Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies


{kind=link}
{kind=link}
Abstract
1. Introduction
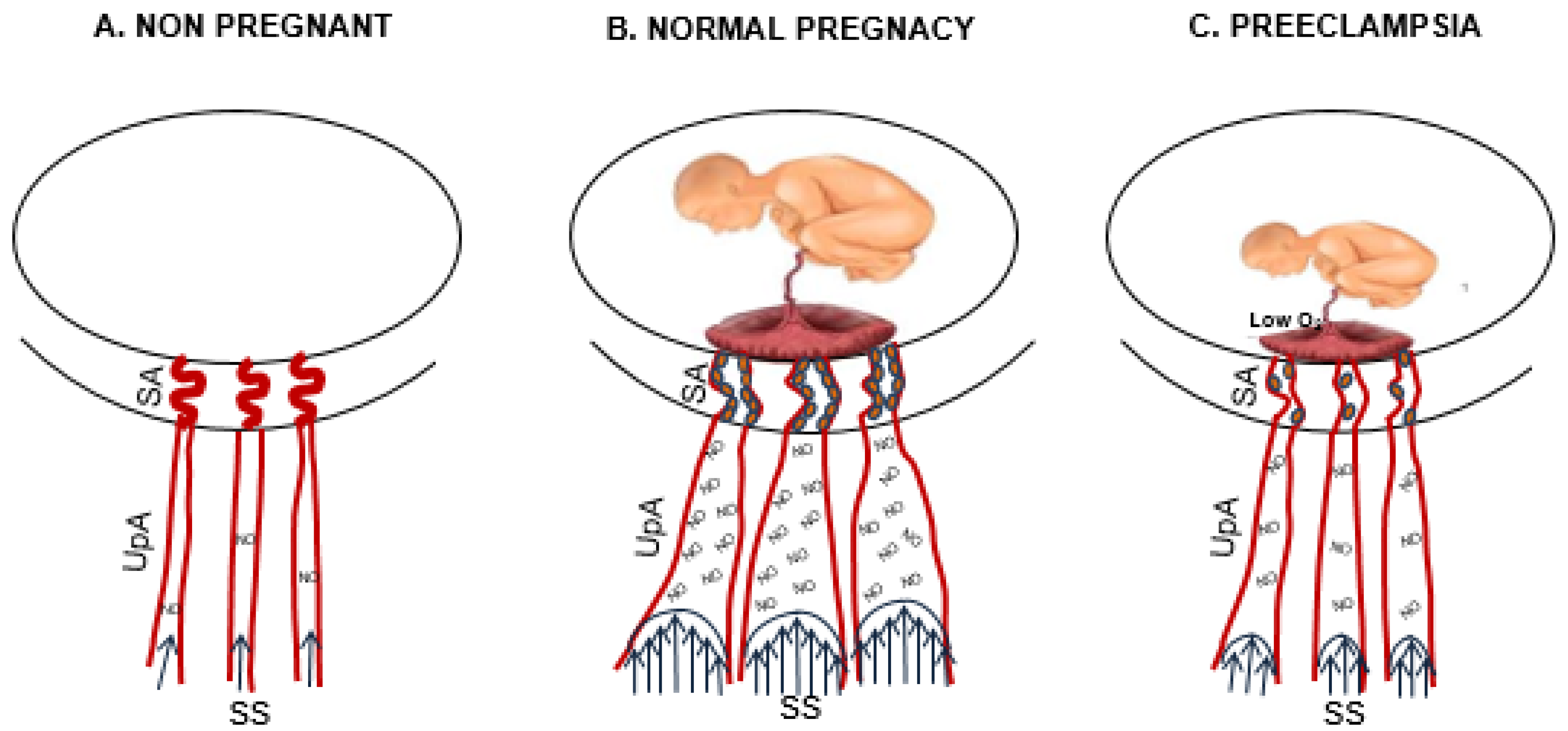
2. Uterine Circulation during Pregnancy
3. Maternal Gestational Uterine Vascular Remodeling
4. Estrogens in Pregnancy
Estrogen Receptors (ERs)
5. Estrogen Influences on Maternal Uterine Vascular Remodeling
5.1. Estrogens Induce Cytotrophoblast Invasion of Spiral Arteries
5.2. Estrogenic Stimulation of VEGF Signaling
5.3. Estrogen Effects on Matrix Metalloproteinases (MMPs)
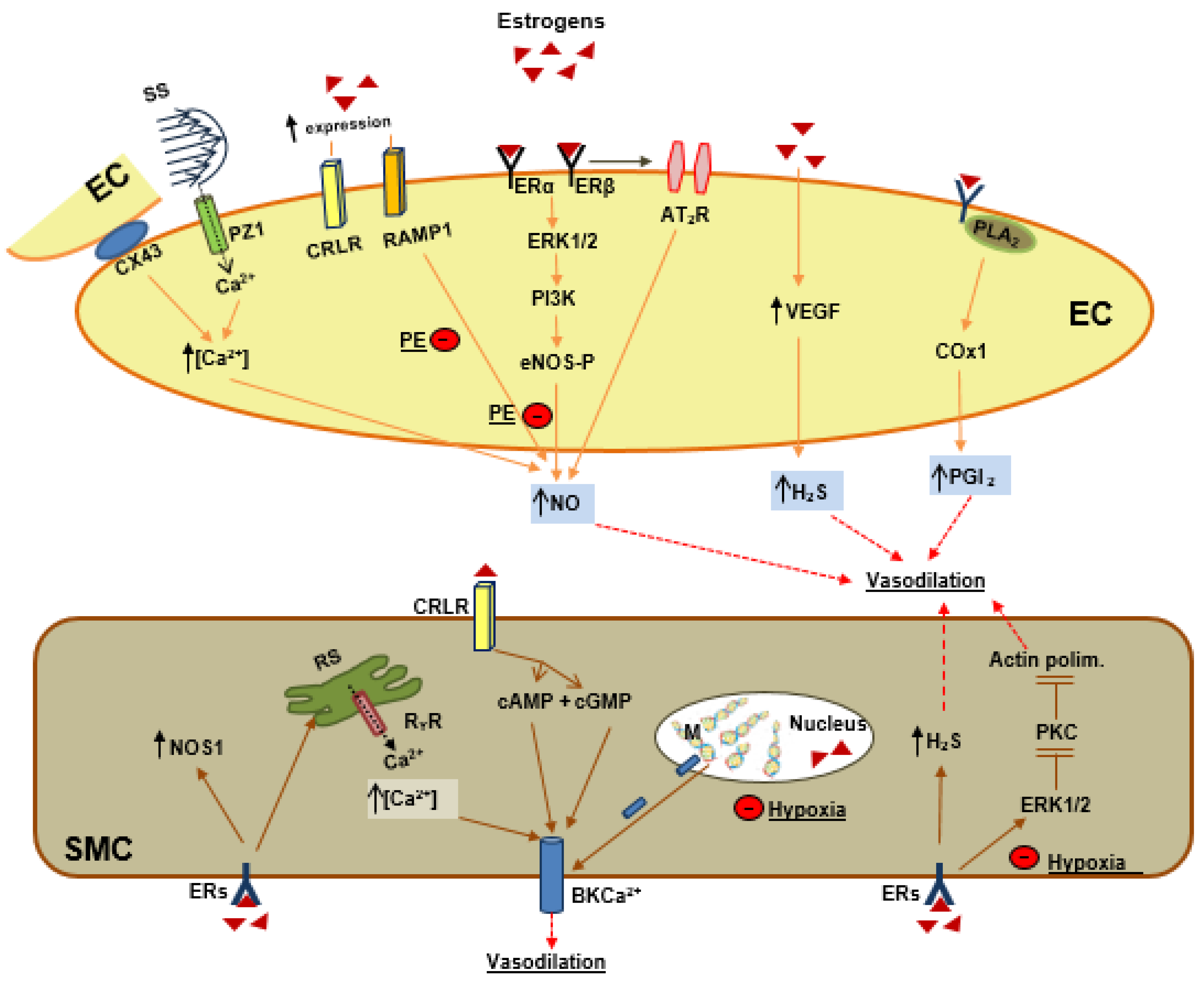
6. Estrogens and Vascular Tone Regulation
6.1. Estrogen and Endothelial Signaling
6.2. Estrogens as Endogenous Vasodilator Peptides
6.3. Estrogens and Smooth Muscle Cells Signaling
7. Preeclampsia
7.1. Preeclampsia is Associated with Lower Estrogen
7.2. Preeclampsia and Vascular Tone
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Palmer, S.K.; Zamudio, S.; Coffin, C.; Parker, S.; Stamm, E.; Moore, L.G. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet. Gynecol. 1992, 80, 1000–1006. [Google Scholar]
- Gerretsen, G.; Huisjes, H.J.; Elema, J.D. The role of the spiral artieries in the placental bed in relation to preeclampsia and fetal growth retardation. Br. J. Obstet. Gynaecol. 1981, 88, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.; Peeters, L.L.H. Pathogenesis of pre-eclampsia: A comprehensive model. Obstet. Gynecol. Surv. 1998, 63, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Lydakis, C.; Beevers, M.; Beevers, D.G.; Lip, G.Y. The prevalence of pre-eclampsia and obstetric outcome in pregnancies of normotensive and hypertensive women attending a hospital specialist clinic. Int. J. Clin. Pract. 2001, 55, 361–367. [Google Scholar] [PubMed]
- Osol, G.; Mandalà, M. Maternal Uterine Vascular Remodeling During Pregnancy. Physiology 2009, 24, 58–71. [Google Scholar] [CrossRef]
- Ueland, K. Maternal cardiovascular dynamics. VII. Intrapartum blood volume changes. Am. J. Obstet. Gynecol. 1976, 126, 671–677. [Google Scholar] [CrossRef]
- Rockwell, L.C.; Vargas, E.; Moore, L.G. Human physiological adaptation to pregnancy: Inter- and intraspecific perspectives. Am. J. Hum. Biol. 2003, 15, 330–341. [Google Scholar] [CrossRef]
- Metcalfe, J.; Ueland, K. Maternal cardiovascular adjustments to preg- nancy. Prog. Cardiovasc. Dis. 1974, 16, 363–374. [Google Scholar] [CrossRef]
- Genbacev, O.D.; Prakobphol, A.; Foulk, R.A.; Krtolica, A.R.; Ilic, D.; Singer, M.S.; Yang, Z.Q.; Kiessling, L.L.; Rosen, S.D.; Fisher, S.J. Trophoblast L-selectin-mediated adhesion at the maternal-fetal interface. Science 2003, 299, 405–408. [Google Scholar] [CrossRef]
- Zhou, Y.; Fisher, S.J.; Janatpour, M.; Genbacev, O.; Dejana, E.; Wheelock, M.; Damsky, C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J. Clin. Invest. 1997, 99, 2139–2151. [Google Scholar] [CrossRef]
- Thaler, I.; Manor, D.; Itskovitz, J.; Rottem, S.; Levit, N.; Timor-Tritsch, I.; Brandes, J.M. Changes in uterine blood flow during human pregnancy. Am. J. Obstet. Gynecol. 1990, 162, 121–125. [Google Scholar] [CrossRef]
- Bjellin, L.; Sjoquist, P.O.; Carter, A.M. Uterine, maternal placental and ovarian blood flow throughout pregnancy in the guinea pig. Zeitschrift fur Geburtshilfe und Perinatologie 1975, 179, 179–187. [Google Scholar] [PubMed]
- Dowell, R.T.; Kauer, C.D. Maternal hemodynamics and uteroplacental blood flow throughout gesta- tion in conscious rats. Meth. Find. Exp. Clin. Pharmacol. 1997, 19, 613–625. [Google Scholar]
- Lees, M.H.; Hill, J.D.; Ochsner, A.J.; Thomas, C.L., 3rd; Novy, M.J. Maternal placental and myometrial blood flow of the rhesus monkey during uterine contractions. Am. J. Obstet. Gynecol. 1971, 110, 68–81. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Morriss, F.H., Jr.; Makowski, E.L.; Meschia, G.; Battaglia, F.C. Circulatory changes in the reproductive tissues of ewes during pregnancy. Gynecol. Invest. 1974, 5, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Lunell, N.O.; Nylund, L.E.; Lewander, R.; Sarby, B. Uteroplacental blood flow in pre-eclampsia measurements with indium-113m and a computer-linked camera. Clin. Exp. Hypertens. B 1982, 1, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, S.; Palmer, S.K.; Droma, T.; Stamm, E.; Coffin, C.; Moore, L.G. Effect of altitude on uterine artery blood flow during normal pregnancy. J. Appl. Physiol. 1995, 79, 7–14. [Google Scholar] [CrossRef]
- Chang, K.; Xiao, D.; Huang, X.; Xue, Z.; Yang, S.; Longo, L.D.; Zhang, L. Chronic hypoxia inhibits sex steroid hormone-mediated attenuation of ovine uterine arterial myogenic tone in pregnancy. Hypertension 2010, 56, 750–757. [Google Scholar] [CrossRef][Green Version]
- Charles, S.M.; Julian, C.G.; Vargas, E.; Moore, L.G. Higher estrogen levels during pregnancy in Andean than European residents of high altitude suggest differences in aromatase activity. J. Clin. Endocrinol. Metab. 2014, 99, 2908–2916. [Google Scholar] [CrossRef]
- Julian, C.G.; Wilson, M.J.; Lopez, M.; Yamashiro, H.; Tellez, W.; Rodriguez, A.; Bigham, A.W.; Shriver, M.D.; Rodriguez, C.; Vargas, E.; et al. Augmented uterine artery blood flow and oxygen delivery protect Andeans from altitude-associated reductions in fetal growth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1564–R1575. [Google Scholar] [CrossRef]
- Mucci, L.A.; Lagiou, P.; Tamimi, R.M.; Hsieh, C.C.; Adami, H.O.; Trichopoulos, D. Pregnancy estriol, estradiol, progesterone and prolactin in relation to birth weight and other birth size variables (United States). Cancer Causes Control 2003, 14, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.M.L.; Ziegler, W.F.; Leavitt, T.; Badger, G.J. Uterine artery hemodynamic adaptations through the menstrual cycle into early pregnancy. Obstet. Gynecol. 2002, 99, 620–624. [Google Scholar] [PubMed]
- Magness, R.R.; Phernetton, T.M.; Gibson, T.C.; Chen, D.B. Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol-17beta-treated, intact follicular and pregnant sheep. J. Physiol. 2005, 15, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Magness, R.R.; Rosenfeld, C.R. Local and systemic estradiol-17 beta: Effects on uterine and systemic vasodilation. Am. J. Physiol. Endocrinol. Metab. 1989, 256, E536–E542. [Google Scholar] [CrossRef] [PubMed]
- Mandalà, M.; Osol, G. Physiological remodelling of the maternal uterine circulation during pregnancy. Basic Clin. Pharmacol. Toxicol. 2012, 110, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Mabie, W.C.; DiSessa, T.G.; Crocker, L.G.; Sibai, B.; Arheart, K.L. A longitudinal study of cardiac output in normal human pregnancy. Am. J. Obstet. Gynecol. 1994, 170, 849–856. [Google Scholar] [CrossRef]
- Mone, S.M.; Sanders, S.; Colan, S.D. Control mechanisms for physiological hypertrophy of pregnancy. Circulation 1996, 94, 667–672. [Google Scholar] [CrossRef]
- Caulin-Glaser, T.; Garcıa-Cardena, G.; Sarrel, P.; Sessa, W.C.; Bender, J.R. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ. Res. 1997, 81, 885–892. [Google Scholar] [CrossRef]
- Storment, J.M.; Meyer, M.; Osol, G. Estrogen augments the vaso- dilatory effects of vascular endothelial growth factor in the uterine circulation of the rat. Am. J. Obstet. Gynecol. 2000, 183, 449–453. [Google Scholar] [CrossRef]
- Stice, S.L.; Ford, S.P.; Rosazza, J.P.; Van Orden, D.E. Interaction of 4- hydroxylated estradiol and potential-sensitive Ca2+ channels in altering uterine blood flow during the estrous cycle and early pregnancy in gilts. Biol. Reprod. 1987, 36, 369–375. [Google Scholar] [CrossRef]
- Salom, J.B.; Burguete, M.C.; Pèrez-Asensio, F.J.; Torregrosa, G.; Alborch, E. Relaxant effects of 17-beta-estradiol in cerebral arteries through Ca(2+) entry inhibition. J. Cereb. Blood Flow Metab. 2001, 21, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Li, W.; Tran, V.; Khalil, R.A. EMMPRIN-mediated induction of uterine and vascular matrix metalloproteinases during pregnancy and in response to estrogen and progesterone. Biochem. Pharmacol. 2013, 15, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Shifren, J.L.; Tseng, J.F.; Zaloudek, C.J.; Ryan, I.P.; Meng, Y.G.; Ferrara, N.; Jaffe, R.B.; Taylor, R.N. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: Implications for angiogenesis during the menstrual cycle and in the patho- genesis of endometriosis. J. Clin. Endocrinol. Metab. 1996, 81, 3112–3118. [Google Scholar]
- Mueller, M.D.; Vigne, J.L.; Minchenko, A.; Lebovic, D.I.; Leitman, D.C.; Taylor, R.N. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta. Proc. Natl. Acad. Sci. USA. 2000, 97, 10972–10977. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Snipe, J.; Krysiak, O.; Schonfelder, G.; Resnick, M.A. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol. Cell Biol. 2007, 27, 2590–2600. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gargett, C.E.; Zaitseva, M.; Bucak, K.; Chu, S.; Fuller, P.J.; Rogers, P.A.W. 17Beta-estradiol up-regulates vascular endothelial growth factor receptor-2 expression in human myometrial microvascular en- dothelial cells: Role of estrogen receptor-alpha and –beta. J. Clin. Endocrinol. Metab. 2002, 87, 4341–4349. [Google Scholar] [CrossRef]
- Holmes, D.I.R.; Zachary, I. Placental growth factor induces FosB and c-Fos gene expression via Flt-1 receptors. FEBS Lett. 2004, 557, 93–98. [Google Scholar] [CrossRef]
- Rauschemberger, M.B.; Sandoval, M.J.; Massheimer, V.L. Cellular and molecular actions displayed by estrone on vascular endothelium. Mol. Cell Endocrinol. 2011, 339, 136–143. [Google Scholar] [CrossRef]
- Torgersen, K.L.; Curran, C.A. A systematic approach to the physiologic adaptations of pregnancy. Crit. Care Nurs. 2006, 29, 2–19. [Google Scholar] [CrossRef]
- Chang, J.; Streitman, D. Physiologic adaptations to pregnancy. Neurol. Clin. 2012, 30, 781–789. [Google Scholar] [CrossRef]
- Longo, L.D. Maternal blood volume and cardiac output during pregnancy: A hypothesis of endocrinologic control. Am. J. Physiol. 1983, 245, R720–R729. [Google Scholar] [CrossRef]
- Cipolla, M.; Osol, G. Hypertrophic and hyperplastic effects of pregnancy on the rat uterine arterial wall. Am. J. Obstet. Gynecol. 1994, 171, 805–811. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Robertson, W.B.; Brosens, I.; Dixon, G. Review article: Trophoblast invasion and the establishment of haemochorial placentation in man and laboratory animals. Placenta 1981, 2, 71–91. [Google Scholar] [CrossRef]
- Wallace, A.E.; Fraser, R.; Cartwright, J.E. Extravillous trophoblast and decidual natural killer cells: A remodelling partnership. Hum. Reprod. Update 2012, 18, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Whitley, G.S.J.; Cartwright, J.E. Trophoblast-mediated spiral artery remodelling: A role for apoptosis. J. Anat. 2009, 215, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kliman, H.J. Trophoblast to Human Placenta; Academic Press: San Diego, CA, USA, 1999; pp. 834–846. [Google Scholar]
- Moll, W. Structure adaptation and blood flow control in the uterine arterial system after hemochorial placentation. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 110, S19–S27. [Google Scholar] [CrossRef]
- Page, K.L.; Celia, G.; Leddy, G.; Taatjes, D.J.; Osol, G. Structural remodeling of rat uterine veins in preg- nancy. Am. J. Obstet. Gynecol. 2002, 187, 1647–1652. [Google Scholar] [CrossRef]
- Gokina, N.I.; Mandalà, M.; Osol, G. Induction of localized differences in rat uterine radial artery behav- ior and structure during gestation. Am. J. Obstet. Gynecol. 2003, 189, 1489–1493. [Google Scholar] [CrossRef]
- Osol, G.; Cipolla, M. Pregnancy-induced changes in the three-dimensional mechanical properties of pressurized rat uteroplacental (radial) arteries. Am. J. Obstet. Gynecol. 1993, 168, 268–274. [Google Scholar] [CrossRef]
- Poston, L. The control of blood flow to the placenta. Exp. Physiol. 1997, 82, 377–387. [Google Scholar] [CrossRef]
- Griendling, K.K.; Fuller, E.O.; Cox, R.H. Pregnancy- induced changes in sheep uterine and carotid arteries. Am. J. Physiol. Heart Circ. Physiol. 1985, 248, H658–H665. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.M.; Huda, S.S.; Sattar, N.; Fraser, R.; Connell, J.M.C.; Davies, E. Depot-specific steroidogenic gene transcription in hu- man adipose tissue. Clin. Endocrinol. (Oxf). 2008, 69, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Devroey, P.; Camus, M.; Palermo, G.; Smitz, J.; Van Waesberghe, L.; Wisanto, A.; Wijbo, I.; Van Steirteghem, A.C. Placental production of estradiol and progesterone after oocyte donation in patients with primary ovarian failure. Am. J. Obstet. Gynecol. 1990, 162, 66–70. [Google Scholar] [CrossRef]
- Oakey, R.E. The progressive increase in estrogen production in human pregnancy: An appraisal of the factors responsible. Vitam. Horm. 1970, 28, 1–36. [Google Scholar] [PubMed]
- Bausero, P.; Ben-Mahdi, M.; Mazucatelli, J.; Bloy, C.; Perrot-Applanat, M. Vascular endothelial growth factor is modulated in vascular muscle cells by estradiol, tamoxifen, and hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2033–H2042. [Google Scholar] [CrossRef] [PubMed]
- Calzada-Mendoza, C.C.; Sanchez, E.C.; Campos, R.R.; Becerril, A.M.; Madrigal, E.B.; Sierra, A.R.; Mendez, E.B.; Ocharan, E.H.; Herrera, N.G.; Ceballos-Reyes, G. Differential aromatase (CYP19) expression in human arteries from normal and neoplasic uterus: An immunohistochemical and in situ hybridization study. Front. Biosci. 2006, 11, 389–393. [Google Scholar]
- Jobe, S.O.; Ramadoss, J.; Wargin, A.J.; Magness, R.R. Estradiol-17b and its cytochrome P450- and catechol-O-methyltransferase-derived metabolites selectively stimulate production of prostacyclin in uterine artery endothelial cells: Role of estrogen receptor-a versus estrogen receptor-b. Hypertension 2013, 61, 509–518. [Google Scholar] [CrossRef]
- Peter, M.; Dorr, H.G.; Sippell, W.G. Changes in the concentrations of dehydroepiandrosterone sulfate and estriol in maternal plasma during pregnancy: A longitudinal study in healthy women throughout gestation and at term. Horm. Res. 1994, 42, 278–281. [Google Scholar] [CrossRef]
- Rivarola, M.A.; Forest, M.; Migeon, C.J. Testosterone, androstenedione and dehydroepiandrosterone in plasma during pregnancy and at delivery: Concentration and protein binding. J. Clin. Endocrinol. Metab. 1968, 28, 34–40. [Google Scholar] [CrossRef]
- Dubey, R.K.; Tofovic, S.P.; Jackson, E.K. Cardiovascular pharmacology of estradiol metabolites. J. Pharmacol. Exp. Ther. 2004, 308, 403–409. [Google Scholar] [CrossRef]
- Mullis, P.E.; Yoshimura, N.; Kuhlmann, B.; Lippuner, K.; Jaeger, P.; Harada, H. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: Impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J. Clin. Endocrinol. Metab. 1997, 82, 1739–1745. [Google Scholar] [CrossRef]
- Ludwikowski, B.; Heger, S.; Datz, N.; Richter-Unruh, A.; Gonzàlez, R. Aromatase deficiency: Rare cause of virilization. Eur. J. Pediatr. Surg. 2013, 23, 418–422. [Google Scholar] [CrossRef]
- Levitz, M.; Young, B.K. Estrogens in pregnancy. Vitam. Horm. 1977, 35, 109–147. [Google Scholar]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and laboratory studies: A reference table for clinicians. Obstet. Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.; Alcorn, J.L.; Kunczt, C.; Mendelson, C.R. Characterization of the regulatory regions of the human aromatase (P450arom) gene involved in placenta-specific expression. Mol. Endocrinol. 1998, 12, 1764–1777. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Petz, L.N.; Nardulli, A.M. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors alpha and beta. J. Biol. Chem. 2005, 280, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Isaacs, G.D.; Diehl, A.G.; Sun, M.; Cheung, E.; Ranish, J.A.; Kraus, W.L. Multiple sequence-specific DNA-binding proteins mediate estrogen receptor signaling through a tethering pathway. Mol. Endocrinol. 2011, 25, 564–574. [Google Scholar] [CrossRef]
- Simoncini, T.; Genazzani, A.R.; Liao, J.K. Non genomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene. Circulation 2002, 105, 368–1373. [Google Scholar] [CrossRef]
- Tessier, C.; Deb, S.; Prigent-Tessier, A.; Ferguson-Gottschall, S.; Gibori, G.B.; Shiu, R.P.; Gibori, G. Estrogen receptors alpha and beta in rat decidua cells: Cell-specific expression and differential regulation by steroid hormones and prolactin. Endocrinology 2000, 141, 3842–3851. [Google Scholar] [CrossRef]
- Bukovsky, A.; Caudle, M.R.; Cekanova, M.; Fernando, R.I.; Wimalasena, J.; Foster, J.S.; Henley, D.C.; Elder, R.F. Placental expression of estrogen receptor beta and its hormone binding variant–comparison with estrogen receptor alpha and a role for estrogen receptors in asymmetric division and differentiation of estrogen-dependent cells. Reprod. Biol. Endocrinol. 2003, 1, 36. [Google Scholar] [CrossRef]
- Perrot-Applanat, M.; Deng, M.; Fernandez, H.; Lelaidier, C.; Meduri, G.; Bouchard, P. Immunohistochemical localization of estradiol and progesterone receptors in human uterus throughout preg- nancy: Expression in endometrial blood vessels. J. Clin. Endocrinol. Metab. 1994, 78, 216–224. [Google Scholar] [PubMed]
- Byers, M.J.; Zangl, A.; Phernetton, T.M.; Lopez, G.; Chen, D.B.; Magness, R.R. Endothelial vasodilator production by ovine uterine and systemic arteries: Ovarian steroid and pregnancy control of Eralpha and Erbeta levels. J. Physiol. 2005, 565, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Barnea, E.R.; MacLusky, N.J.; Naftolin, F. Kinetics of catechol estrogen-estrogen receptor dissociation: A possible factor un- derlying differences in catechol estrogen biological activity. Steroids 1983, 41, 643–656. [Google Scholar] [CrossRef]
- Liao, W.X.; Magness, R.R.; Chen, D.B. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol. Reprod. 2005, 72, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Tropea, T.; De Francesco, E.M.; Rigiracciolo, D.; Maggiolini, M.; Wareing, M.; Osol, G.; Mandalà, M. Pregnancy Augments G Protein Estrogen Receptor (GPER) Induced Vasodilation in Rat Uterine Arteries via the Nitric Oxide - cGMP Signaling Pathway. PLoS ONE 2015, 10, e0141997. [Google Scholar] [CrossRef]
- Iruela-Arispe, M.; Rodriguez-Manzaneque, J.C.; Abu-Jawdeh, G. Endometrial endothelial cells express estrogen and progesterone receptors and exhibit a tissue specific response to angiogenic growth factors. Microcirculation 1999, 6, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.B.; Jobe, S.O.; Ramadoss, J.; Magness, R.R. Estrogen receptor-α and estrogen receptor-β in the uterine vascular endothelium during pregnancy: Functional implications for regulating uterine blood flow. Semin. Reprod. Med. 2012, 30, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Yang, S.; Zhang, L. Direct chronic effect of steroid hormones in attenuating uterine arterial myogenic tone: Role of protein kinase c/extracellular signal-regulated kinase 1/2. Hypertension 2009, 54, 352–358. [Google Scholar] [CrossRef]
- Xiao, D.; Huang, X.; Yang, S.; Longo, L.D.; Zhang, L. Pregnancy downregulates actin polymerization and pressure-dependent myogenic tone in ovine uterine arteries. Hypertension 2010, 56, 1009–1015. [Google Scholar] [CrossRef]
- Lydrup, M.L.; Fernö, M. Correlation between estrogen receptor alpha expression, collagen content and stiffness in human uterine arteries. Acta Obstet. Gynecol. Scand. 2003, 82, 610–615. [Google Scholar] [CrossRef]
- van der Heijden, O.W.; Essers, Y.P.; Fazzi, G.; Peeters, L.L.; De Mey, J.G.; van Eys, G.J. Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase-deficient mice. Biol. Reprod. 2005, 72, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Makinoda, S.; Moll, W. Deoxyribonucleic acid synthesis in mesometrial arteries of guinea pigs during oestrous cycle, pregnancy and treatment with oestradiol benzoate. Placenta 1986, 7, 189–198. [Google Scholar] [CrossRef]
- Moll, W.; Nienartowicz, A.; Hees, H.; Wrobel, K.H.; Lenz, A. Blood flow regulation in the uteroplacental arteries. Troph Res. 1988, 3, 83–96. [Google Scholar]
- Chang, K.; Lubo, Z. Review article: Steroid hormones and uterine vascular adaptation to pregnancy. Reprod. Sci. 2008, 15, 336–348. [Google Scholar] [CrossRef]
- Bonagura, T.W.; Pepe, G.J.; Enders, A.C.; Albrecht, E.D. Suppression of Extravillous Trophoblast Vascular Endothelial Growth Factor Expression and Uterine Spiral Artery Invasion by Estrogen during Early Baboon Pregnancy. Endocrinology 2008, 149, 5078–5087. [Google Scholar] [CrossRef]
- Robson, A.; Harris, L.K.; Innes, B.A.; Lash, G.E.; Aljunaidy, M.M.; Aplin, J.D.; Baker, P.N.; Robson, S.C.; Bulmer, J.N. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. 2012, 26, 4876–4885. [Google Scholar] [CrossRef] [PubMed]
- Lash, G.E.; Pitman, H.; Morgan, H.L.; Innes, B.A.; Agwu, C.N.; Bulmer, J.N. Decidual macrophages: Key regulators of vascular remodeling in human pregnancy. J. Leukoc. Biol. 2016, 100, 315–325. [Google Scholar] [CrossRef]
- Gibson, D.A.; Esnal-Zufiaurre, A.; Bajo-Santos, C.; Collins, F.; Critchley, H.O.D.; Saunders, P.T.K. Profiling the expression and function of oestrogen receptor isoform ER46 in human endometrial tissues and uterine natural killer cells. Hum. Reprod. 2020. [Google Scholar] [CrossRef]
- Brownlee, R.D.; Langille, B.L. Arterial adaptations to altered blood flow. Can. J. Physiol. Pharmacol. 1991, 69, 978–983. [Google Scholar]
- Tarhouni, K.; Guihot, A.L.; Freidja, M.L.; Toutain, B.; Henrion, B.; Baufreton, C.; Pinaud, F.; Procaccio, V.; Grimaud, L.; Ayer, A.; et al. Key role of estrogens and endothelial estrogen receptor α in blood flow-mediated remodeling of resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 605–611. [Google Scholar] [CrossRef]
- Zarins, C.K.; Zatina, M.A.; Giddens, D.P.; Ku, D.N.; Glagov, S. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J. Vasc. Surg. 1987, 5, 413–420. [Google Scholar] [CrossRef]
- Ni, Y.; May, V.; Brees, K.; Osol, G. Pregnancy augments uteroplacental vascular endothelial growth factor gene expression and vasodilator effects. Am. J. Physiol. 1997, 273, H938–H944. [Google Scholar] [CrossRef] [PubMed]
- Greb, R.R.; Heikinheimo, O.; Williams, R.F.; Hodgen, G.D.; Goodman, A.L. Vascular endothelial growth factor in primate endometrium is regulated by oestrogen-receptor and progesterone-receptor ligands in vivo. Hum. Reprod. 1997, 12, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Klein, C.; Ealy, A.D.; Sharp, D.C. Conceptus-mediated endometrial vascular changes during early pregnancy in mares: An anatomic, histomorphometric, and vascular endothelial growth factor receptor system immunolocalization and gene expression study. Reproduction 2011, 142, 593–603. [Google Scholar] [CrossRef][Green Version]
- Rockwell, L.C.; Pillai, S.; Olson, C.E.; Koos, R.D. Inhibition of vascular endothelial growth factor/vascular permeability factor action blocks estrogen-induced uterine edema and implantation in rodents. Biol. Reprod. 2002, 67, 1804–1810. [Google Scholar] [CrossRef]
- Hervè, M.A.; Meduri, G.; Petit, F.G.; Domet, T.S.; Lazennec, G.; Mourah, S.; Perrot-Applanat, M. Regulation of the vascular endothelial growth factor (VEGF) receptor Flk-1/KDR by estradiol through VEGF in uterus. J. Endocrinol. 2006, 188, 91–99. [Google Scholar] [CrossRef][Green Version]
- Boeldt, D.S.; Grummer, M.A.; Magness, R.R.; Bird, I.M. Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC. J. Endocrinol. 2014, 223, 1–11. [Google Scholar] [CrossRef]
- Mehta, V.; Abi-Nader, K.N.; Shangaris, P.; Shaw, S.W.; Filippi, E.; Benjamin, E.; Boyd, M.; Peebles, D.M.; Martin, J.; Zachary, I.; et al. Local over-expression of VEGF-DΔNΔC in the uterine arteries of pregnant sheep results in long-term changes in uterine artery contractility and angiogenesis. PLoS ONE 2014, 9, e100021. [Google Scholar] [CrossRef]
- Hale, S.A.; Weger, L.; Mandalà, M.; Osol, G. Reduced NO signaling during pregnancy attenuates outward uterine artery remodeling by altering MMP expression and collagen and elastin deposition. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1266–H1275. [Google Scholar] [CrossRef]
- Kelly, B.A.; Bond, B.C.; Poston, L. Gestational profile of matrix metalloproteinases in rat uterine artery. Mol. Hum. Reprod. 2003, 9, 351–358. [Google Scholar] [CrossRef]
- Schafer-Somi, S.; Ali Aksoy, O.; Patzl, M.; Findik, M.; Erunal-Maral, N.; Beceriklisoy, H.B.; Polat, B.; Aslan, S. The activity of matrix metalloproteinase-2 and -9 in serum of pregnant and non-pregnant bitches. Reprod. Domest. Anim. 2005, 40, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Dumont, O.; Loufrani, L.; Henrion, D. Key role of the NO-pathway and matrix metalloprotease-9 in high blood flow-induced remodeling of rat resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 317–324. [Google Scholar] [CrossRef] [PubMed]
- John, L.; Ko, N.L.; Gokin, A.; Gokina, N.; Mandalà, M.; Osol, G. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1019–H1026. [Google Scholar] [CrossRef] [PubMed]
- Morschauser, T.J.; Ramadoss, J.; Koch, J.M.; Yi, F.X.; Lopez, G.E.; Bird, I.M.; Magness, R.R. Local effects of pregnancy on connexin proteins that mediate Ca2+-associated uterine endothelial NO synthesis. Hypertension 2014, 63, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.B.; Bird, I.M.; Zheng, J.; Magness, R.R. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology 2004, 145, 113–125. [Google Scholar] [CrossRef]
- Pastore, M.B.; Talwar, S.; Conley, M.R.; Magness, R.R. Identification of Differential ER-Alpha Versus ER-Beta Mediated Activation of eNOS in Ovine Uterine Artery Endothelial Cells. Biol. Reprod. 2016, 94, 139. [Google Scholar] [CrossRef]
- Pang, Y.; Thomas, P. Additive effects of low concentrations of estradiol-17β and progesterone on nitric oxide production by human vascular endothelial cells through shared signaling pathways. J. Steroid Biochem. Mol. Biol. 2017, 165, 258–267. [Google Scholar] [CrossRef]
- Landeros, R.V.; Pastore, M.B.; Magness, R.R. Effects of the Catechol and Methoxy Metabolites of 17β-Estradiol on Nitric Oxide Production by Ovine Uterine Artery Endothelial Cells. Reprod. Sci. 2019, 26, 459–468. [Google Scholar] [CrossRef]
- Mishra, J.S.; Te Riele, G.M.; Qi, Q.R.; Lechuga, T.J.; Gopalakrishnan, K.; Chen, D.B.; Kumar, S. Estrogen Receptor-β Mediates Estradiol-Induced Pregnancy-Specific Uterine Artery Endothelial Cell Angiotensin Type-2 Receptor Expression. Hypertension 2019, 74, 967–974. [Google Scholar] [CrossRef]
- Lechuga, T.J.; Qi, Q.R.; Magness, R.R.; Chen, D.B. Ovine uterine artery hydrogen sulfide biosynthesis in vivo: Effects of ovarian cycle and pregnancy. Biol. Reprod. 2019, 100, 1630–1636. [Google Scholar] [CrossRef]
- Zhang, H.H.; Chen, J.C.; Sheibani, L.; Lechuga, T.J.; Chen, D.B. Pregnancy Augments VEGF-Stimulated In Vitro Angiogenesis and Vasodilator (NO and H2S) Production in Human Uterine Artery Endothelial Cells. J. Clin. Endocrinol. Metab. 2017, 102, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Gangula, P.R.; Thota, C.; Wimalawansa, S.J.; Bukoski, R.D.; Yallampalli, C. Mechanisms involved in calcitonin gene-related Peptide-induced relaxation in pregnant rat uterine artery. Biol. Reprod. 2003, 69, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.R.; Yallampalli, U.; Gangula, P.R.; Reed, L.; Sathishkumar, K.; Gao, H.; Chauhan, M.; Yallampalli, C. Adrenomedullin relaxes rat uterine artery: Mechanisms and influence of pregnancy and estradiol. Endocrinology 2010, 151, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.L.; Green, K.E.; Vegiragu, S.; Hankins, G.D.; Martin, E.; Chauhan, M.; Thota, C.; Yallampalli, C. Evidence for decreased calcitonin gene-related peptide (CGRP) receptors and compromised responsiveness to CGRP of fetoplacental vessels in preeclamptic pregnancies. J. Clin. Endocrinol. Metab. 2005, 90, 2336–2343. [Google Scholar] [CrossRef]
- Scott, P.A.; Tremblay, A.; Brochu, M.; St-Louis, J. Vasorelaxant action of 17 -estradiol in rat uterine arteries: Role of nitric oxide synthases and estrogen receptors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3713–H3719. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Roy, T. Large conductance Ca2+-activated and voltage-activated K+ channels contribute to the rise and maintenance of estrogen-induced uterine vasodilation and maintenance of blood pressure. Endocrinology 2012, 153, 6012–6020. [Google Scholar] [CrossRef]
- Hu, X.Q.; Xiao, D.; Zhu, R.; Huang, X.; Yang, S.; Wilson, S.; Zhang, L. Pregnancy upregulates large-conductance Ca(2+)-activated K(+) channel activity and attenuates myogenic tone in uterine arteries. Hypertension 2011, 58, 1132–1139. [Google Scholar] [CrossRef]
- Hu, X.Q.; Song, R.; Romero, M.; Dasgupta, C.; Huang, X.; Holguin, M.A.; Williams, V.; Xiao, D.; Wilson, S.M.; Zhang, L. Pregnancy Increases Ca2+ Sparks/Spontaneous Transient Outward Currents and Reduces Uterine Arterial Myogenic Tone. Hypertension 2019, 73, 691–702. [Google Scholar] [CrossRef]
- Hu, X.Q.; Dasgupta, C.; Chen, M.; Xiao, D.; Huang, X.; Han, L.; Yang, S.; Xu, Z.; Zhang, L. Pregnancy Reprograms Large-Conductance Ca2+-Activated K+ Channel in Uterine Arteries: Roles of Ten-Eleven Translocation Methylcytosine Dioxygenase 1-Mediated Active Demethylation. Hypertension 2017, 69, 1181–1191. [Google Scholar] [CrossRef]
- Pertegal, M.; Fenoy, F.J.; Hernàndez, M.; Mendiola, J.; Delgado, J.L.; Bonacasa, B.; Corno, A.; Lòpez, B.; Bosch, V.; Hernàndez, I. Fetal Val108/158Met catechol-O-methyltransferase (COMT) polymorphism and placental COMT activity are associated with the development of preeclampsia. Fertil. Steril. 2016, 105, 134–143. [Google Scholar] [CrossRef]
- Shen, Z.; Wu, Y.; Chen, X.; Chang, X.; Zhou, Q.; Zhou, J.; Ying, H.; Zheng, J.; Duan, T.; Wang, K. Decreased maternal serum 2-methoxyestradiol levels are associated with the development of preeclampsia. Cell Physiol. Biochem. 2014, 34, 2189–2199. [Google Scholar] [CrossRef]
- Hahnel, M.E.; Martin, J.D.; Michael, C.A.; Hahnel, R. Metabolism of androstenedione by placental microsomes in pregnancy hypertension. Clin. Chim. Acta 1989, 181, 103–108. [Google Scholar] [CrossRef]
- Shimodaira, M.; Nakayama, T.; Sato, I.; Sato, N.; Izawa, N.; Mizutani, Y.; Furuya, K.; Yamamoto, T. Estrogen synthesis genes CYP19A1, HSD3B1, and HSD3B2 in hypertensive disorders of pregnancy. Endocrine 2012, 42, 700–707. [Google Scholar] [CrossRef]
- Morisset, A.S.; Dubè, M.C.; Drolet, R.; Pelletier, M.; Labrie, F.; Luu-The, V.; Tremblay, Y.; Robitaille, J.; John Weisnagel, S.; Tchernof, A. Androgens in the maternal and fetal circulation: Association with insulin resistance. J. Matern. Fetal Neonatal Med. 2013, 26, 513–519. [Google Scholar] [CrossRef]
- Nestler, J.E. Modulation of aromatase and P450 cholesterol sidechain cleavage enzyme activities of human placental cytotrophoblasts by insulin and insulin-like growth factor I. Endocrinology 1987, 121, 1845–1852. [Google Scholar] [CrossRef]
- Turgut, A.; Ozler, A.; Goruk, N.Y.; Tunç, S.Y.; Sak, M.E.; Evsen, M.S.; Evliyaoglu, O.; Gul, T. Serum levels of the adipokines, free fatty acids, and oxidative stress markers in obese and non-obese preeclamptic patients. Clin. Exp. Obstet. Gynecol. 2015, 42, 473–479. [Google Scholar]
- Coya, R.; Martul, P.; Algorta, J.; Aniel-Quiroga, M.A.; Busturia, M.A.; Senaris, R. Effect of leptin on the regulation of placental hormone secretion in cultured human placental cells. Gynecol. Endocrinol. 2006, 22, 620–626. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Lefevre, G.; Alfaidy, N.; Nahed, R.A.; Vincent, J.; Oudinet, J.P.; Pianos, A.; Cambourg, A.; Rozenberg, P.; et al. Abnormal steroidogenesis and aromatase activity in preeclampsia. Placenta 2018, 69, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sepulveda, A.; Monteiro, L.J.; Dobierzewska, A.; España-Perrot, P.P.; Venegas-Araneda, P.; Guzmán-Rojas, A.M.; González, M.I.; Palominos-Rivera, M.; Irarrazabal, C.E.; Figueroa-Diesel, H.; et al. Placental Aromatase Is Deficient in Placental Ischemia and Preeclampsia. PLoS ONE 2015, 7, e0139682. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Mendelson, C.R. USF1 and USF2 mediate inhibition of human trophoblast differentiation and CYP19 gene expression by Mash-2 and hypoxia. Mol. Cell Biol. 2003, 23, 6117–6128. [Google Scholar] [CrossRef] [PubMed]
- Hertig, A.; Liere, P.; Chabbert-Buffet, N.; Fort, J.; Pianos, A.; Eychenne, B.; Cambourg, A.; Schumacher, M.; Berkane, N.; Lefevre, G.; et al. Steroid profiling in preeclamptic women: Evidence for aromatase deficiency. Am. J. Obstet. Gynecol. 2010, 203, 477. [Google Scholar] [CrossRef] [PubMed]
- Aberdeen, G.W.; Baschat, A.A.; Harman, C.R.; Weiner, C.P.; Langenberg, P.W.; Pepe, G.J.; Albrecht, E.D. Uterine and fetal blood flow indexes and fetal growth assessment after chronic estrogen suppression in the second half of baboon pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H881–H889. [Google Scholar] [CrossRef]
- Stamilio, D.M.; Sehdev, H.M.; Morgan, M.A.; Propert, K.; Macones, G.A. Can antenatal clinical and biochemical markers predict the development of severe preeclampsia? Am. J. Obstet. Gynecol. 2000, 182, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Babic, G.M.; Markovic, S.D.; Varjacic, M.; Djordjevic, N.Z.; Nikolic, T.; Stojic, I.; Jakovljevic, V. Estradiol decreases blood pressure in association with redox regulation in preeclampsia. Clin. Exp. Hypertens. 2018, 40, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Djordjević, N.Z.; Babić, G.M.; Marković, S.D.; Ognjanović, B.I.; Stajn, A.S.; Saicić, Z.S. The antioxidative effect of estradiol therapy on erythrocytes in women with preeclampsia. Reprod. Toxicol. 2010, 29, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Bukovsky, A.; Cekanova, M.; Caudle, M.R.; Wimalasena, J.; Foster, J.S.; Henley, D.C.; Elder, R.F. Expression and localization of estrogen receptor-alpha protein in normal and abnormal term placentae and stimulation of trophoblast differentiation by estradiol. Reprod. Biol. Endocrinol. 2003, 1, 13. [Google Scholar] [CrossRef]
- Malassine, A.; Cronier, L. Hormones and human trophoblast differentiation: A review. Endocrine 2002, 19, 3–11. [Google Scholar] [CrossRef]
- Dasgupta, C.; Chen, M.; Zhang, H.; Yang, S.; Zhang, L. Chronic hypoxia during gestation causes epigenetic repression of the estrogen receptor-a gene in ovine uterine arteries via heightened promoter methylation. Hypertension 2012, 60, 697–704. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molvarec, A.; Vèr, A.; Fekete, A.; Rosta, K.; Derzbach, L.; Derzsy, Z.; Karàdi, I.; Rigò, J., Jr. Association between estrogen receptor alpha (ESR1) gene polymorphisms and severe preeclampsia. Hypertens. Res. 2007, 30, 205–211. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Bai, H.; Liu, X.; Fan, P.; Liu, R.; Huang, Y.; Wang, X.; He, G.; Liu, Y.; Liu, B. Genotype distribution of estrogen receptor alpha polymorphisms in pregnant women from healthy and preeclampsia populations and its relation to blood pressure levels. Clin. Chem. Lab. Med. 2009, 47, 391–397. [Google Scholar] [CrossRef]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts Up-Regulate Soluble Fms-Like Tyrosine Kinase-1 Expression under Reduced Oxygen: An Implication for the Placental Vascular Development and the Pathophysiology of Preeclampsia. Endocrinology 2004, 145, 4838–4845. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Brockelsby, J.; Hayman, R.; Ahmed, A.; Warren, A.; Johnson, I.; Baker, P. VEGF via VEGF receptor-1 (Flt-1) mimics preeclamptic plasma in inhibiting uterine blood vessel relaxation in pregnancy: Implications in the pathogenesis of preeclampsia. Lab. Invest. 1999, 79, 1101–1111. [Google Scholar] [PubMed]
- Khankin, E.V.; Mandalà, M.; Colton, I.; KarumanchI, S.A.; Osol, G. Hemodynamic, vascular, and reproductive impact of FMS-like tyrosine kinase 1 (FLT1) blockade on the uteroplacental circulation during normal mouse pregnancy. Biol. Reprod. 2012, 86, 57. [Google Scholar] [CrossRef] [PubMed]
- Savvidou, M.D.; Yu, C.K.; Harland, L.C.; Hingorani, A.D.; Nicolaides, K.H. Maternal serum concentration of soluble fms-like tyrosine kinase 1 and vascular endothelial growth factor in women with abnormal uterine artery Doppler and in those with fetal growth restriction. Am. J. Obstet. Gynecol. 2006, 195, 1668–1673. [Google Scholar] [CrossRef]
- Schlembach, D.; Wallner, W.; Sengenberger, R.; Stiegler, E.; Mörtl, M.; Beckmann, M.W.; Lang, U. Angiogenic growth factor levels in maternal and fetal blood: Correlation with Doppler ultrasound parameters in pregnancies complicated by pre-eclampsia and intrauterine growth restriction. Ultrasound Obstet. Gynecol. 2007, 29, 407–413. [Google Scholar] [CrossRef]
- Lin, C.; He, H.; Cui, N.; Ren, Z.; Zhu, M.; Khalil, R.A. Decreased uterine vascularization and uterine arterial expansive remodeling with reduced matrix metalloproteinase-2 and -9 in hypertensive pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H165–H180. [Google Scholar] [CrossRef]
- Li, W.; Mata, K.M.; Mazzuza, M.Q.; Khalil, R.A. Altered matrix metalloproteinase-2 and -9 expression/activity links placental ischemia and anti-angiogenic sFlt-1 to uteroplacental and vascular remodeling and collagen deposition in hypertensive pregnancy. Biochem. Pharmacol. 2014, 89, 370–385. [Google Scholar] [CrossRef]
- Ren, Z.; Cui, N.; Zhu, M.; Khalil, R.A. Placental growth factor reverses decreased vascular and uteroplacental MMP-2 and MMP-9 and increased MMP-1 and MMP-7 and collagen types I and iV in hypertensive pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H33–H47. [Google Scholar] [CrossRef]
- Kublickiene, K.R.; Lindblom, B.; Krüger, K.; Nisell, H. Preeclampsia: Evidence for impaired shear stress-mediated nitric oxide release in uterine circulation. Am. J. Obstet. Gynecol. 2000, 183, 160–166. [Google Scholar] [CrossRef]
- He, M.; Li, F.; Yang, M.; Fan, Y.; Beejadhursing, R.; Xie, Y.; Zhou, Y.; Deng, D. Impairment of BKca channels in human placental chorionic plate arteries is potentially relevant to the development of preeclampsia. Hypertens. Res. 2018, 41, 126–134. [Google Scholar] [CrossRef]
- Pascoal, I.F.; Lindheimer, M.D.; Nalbantian-Brandt, C.; Umans, J.G. Preeclampsia selectively impairs endothelium-dependent relaxation and leads to oscillatory activity in small omental arteries. J. Clin. Invest. 1998, 101, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Zhang, L. Chronic hypoxia differentially up-regulates protein kinase C-mediated ovine uterine arterial contraction via actin polymerization signaling in pregnancy. Biol. Reprod. 2012, 87, 142. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhu, R.; Zhang, L. Gestational hypoxia up-regulates protein kinase C and inhibits calcium-activated potassium channels in ovine uterine arteries. Int. J. Med. Sci. 2014, 11, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Hu, X.Q.; Xiao, D.; Yang, S.; Wilson, S.M.; Longo, L.D.; Zhang, L. Chronic Hypoxia Inhibits Pregnancy-Induced Upregulation of SKCa Channel Expression and Function in Uterine Arteries. Hypertension 2013, 62, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xiao, D.; Hu, X.Q.; Dasgupta, C.; Yang, S.; Zhang, L. Hypoxia Represses ER-α Expression and Inhibits Estrogen-Induced Regulation of Ca2+-Activated K+ Channel Activity and Myogenic Tone in Ovine Uterine Arteries: Causal Role of DNA Methylation. Hypertension 2015, 66, 44–51. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandalà, M. Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies. Int. J. Mol. Sci. 2020, 21, 2592. https://doi.org/10.3390/ijms21072592
Mandalà M. Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies. International Journal of Molecular Sciences. 2020; 21(7):2592. https://doi.org/10.3390/ijms21072592
Chicago/Turabian StyleMandalà, Maurizio. 2020. "Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies" International Journal of Molecular Sciences 21, no. 7: 2592. https://doi.org/10.3390/ijms21072592
APA StyleMandalà, M. (2020). Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies. International Journal of Molecular Sciences, 21(7), 2592. https://doi.org/10.3390/ijms21072592