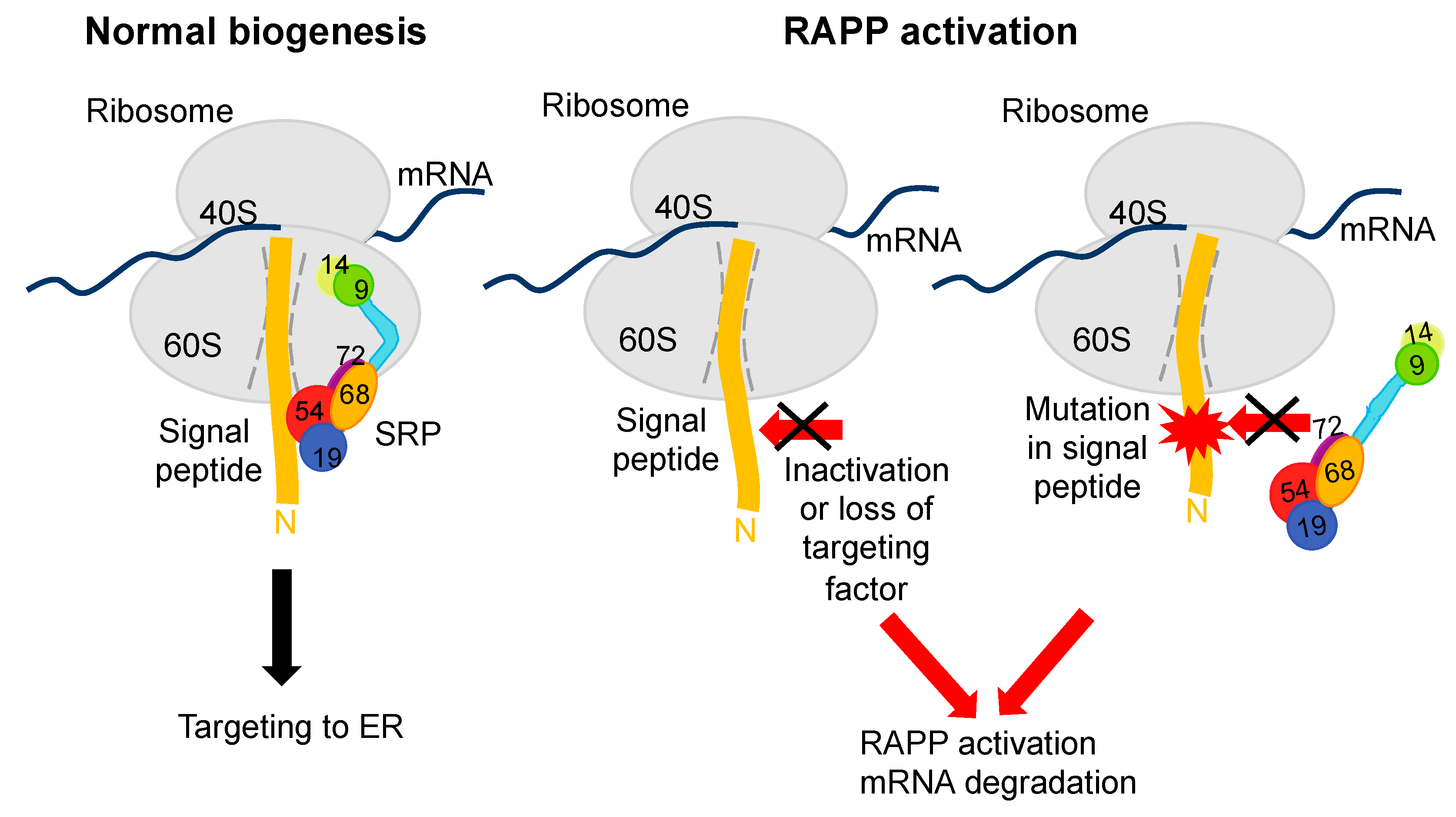
2.2. Signal Recognition Particle (SRP) Binds Signal Peptides and Targets Ribosomes to the ER Membrane
3. Quality Control of mRNAs and Proteins during Translation
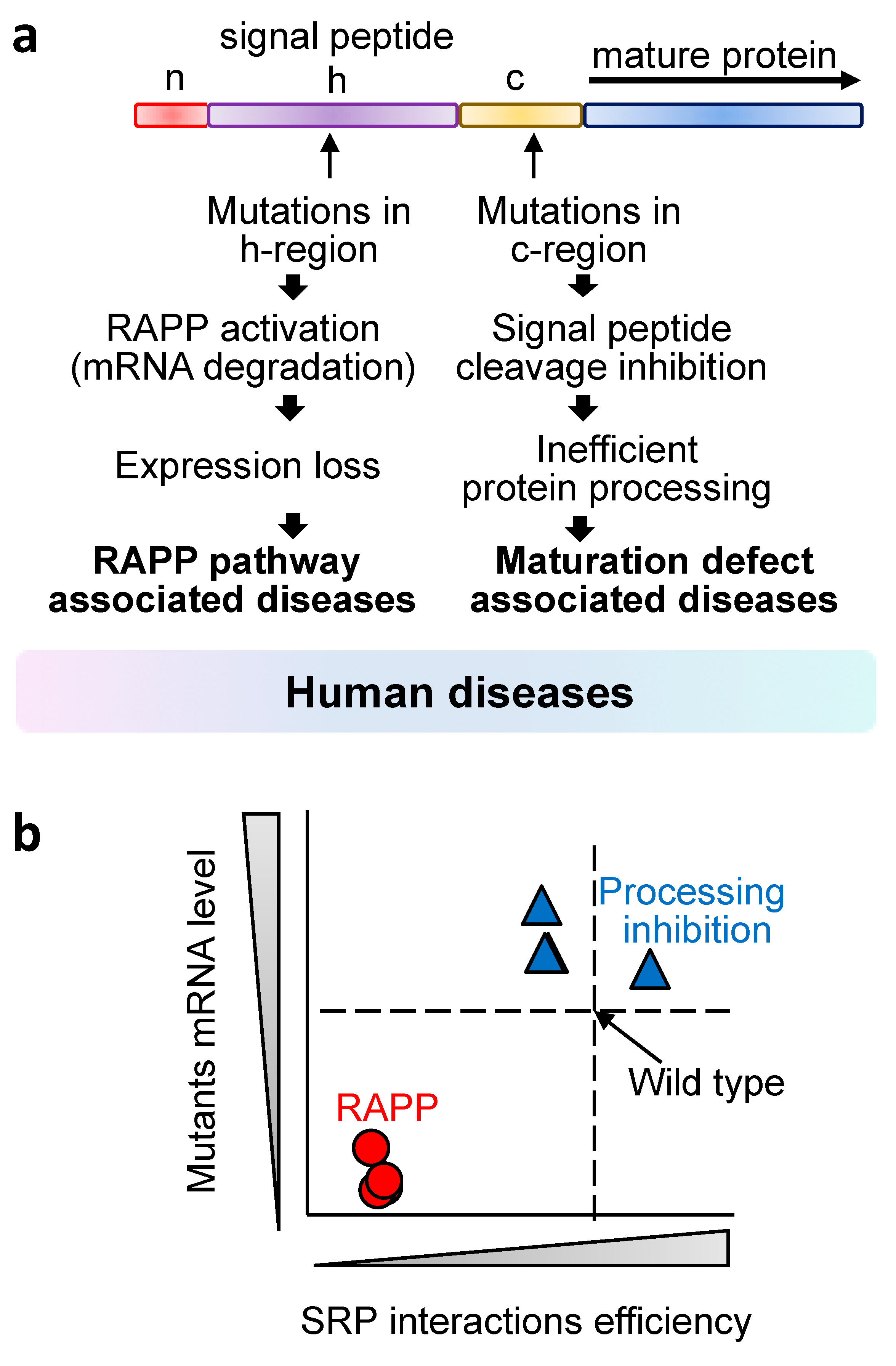
4. Defective SRP, Mutations in Secretory Proteins and Human Diseases
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SRP | Signal Recognition Particle |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum associated degradation |
| UPR | Unfolded protein response |
| NMD | Nonsense-mediated decay |
| NGD | No-go-decay |
| NSD | Non-stop decay |
| RAPP | Regulation of Aberrant Protein Production |
References
- Karamyshev, A.L.; Karamysheva, Z.N. Lost in Translation: Ribosome-Associated mRNA and Protein Quality Controls. Front. Genet. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ibrahimi, I.; Blobel, G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981, 91, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Wild, K.; Halic, M.; Sinning, I.; Beckmann, R. SRP meets the ribosome. Nat. Struct. Mol. Biol. 2004, 11, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Schuldiner, M.; Metz, J.; Schmid, V.; Denic, V.; Rakwalska, M.; Schmitt, H.D.; Schwappach, B.; Weissman, J.S. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 2008, 134, 634–645. [Google Scholar] [CrossRef]
- Stefanovic, S.; Hegde, R.S. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell 2007, 128, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Aviram, N.; Ast, T.; Costa, E.A.; Arakel, E.C.; Chuartzman, S.G.; Jan, C.H.; Hassdenteufel, S.; Dudek, J.; Jung, M.; Schorr, S.; et al. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 2016, 540, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ast, T.; Cohen, G.; Schuldiner, M. A network of cytosolic factors targets SRP-independent proteins to the endoplasmic reticulum. Cell 2013, 152, 1134–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamyshev, A.L.; Patrick, A.E.; Karamysheva, Z.N.; Griesemer, D.S.; Hudson, H.; Tjon-Kon-Sang, S.; Nilsson, I.; Otto, H.; Liu, Q.; Rospert, S.; et al. Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell 2014, 156, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975, 67, 835–851. [Google Scholar] [CrossRef] [Green Version]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 1975, 67, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Walter, P.; Chang, C.N.; Goldman, B.M.; Erickson, A.H.; Lingappa, V.R. Translocation of proteins across membranes: The signal hypothesis and beyond. Symp. Soc. Exp. Biol. 1979, 33, 9–36. [Google Scholar] [PubMed]
- Matlin, K.S. Spatial expression of the genome: The signal hypothesis at forty. Nat. Rev. Mol. Cell Biol. 2011, 12, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Signal sequences. The limits of variation. J. Mol. Biol. 1985, 184, 99–105. [Google Scholar] [CrossRef]
- Von Heijne, G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef]
- Von Heijne, G. Patterns of amino acids near signal-sequence cleavage sites. Eur. J. Biochem. 1983, 133, 17–21. [Google Scholar] [CrossRef]
- Nesmeyanova, M.A.; Tsfasman, I.M.; Karamyshev, A.L.; Suzina, N.E. Secretion of the overproduced periplasmic PhoA protein into the medium and accumulation of its precursor in phoA-transformed Escherichia coli strains: Involvement of outer membrane vesicles. World J. Microbiol. Biotechnol. 1991, 7, 394–406. [Google Scholar] [CrossRef]
- Nesmeyanova, M.A.; Kalinin, A.E.; Karamyshev, A.L.; Mikhaleva, N.I.; Krupyanko, V.I. Overproduction, secretion, isolation and properties of recombinant alkaline phosphatase encoded in Escherichia coli. Process Biochem. 1997, 32, 1–7. [Google Scholar] [CrossRef]
- Nesmeyanova, M.A.; Karamyshev, A.L.; Karamysheva, Z.N.; Kalinin, A.E.; Ksenzenko, V.N.; Kajava, A.V. Positively charged lysine at the N-terminus of the signal peptide of the Escherichia coli alkaline phosphatase provides the secretion efficiency and is involved in the interaction with anionic phospholipids. FEBS Lett. 1997, 403, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Karamyshev, A.L.; Karamysheva, Z.N.; Kajava, A.V.; Ksenzenko, V.N.; Nesmeyanova, M.A. Processing of Escherichia coli alkaline phosphatase: Role of the primary structure of the signal peptide cleavage region. J. Mol. Biol. 1998, 277, 859–870. [Google Scholar] [CrossRef]
- Kalinin, A.E.; Karamyshev, A.L.; Nesmeianova, M.A. Disruption of processing of alkaline phosphatase as a result of single amino acid changes affects the composition and metabolism of phospholipids from Escherichia coli, secreting mutant proteins. Biokhimiia 1996, 61, 100–109. [Google Scholar] [PubMed]
- Nilsson, I.; Lara, P.; Hessa, T.; Johnson, A.E.; von Heijne, G.; Karamyshev, A.L. The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J. Mol. Biol. 2015, 427, 1191–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinarbasi, E.S.; Karamyshev, A.L.; Tikhonova, E.B.; Wu, I.H.; Hudson, H.; Thomas, P.J. Pathogenic Signal Sequence Mutations in Progranulin Disrupt SRP Interactions Required for mRNA Stability. Cell Rep. 2018, 23, 2844–2851. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, E.B.; Karamysheva, Z.N.; von Heijne, G.; Karamyshev, A.L. Silencing of Aberrant Secretory Protein Expression by Disease-Associated Mutations. J. Mol. Biol. 2019, 431, 2567–2580. [Google Scholar] [CrossRef]
- Karamyshev, A.L.; Johnson, A.E. Selective SecA association with signal sequences in ribosome-bound nascent chains: A potential role for SecA in ribosome targeting to the bacterial membrane. J. Biol. Chem. 2005, 280, 37930–37940. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, R.; Knupffer, L.; Origi, A.; Asti, R.; Koch, H.G. Co-translational protein targeting in bacteria. FEMS Microbiol. Lett. 2018, 365, fny095. [Google Scholar] [CrossRef]
- Wang, S.; Yang, C.I.; Shan, S.O. SecA mediates cotranslational targeting and translocation of an inner membrane protein. J. Cell Biol. 2017, 216, 3639–3653. [Google Scholar] [CrossRef]
- Huber, D.; Jamshad, M.; Hanmer, R.; Schibich, D.; Doring, K.; Marcomini, I.; Kramer, G.; Bukau, B. SecA Cotranslationally Interacts with Nascent Substrate Proteins In Vivo. J. Bacteriol. 2017, 199, e00622-16. [Google Scholar] [CrossRef] [Green Version]
- Knupffer, L.; Fehrenbach, C.; Denks, K.; Erichsen, V.; Petriman, N.A.; Koch, H.G. Molecular Mimicry of SecA and Signal Recognition Particle Binding to the Bacterial Ribosome. mBio 2019, 10, e01317-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.D.; Bernstein, H.D.; Walter, P. Interaction of E. coli Ffh/4.5S ribonucleoprotein and FtsY mimics that of mammalian signal recognition particle and its receptor. Nature 1994, 367, 657–659. [Google Scholar] [CrossRef]
- Krieg, U.C.; Walter, P.; Johnson, A.E. Photocrosslinking of the signal sequence of nascent preprolactin to the 54-kilodalton polypeptide of the signal recognition particle. Proc. Natl. Acad. Sci. USA 1986, 83, 8604–8608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzchalia, T.V.; Wiedmann, M.; Girshovich, A.S.; Bochkareva, E.S.; Bielka, H.; Rapoport, T.A. The signal sequence of nascent preprolactin interacts with the 54K polypeptide of the signal recognition particle. Nature 1986, 320, 634–636. [Google Scholar] [CrossRef]
- Noriega, T.R.; Tsai, A.; Elvekrog, M.M.; Petrov, A.; Neher, S.B.; Chen, J.; Bradshaw, N.; Puglisi, J.D.; Walter, P. Signal recognition particle-ribosome binding is sensitive to nascent chain length. J. Biol. Chem. 2014, 289, 19294–19305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.J.; Chen, J.C.; Miao, Y.; Shao, Y.; Lin, J.; Bock, P.E.; Johnson, A.E. Signal recognition particle binds to ribosome-bound signal sequences with fluorescence-detected subnanomolar affinity that does not diminish as the nascent chain lengthens. J. Biol. Chem. 2003, 278, 18628–18637. [Google Scholar] [CrossRef] [Green Version]
- Halic, M.; Becker, T.; Pool, M.R.; Spahn, C.M.; Grassucci, R.A.; Frank, J.; Beckmann, R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 2004, 427, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Pfeffer, S.; Lee, P.H.; Jung, M.; Cavalie, A.; Helms, V.; Forster, F.; Zimmermann, R. Protein transport into the human endoplasmic reticulum. J. Mol. Biol. 2015, 427, 1159–1175. [Google Scholar] [CrossRef]
- Akopian, D.; Shen, K.; Zhang, X.; Shan, S.O. Signal recognition particle: An essential protein-targeting machine. Ann. Rev. Biochem. 2013, 82, 693–721. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Hegde, R.S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 2016, 41, 91–99. [Google Scholar] [CrossRef]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heck, J.W.; Cheung, S.K.; Hampton, R.Y. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 2010, 107, 1106–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Ann. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, C.J.; Green, R. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 2012, 19, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef]
- Welch, E.M.; Jacobson, A. An internal open reading frame triggers nonsense-mediated decay of the yeast SPT10 mRNA. EMBO J. 1999, 18, 6134–6145. [Google Scholar] [CrossRef] [Green Version]
- Doma, M.K.; Parker, R. RNA quality control in eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Popp, M.W.; Maquat, L.E. Organizing principles of mammalian nonsense-mediated mRNA decay. Ann. Rev. Genet. 2013, 47, 139–165. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, T.; Kuroha, K.; Kudo, K.; Makino, S.; Inoue, E.; Kashima, I.; Inada, T. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3’ end of aberrant mRNA. Mol. Cell 2012, 46, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Dimitrova, L.N.; Kuroha, K.; Tatematsu, T.; Inada, T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 2009, 284, 10343–10352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtson, M.H.; Joazeiro, C.A. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 2010, 467, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Stewart-Ornstein, J.; Wong, D.; Larson, A.; Williams, C.C.; Li, G.W.; Zhou, S.; King, D.; Shen, P.S.; Weibezahn, J.; et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012, 151, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duttler, S.; Pechmann, S.; Frydman, J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell 2013, 50, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.P.; Green, R. Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol. 2018, 19, 526–541. [Google Scholar] [CrossRef]
- Wolin, S.L.; Maquat, L.E. Cellular RNA surveillance in health and disease. Science 2019, 366, 822–827. [Google Scholar] [CrossRef]
- Inada, T. The Ribosome as a Platform for mRNA and Nascent Polypeptide Quality Control. Trends Biochem. Sci. 2017, 42, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, K.; Izawa, T.; Inada, T. Recent Progress on the Molecular Mechanism of Quality Controls Induced by Ribosome Stalling. Front. Genet. 2018, 9, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inada, T. Quality controls induced by aberrant translation. Nucleic Acids Res. 2020, 48, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juszkiewicz, S.; Hegde, R.S. Quality Control of Orphaned Proteins. Mol. Cell 2018, 71, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, R.S.; Zavodszky, E. Recognition and Degradation of Mislocalized Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a033902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, S.E.; Maduka, A.O.; Inada, T.; Silva, G.M. Expanding Role of Ubiquitin in Translational Control. Int. J. Mol. Sci. 2020, 21, 1151. [Google Scholar] [CrossRef] [Green Version]
- Karamysheva, Z.N.; Tikhonova, E.B.; Karamyshev, A.L. Granulin in Frontotemporal Lobar Degeneration: Molecular Mechanisms of the Disease. Front. Neurosci. 2019, 13, 395. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Luhrmann, R.; Tuschl, T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Smith, S.K.; Hannon, G.J.; Joshua-Tor, L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 2004, 305, 1434–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolia, N.H.; Joshua-Tor, L. Slicer and the argonautes. Nat. Chem. Biol. 2007, 3, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Carapito, R.; Konantz, M.; Paillard, C.; Miao, Z.; Pichot, A.; Leduc, M.S.; Yang, Y.; Bergstrom, K.L.; Mahoney, D.H.; Shardy, D.L.; et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J. Clin. Investig. 2017, 127, 4090–4103. [Google Scholar] [CrossRef] [PubMed]
- Bellanne-Chantelot, C.; Schmaltz-Panneau, B.; Marty, C.; Fenneteau, O.; Callebaut, I.; Clauin, S.; Docet, A.; Damaj, G.L.; Leblanc, T.; Pellier, I.; et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood 2018, 132, 1318–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, T.; Walter, P. Reciprocal stimulation of GTP hydrolysis by two directly interacting GTPases. Science 1995, 269, 1422–1424. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Walne, A.J.; Plagnol, V.; Velangi, M.; Ho, A.; Hossain, U.; Vulliamy, T.; Dokal, I. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am. J. Hum. Genet. 2012, 90, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Muhlen, C.A.; Tan, E.M. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin. Arthritis Rheum. 1995, 24, 323–358. [Google Scholar] [CrossRef]
- Kao, A.H.; Lacomis, D.; Lucas, M.; Fertig, N.; Oddis, C.V. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004, 50, 209–215. [Google Scholar] [CrossRef]
- Benveniste, O.; Drouot, L.; Jouen, F.; Charuel, J.L.; Bloch-Queyrat, C.; Behin, A.; Amoura, Z.; Marie, I.; Guiguet, M.; Eymard, B.; et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum. 2011, 63, 1961–1971. [Google Scholar] [CrossRef]
- Kusumoto, T.; Okamori, S.; Masuzawa, K.; Asakura, T.; Nishina, N.; Chubachi, S.; Naoki, K.; Fukunaga, K.; Betsuyaku, T. Development of Necrotizing Myopathy Following Interstitial Lung Disease with Anti-signal Recognition Particle Antibody. Intern Med. 2018, 57, 2045–2049. [Google Scholar] [CrossRef] [Green Version]
- Tanino, Y. Interstitial Lung Disease Is a Possible Manifestation of Anti-signal Recognition Particle Antibody Syndrome. Intern Med. 2018, 57, 1957–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanaoka, H.; Kaneko, Y.; Suzuki, S.; Takada, T.; Hirakata, M.; Takeuchi, T.; Kuwana, M. A unique case of polymyositis with anti-signal recognition particle antibody complicated by subacute interstitial lung disease and subluxing arthropathy, resembling anti-synthetase syndrome. Mod. Rheumatol. 2016, 26, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Kabuto, M.; Fujimoto, N.; Hamaguchi, Y.; Tanaka, T. Anti-signal recognition particle antibody-positive polymyositis in a patient with Sjogren’s syndrome showing various types of annular erythema: Positive correlation between the activities of annular erythema and myositis. J. Dermatol. 2016, 43, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, H.; Kaneko, Y.; Suzuki, S.; Takada, T.; Hirakata, M.; Takeuchi, T.; Kuwana, M. Anti-signal recognition particle antibody in patients without inflammatory myopathy: A survey of 6180 patients with connective tissue diseases. Scand. J. Rheumatol. 2016, 45, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Fujii, K.; Gupta, V.; Hata, H.; Koizumu, H.; Hoshikawa, M.; Naruki, S.; Miyata, Y.; Takahashi, I.; Miyazawa, T.; et al. Identification of key modules and hub genes for small-cell lung carcinoma and large-cell neuroendocrine lung carcinoma by weighted gene co-expression network analysis of clinical tissue-proteomes. PLoS ONE 2019, 14, e0217105. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J.; et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 2017, 170, 352–366. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Muller, L.; Wullich, B. Protein transport into the endoplasmic reticulum: Mechanisms and pathologies. Trends Mol. Med. 2006, 12, 567–573. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Lin, W.J.; Salton, S.R. The regulated secretory pathway and human disease: Insights from gene variants and single nucleotide polymorphisms. Front. Endocrinol. 2013, 4, 96. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Castro-Fernandez, C.; Maya-Nunez, G.; Conn, P.M. Beyond the signal sequence: Protein routing in health and disease. Endocr. Rev. 2005, 26, 479–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarjanazi, H.; Savas, S.; Pabalan, N.; Dennis, J.W.; Ozcelik, H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins 2008, 70, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; von Schantz, C.; Peltonen, L.; Jalanko, A. A novel aspartylglucosaminuria mutation affects translocation of aspartylglucosaminidase. Hum. Mutat. 2004, 24, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, M.; Regis, S.; Tappino, B.; Rosano, C.; Assereto, S.; Corsolini, F.; Di Rocco, M.; Filocamo, M. Molecular analysis and characterization of nine novel CTSK mutations in twelve patients affected by pycnodysostosis. Mutation in brief #961. Online. Hum. Mutat. 2007, 28, 524. [Google Scholar] [PubMed]
- Fujita, Y.; Nakata, K.; Yasui, N.; Matsui, Y.; Kataoka, E.; Hiroshima, K.; Shiba, R.I.; Ochi, T. Novel mutations of the cathepsin K gene in patients with pycnodysostosis and their characterization. J. Clin. Endocrinol. Metab. 2000, 85, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Seppen, J.; Steenken, E.; Lindhout, D.; Bosma, P.J.; Elferink, R.P. A mutation which disrupts the hydrophobic core of the signal peptide of bilirubin UDP-glucuronosyltransferase, an endoplasmic reticulum membrane protein, causes Crigler-Najjar type II. FEBS Lett. 1996, 390, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Fingerhut, A.; Reutrakul, S.; Knuedeler, S.D.; Moeller, L.C.; Greenlee, C.; Refetoff, S.; Janssen, O.E. Partial deficiency of thyroxine-binding globulin-Allentown is due to a mutation in the signal peptide. J. Clin. Endocrinol. Metab. 2004, 89, 2477–2483. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, S.; Xu, S.Y.; Caballero, M.; Salcedo, M.; La, O.A.; Wedemann, H.; Gal, A. A missense point mutation (Leu13Arg) of the Norrie disease gene in a large Cuban kindred with Norrie disease. Hum. Mol. Genet. 1994, 3, 655–656. [Google Scholar] [CrossRef]
- Sunthornthepvarakul, T.; Churesigaew, S.; Ngowngarmratana, S. A novel mutation of the signal peptide of the preproparathyroid hormone gene associated with autosomal recessive familial isolated hypoparathyroidism. J. Clin. Endocrinol. Metab. 1999, 84, 3792–3796. [Google Scholar] [CrossRef]
- Arnold, A.; Horst, S.A.; Gardella, T.J.; Baba, H.; Levine, M.A.; Kronenberg, H.M. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. J. Clin. Investig. 1990, 86, 1084–1087. [Google Scholar] [CrossRef]
- Chow, K.M.; Szeto, C.C.; Poon, P.; Lau, W.Y.; Lai, F.M.; Li, P.K. Transforming growth factor-beta1 gene polymorphism in renal transplant recipients. Ren. Fail. 2005, 27, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Lacha, J.; Hubacek, J.A.; Potmesil, P.; Viklicky, O.; Malek, I.; Vitko, S. TGF-beta I gene polymorphism in heart transplant recipients--effect on renal function. Ann. Transplant. 2001, 6, 39–43. [Google Scholar] [PubMed]
- Yamada, Y.; Miyauchi, A.; Goto, J.; Takagi, Y.; Okuizumi, H.; Kanematsu, M.; Hase, M.; Takai, H.; Harada, A.; Ikeda, K. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J. Bone Miner. Res. 1998, 13, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Beranek, M.; Kankova, K.; Benes, P.; Izakovicova-Holla, L.; Znojil, V.; Hajek, D.; Vlkova, E.; Vacha, J. Polymorphism R25P in the gene encoding transforming growth factor-beta (TGF-beta1) is a newly identified risk factor for proliferative diabetic retinopathy. Am. J. Med. Genet. 2002, 109, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Anjos, S.; Nguyen, A.; Ounissi-Benkalha, H.; Tessier, M.C.; Polychronakos, C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J. Biol. Chem. 2002, 277, 46478–46486. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Lamminen, T.; Pakarinen, P.; Hellman, J.; Manna, P.; Herrera, R.J.; Huhtaniemi, I. A novel Ala−3Thr mutation in the signal peptide of human luteinizing hormone beta-subunit: Potentiation of the inositol phosphate signalling pathway and attenuation of the adenylate cyclase pathway by recombinant variant hormone. Mol. Hum. Reprod. 2002, 8, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Wang, Z.Y.; Dong, N.Z.; Bai, X.; Zhang, W.; Ruan, C.G. A case of deficiency of plasma plasminogen activator inhibitor-1 related to Ala15Thr mutation in its signal peptide. Blood Coagul. Fibrinolysis 2005, 16, 79–84. [Google Scholar] [CrossRef]
- Chen, J.M.; Raguenes, O.; Ferec, C.; Deprez, P.H.; Verellen-Dumoulin, C.; Andriulli, A. The A16V signal peptide cleavage site mutation in the cationic trypsinogen gene and chronic pancreatitis. Gastroenterology 1999, 117, 1508–1509. [Google Scholar] [CrossRef]
- Ikegawa, S.; Nakamura, K.; Nagano, A.; Haga, N.; Nakamura, Y. Mutations in the N-terminal globular domain of the type X collagen gene (COL10A1) in patients with Schmid metaphyseal chondrodysplasia. Hum. Mutat. 1997, 9, 131–135. [Google Scholar] [CrossRef]
- Chan, D.; Ho, M.S.; Cheah, K.S. Aberrant signal peptide cleavage of collagen X in Schmid metaphyseal chondrodysplasia. Implications for the molecular basis of the disease. J. Biol. Chem. 2001, 276, 7992–7997. [Google Scholar] [CrossRef] [Green Version]
- Zschenker, O.; Jung, N.; Rethmeier, J.; Trautwein, S.; Hertel, S.; Zeigler, M.; Ameis, D. Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease. J. Lipid Res. 2001, 42, 1033–1040. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SRP Subunit | Mutation | Disease | References |
|---|---|---|---|
| SRP54 | G113R, T115A, T117del, C118Y, C136Y, A223D, G226E, G274D | Neutropenia and Shwachman-Diamond-like syndrome | [73,74] |
| SRP72 | R207H, truncated T355K due to two nucleotides deletion and frameshift | Aplasia (aplastic anemia), myelodysplasia | [76] |
| Gene (Protein) | Signal Sequence Plus 2 Amino Acid Residues (Cleavage Site is Underlined) 2 | Mutation | mRNA Expression 3 | Disease or Note | References |
|---|---|---|---|---|---|
| GRN (granulin) | MWTLVSWVALTAGLVAG TR MWTLVSWVDLTAGLVAG TR MWTLVSRVALTAGLVAG TR MWTLLSWVALTAGLVAG TR | Wild-type A9D W7R V5L | +++++ ++ + +++++ | Frontotemporal lobar degeneration (FTLD); V5L is a benign polymorphism | [23] |
| AGA (aspartylglucosaminidase) | MARKSNLPVLLVPFLLCQALVRC SS MARKSNLPVLLVPFRLCQALVRC SS | Wild-type L15R | +++++ + | Aspartylglucosaminuria | [24,93] |
| CTSK (cathepsin K) | MWGLKVLLLPVVSFA LY MWGLKVPLLPVVSFA LY MWGLKVLLPPVVSFA LY | Wild-type L7P L9P | +++++ + + | Pycnodysostosis | [24,94,95] |
| UGT1A1 (UDP-glucuronosyltransferase) | MAVESQGGRPLVLGLLLCVLGPVVS HA MAVESQGGRPLVLGRLLCVLGPVVS HA | Wild-type L15R | +++++ ++ | Crigler-Najjar disease | [24,96] |
| SERPINA7 (serpin peptidase inhibitor A7) | MSPFLYLVLLVLGLHATIHC AS MSPFLYLVLLVLGLHATIYC AS | Wild-type H19Y | +++++ ++ | Thyroxine-binding globulin deficiency | [24,97] |
| NDP (Norrie disease protein) | MRKHVLAASFSMLSLLVIMGDTD SK MRKHVLAASFSMRSLLVIMGDTD SK MLSLLVIMGDTD SK | Wild-type L13R Δ11 | +++++ +++ +++ | Norrie disease | [24,98] |
| PTH (parathyroid hormone) | MIPAKDMAKVMIVMLAICFLTKSDG KS MIPAKDMAKVMIVMLAIRFLTKSDG KS MIPAKDMAKVMIVMLAICFLTKPDG KS | Wild-type C18R S23P | +++++ +++ +++ | Hypoparathyroidism | [24,99,100] |
| TGFB1 (transforming growth factor beta 1) | MPPSGLRLLPLLLPLLWLLVLTPGRPAAG LS MPPSGLRLLLLLLPLLWLLVLTPGRPAAG LS MPPSGLRLLPLLLPLLWLLVLTPGPPAAG LS | Wild-type P10L R25P | +++++ ++++ +++ | Renal function decline, osteoporosis, proliferative diabetic retinopathy | [24,101,102,103,104] |
| CTLA4 (cytotoxic T-lymphocyte associated protein 4) | MACLGFQRHKAQLNLATRTWPCTLLFFLLFIPVFC KA MACLGFQRHKAQLNLAARTWPCTLLFFLLFIPVFC KA | Wild-type T17A | +++++ ++++ | Autoimmune disease | [24,105] |
| LHB (luteinizing hormone beta polypeptide) | MEMLQGLLLLLLLSMGGAWA SR MEMLQGLLLLLLLSMGGTWA SR | Wild-type A18T | +++++ ++++ | Hypogonadotropic hypogonadism | [24,106] |
| SERPINE1 (serpin peptidase inhibitor E1) | MQMSPALTCLVLGLALVFGEGSA VH MQMSPALTCLVLGLTLVFGEGSA VH | Wild-type A15T | +++++ ++++ | Fibrinolytic bleeding disorder | [24,107] |
| PRSS1 (serine protease 1) | MNPLLILTFVAAALA AP MNPLLILTFVAAALA VP | Wild-type A16V | +++++ +++++ | Chronic pancreatitis | [24,107,108] |
| COL10A1 (collagen type X alpha 1) | MLPQIPFLLLVSLNLVHG VF MLPQIPFLLLVSLNLVHRVF MLPQIPFLLLVSLNLVHEVF | Wild-type G18R G18E | +++++ +++++ | Schmid metaphyseal chondrodysplasia | [24,109,110] |
| LIPA (lipase A) | MKMRFLGLVVCLVLWPLHSEGSG GKL MKMRFLGLVVCLVLWPLHSEGSR GKL | Wild-type G23R | +++++ +++++ | Wolman disease | [24,111] |
| PRL (prolactin, bovine) | MDSKGSSQKGSRLLLLLVVSNLLLCQGVVS TP MDSKGSSQKGSRLLLLLVVSNLLCQGVVS TP MDSKGSSQKGSRLLLLLVVSNLCQGVVS TP MDSKGSSQKGSRLLLLVVSNLCQGVVS TP MDSKGSSQKGSRLLLVVSNLCQGVVS TP | Wild-type Δ1L Δ2L Δ3L Δ4L | +++++ ++++ +++ ++ + | Artificial mutations | [8,22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karamyshev, A.L.; Tikhonova, E.B.; Karamysheva, Z.N. Translational Control of Secretory Proteins in Health and Disease. Int. J. Mol. Sci. 2020, 21, 2538. https://doi.org/10.3390/ijms21072538
Karamyshev AL, Tikhonova EB, Karamysheva ZN. Translational Control of Secretory Proteins in Health and Disease. International Journal of Molecular Sciences. 2020; 21(7):2538. https://doi.org/10.3390/ijms21072538
Chicago/Turabian StyleKaramyshev, Andrey L., Elena B. Tikhonova, and Zemfira N. Karamysheva. 2020. "Translational Control of Secretory Proteins in Health and Disease" International Journal of Molecular Sciences 21, no. 7: 2538. https://doi.org/10.3390/ijms21072538
APA StyleKaramyshev, A. L., Tikhonova, E. B., & Karamysheva, Z. N. (2020). Translational Control of Secretory Proteins in Health and Disease. International Journal of Molecular Sciences, 21(7), 2538. https://doi.org/10.3390/ijms21072538