JNK Signaling in Stem Cell Self-Renewal and Differentiation

 , ,
, , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
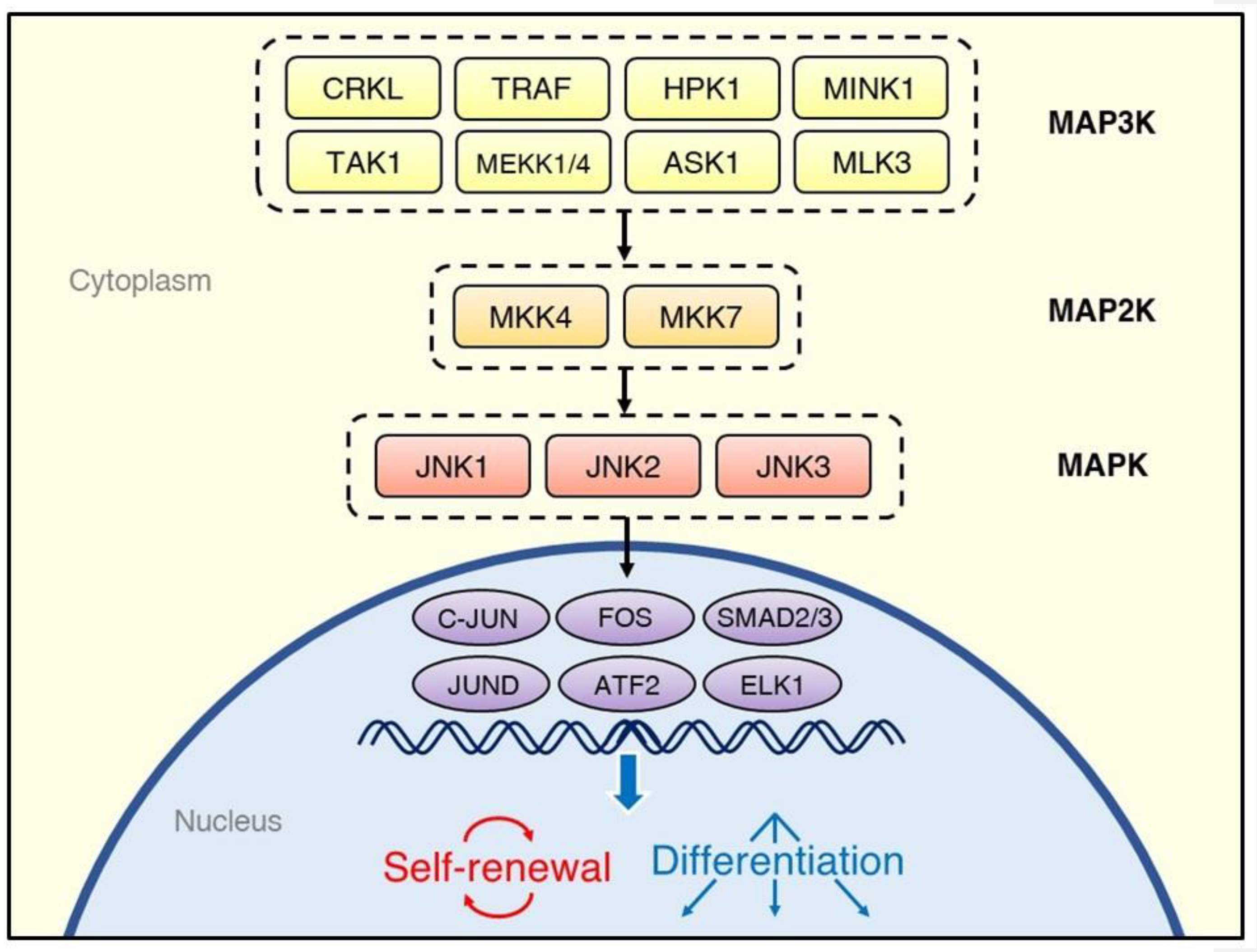
:1. Introduction
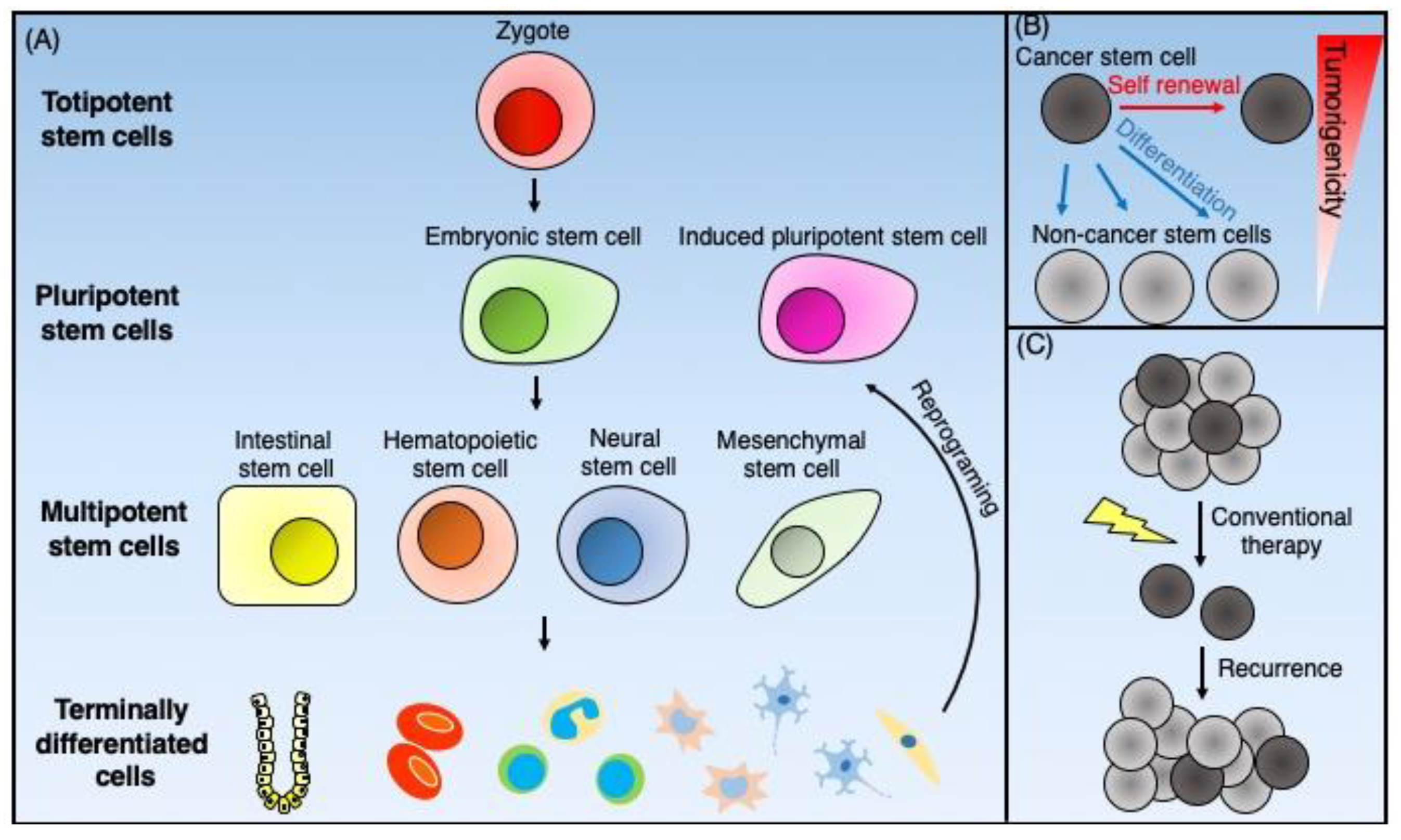
2. Overview of Stem Cells
2.1. Normal Stem Cells
2.2. Cancer Stem Cells
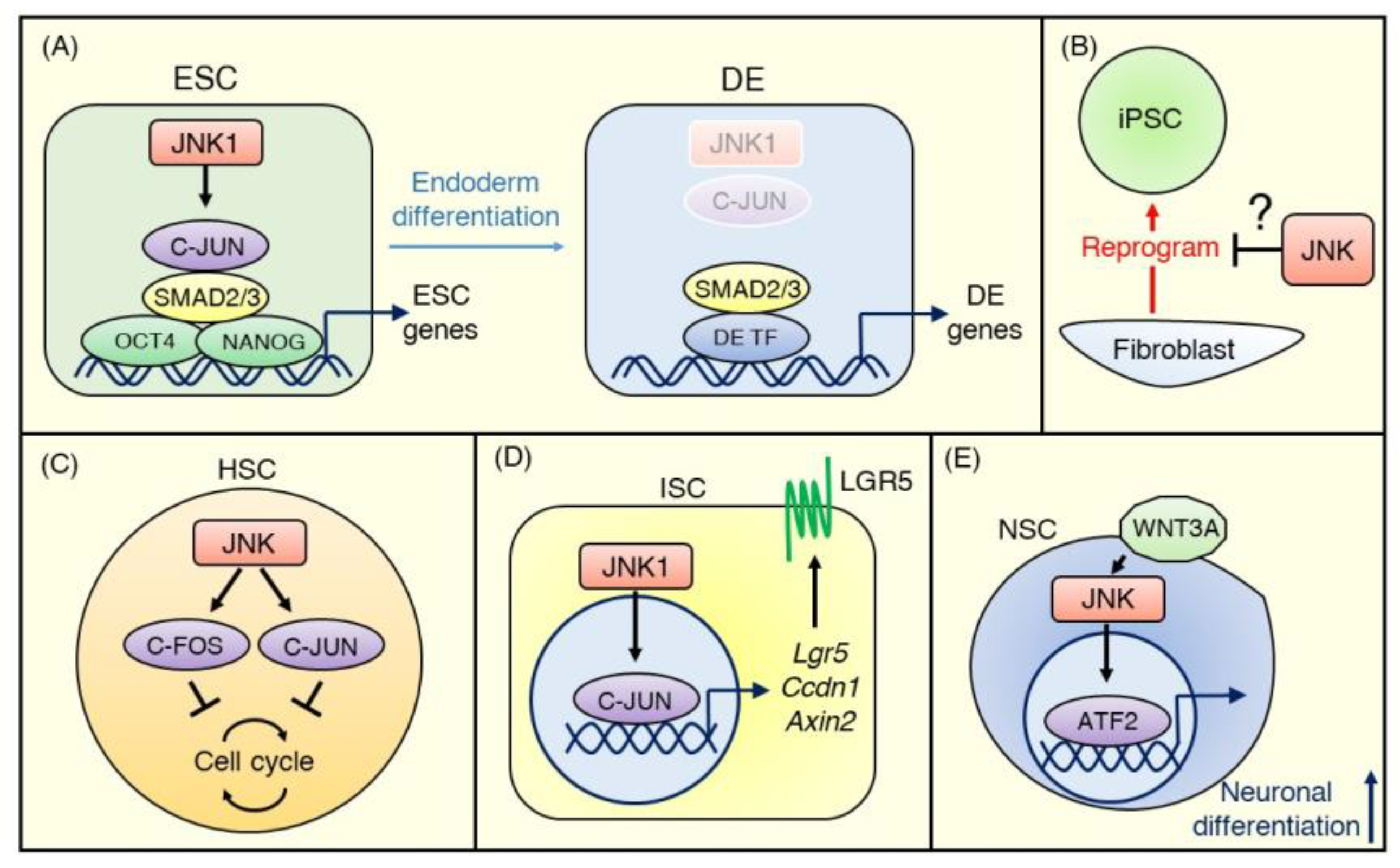
3. JNK Signaling in Pluripotent Stem Cells
3.1. Embryonic Stem Cells
3.2. Induced Pluripotent Stem Cells
4. JNK Signaling in Adult Tissue-Specific Stem Cells
4.1. Hematopoietic Stem Cells
4.2. Intestinal Stem Cells
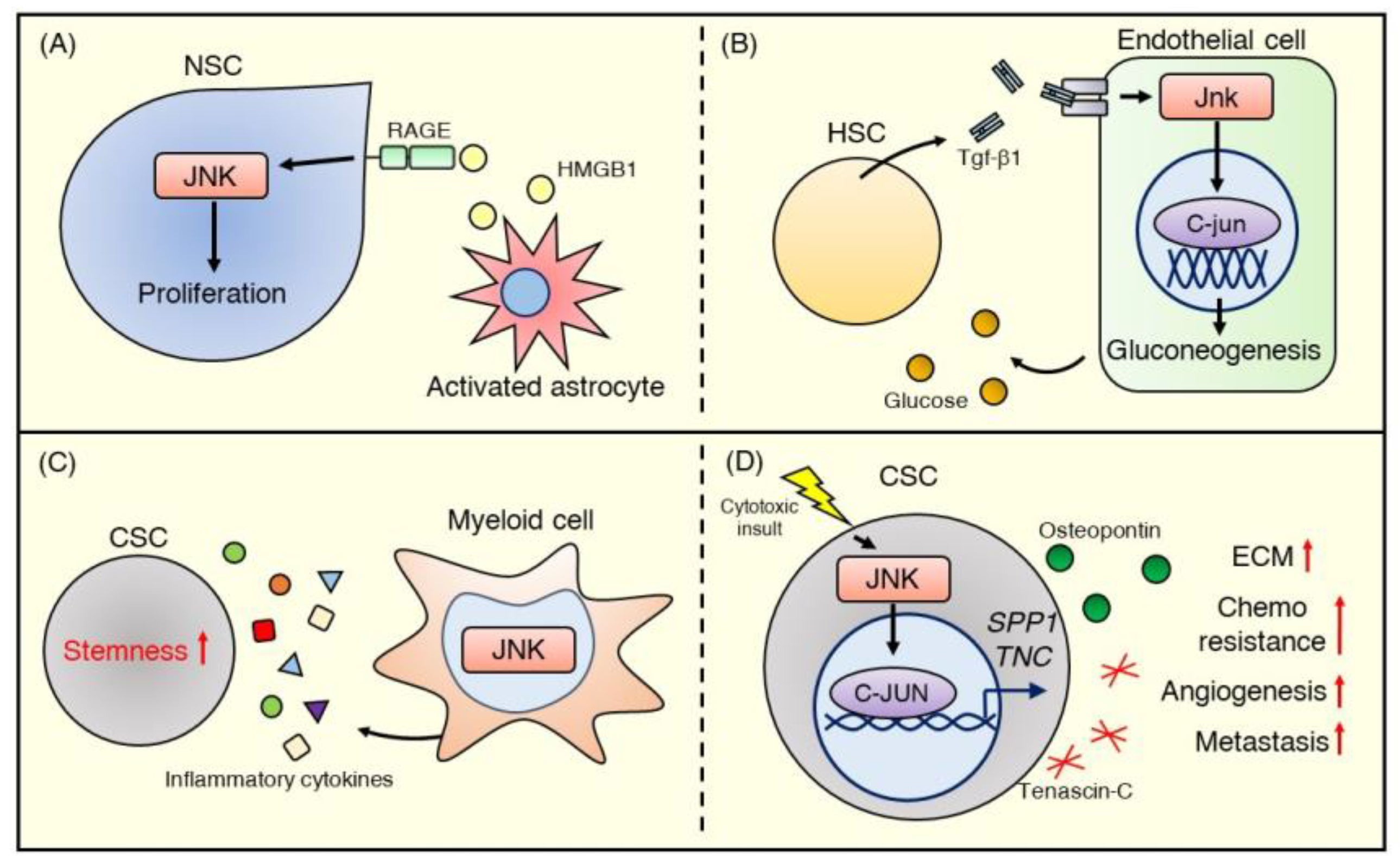
4.3. Neural Stem Cells
5. JNK Signaling in Cancer Stem Cells
6. JNK Signaling in Regulation of Stem Cell Niche Crosstalk
6.1. Crosstalk between the Stem Cell Niche and Normal Stem Cells
6.2. Crosstalk between the Tumor Microenvironment and Cancer Stem Cells
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH1 | Aldehyde dehydrogenase 1 |
| CSCs | Cancer stem cells |
| DE | Definitive endoderm |
| ECM | Extracellular matrix |
| ESCs | Embryonic stem cells |
| hESCs | Human embryonic stem cells |
| HSCs | Hematopoietic stem cells |
| iPSCs | Induced pluripotent stem cells |
| ISCs | Intestinal stem cells |
| JNKKO | Wap-Cre+/−:Trp53LoxP/LoxP:Jnk1LoxP/LoxP:Jnk2LoxP/LoxP |
| JNKs | c-Jun N-terminal kinases |
| JNKWT | Wap-Cre+/−:Trp53LoxP/LoxP |
| MAPK | Mitogen-activated protein kinase |
| mESCs | Murine embryonic stem cells |
| MSCs | Mesenchymal stromal cells |
| NSCs | Neural stem cells |
| SPP1 | Osteopontin |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| TNC | Tenascin C |
References
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Moriguchi, T.; Koyasu, S.; Nishida, E. T lymphocyte activation signals for interleukin-2 production involve activation of MKK6-p38 and MKK7-SAPK/JNK signaling pathways sensitive to cyclosporin A. J. Biol. Chem. 1998, 273, 12378–12382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, S.K.; Falasca, M.; Hu, P.; Schlessinger, J. Phosphatidylinositol 3-kinase mediates epidermal growth factor-induced activation of the c-Jun N-terminal kinase signaling pathway. Mol. Cell. Biol. 1997, 17, 5784–5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururajan, M.; Chui, R.; Karuppannan, A.K.; Ke, J.; Jennings, C.D.; Bondada, S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood 2005, 106, 1382–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Yan, D.P.; Ge, B.X. JNK regulates cell migration through promotion of tyrosine phosphorylation of paxillin. Cell Signal. 2008, 20, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes. Cancer 2017, 8, 682–694. [Google Scholar]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Yang, T.; Xu, Z. The JNK Pathway and Neuronal Migration. J. Genet. Genom. 2007, 34, 957–965. [Google Scholar] [CrossRef]
- Koehler, K.; Mielke, K.; Schunck, M.; Neumann, C.; Herdegen, T.; Proksch, E. Distinct roles of JNK-1 and ERK-2 isoforms in permeability barrier repair and wound healing. Eur. J. Cell Biol. 2011, 90, 565–571. [Google Scholar] [CrossRef]
- Sabapathy, K.; Hu, Y.; Kallunki, T.; Schreiber, M.; David, J.P.; Jochum, W.; Wagner, E.F.; Karin, M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr. Biol. 1999, 11, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Tuncman, G.; Hirosumi, J.; Solinas, G.; Chang, L.F.; Karin, M.; Hotamisligil, G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 10741–10746. [Google Scholar] [CrossRef] [Green Version]
- Cellurale, C.; Sabio, G.; Kennedy, N.J.; Das, M.; Barlow, M.; Sandy, P.; Jacks, T.; Davis, R.J. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol. Cell. Biol. 2011, 31, 1565–1576. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [Green Version]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [Green Version]
- Cobb, M.H.; Goldsmith, E.J. How Map Kinases Are Regulated. J. Boil. Chem. 1995, 270, 14843–14846. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.D.; Gutowski, S.; Sternweis, P.C.; Cobb, M.H. RhoA binds to the amino terminus of MEKK1 and regulates its kinase activity. J. Biol. Chem. 2004, 279, 1872–1877. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.-C.; Shahinian, A.; Speiser, D.; Kraunus, J.; Billia, F.; Wakeham, A.; de la Pompa, J.L.; Ferrick, D.; Hum, B.; Iscove, N.; et al. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 1997, 7, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Bunkoczi, G.; Salah, E.; Filippakopoulos, P.; Fedorov, O.; Muller, S.; Sobott, F.; Parker, S.A.; Zhang, H.; Min, W.; Turk, B.E.; et al. Structural and functional characterization of the human protein kinase ASK1. Structure 2007, 15, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Kiefer, F.; Tibbles, L.A.; Anafi, M.; Janssen, A.; Zanke, B.W.; Lassam, N.; Pawson, T.; Woodgett, J.R.; Iscove, N.N. HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 1996, 15, 7013–7025. [Google Scholar] [CrossRef]
- Teramoto, H.; Coso, O.A.; Miyata, H.; Igishi, T.; Miki, T.; Gutkind, J.S. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 1996, 271, 27225–27228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, Y.; Armstrong, C.G.; Morrice, N.; Paterson, A.; Goedert, M.; Cohen, P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem. J. 2000, 352, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Kyriakis, J.M.; Avruch, J.; Nikolakaki, E.; Woodgett, J.R. Phosphorylation of c-jun mediated by MAP kinases. Nature 1991, 353, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Yazgan, O.; Pfarr, C.M. Regulation of two JunD isoforms by Jun N-terminal kinases. J. Biol. Chem. 2002, 277, 29710–29718. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Campbell, D.; Derijard, B.; Davis, R.J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995, 267, 389–393. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Shore, P.; Sharrocks, A.D.; Davis, R.J. Integration of MAP kinase signal transduction pathways at the serum response element. Science 1995, 269, 403–407. [Google Scholar] [CrossRef]
- Wu, J.; Izpisua Belmonte, J.C. Stem Cells: A Renaissance in Human Biology Research. Cell 2016, 165, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Ganiatsas, S.; Kwee, L.; Fujiwara, Y.; Perkins, A.; Ikeda, T.; Labow, M.A.; Zon, L.I. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 6881–6886. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yang, D.D.; Tournier, C.; Whitmarsh, A.J.; Xu, J.; Davis, R.J.; Flavell, R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature 2000, 405, 91–94. [Google Scholar] [CrossRef]
- Hilberg, F.; Aguzzi, A.; Howells, N.; Wagner, E.F. c-jun is essential for normal mouse development and hepatogenesis. Nature 1993, 365, 179–181. [Google Scholar] [CrossRef]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 2008, 3, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Sancho, R.; Nateri, A.S.; de Vinuesa, A.G.; Aguilera, C.; Nye, E.; Spencer-Dene, B.; Behrens, A. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009, 28, 1843–1854. [Google Scholar] [CrossRef] [Green Version]
- Mundorf, J.; Donohoe, C.D.; McClure, C.D.; Southall, T.D.; Uhlirova, M. Ets21c governs tissue renewal, stress tolerance, and aging in the Drosophila intestine. Cell Rep. 2019, 27, 3019–3033. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef]
- Zhao, W.; Ji, X.; Zhang, F.; Li, L.; Ma, L. Embryonic stem cell markers. Molecules 2012, 17, 6196–6236. [Google Scholar] [CrossRef]
- Mitalipov, S.; Wolf, D. Advances in Biochemical Engineering/Biotechnology; Scheper, T., Martin, U., Eds.; Springer Science+Business Media: Heidelberg, Germany, 2009; pp. 185–199. [Google Scholar]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7698. [Google Scholar] [CrossRef] [Green Version]
- Lumelsky, N.; Blondel, O.; Laeng, P.; Velasco, I.; Ravin, R.; McKay, R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001, 292, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Chinzei, R.; Tanaka, Y.; Shimizu-Saito, K.; Hara, Y.; Kakinuma, S.; Watanabe, M.; Teramoto, K.; Arii, S.; Takase, K.; Sato, C.; et al. Embryoid-body cells derived from a mouse embryonic stem cell line show differentiation into functional hepatocytes. Hepatology 2002, 36, 22–29. [Google Scholar] [CrossRef]
- Brustle, O.; Spiro, A.C.; Karram, K.; Choudhary, K.; Okabe, S.; McKay, R.D. In vitro-generated neural precursors participate in mammalian brain development. Proc. Natl. Acad. Sci. USA 1997, 94, 14809–14814. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, M.; Kataoka, H.; Nishikawa, S.; Matsuyoshi, N.; Nishikawa, S.I. Maturation of embryonic stem cells into endothelial cells in an in vitro model of vasculogenesis. Blood 1999, 93, 1253–1263. [Google Scholar] [CrossRef]
- Klug, M.G.; Soonpaa, M.H.; Koh, G.Y.; Field, L.J. Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J. Clin. Investig. 1996, 98, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.; Firpo, M.; Choi, K.; Wall, C.; Robertson, S.; Kabrun, N.; Keller, G. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature 1997, 386, 488–493. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sato, H.; Kato-Itoh, M.; Goto, T.; Hara, H.; Sanbo, M.; Mizuno, N.; Kobayashi, T.; Yanagida, A.; Umino, A.; et al. Interspecies organogenesis generates autologous functional islets. Nature 2017, 542, 191–196. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Park, T.S.; Bhutto, I.; Zimmerlin, L.; Huo, J.S.; Nagaria, P.; Miller, D.; Rufaihah, A.J.; Talbot, C.; Aguilar, J.; Grebe, R.; et al. Vascular progenitors from cord blood-derived induced pluripotent stem cells possess augmented capacity for regenerating ischemic retinal vasculature. Circulation 2014, 129, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Slack, J.M. Origin of stem cells in organogenesis. Science 2008, 322, 1498–1501. [Google Scholar] [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, R.; Semba, T.; Saya, H.; Arima, Y. Stem cells and epithelial-mesenchymal transition (EMT) in cancer: Biological implications and therapeutic targets. Stem Cells 2016, 34, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [Green Version]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.G.; Lee, J.J.; Jung, D.Y.; Jeon, J.; Heo, H.S.; Kang, H.C.; Shin, J.H.; Cho, Y.S.; Cha, K.J.; Kim, C.G.; et al. Profiling of differentially expressed genes in human stem cells by cDNA microarray. Mol. Cells 2006, 21, 343–355. [Google Scholar]
- Brill, L.M.; Xiong, W.; Lee, K.B.; Ficarro, S.B.; Crain, A.; Xu, Y.; Terskikh, A.; Snyder, E.Y.; Ding, S. Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 2009, 5, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [Green Version]
- Holzberg, D.; Knight, C.G.; Dittrich-Breiholz, O.; Schneider, H.; Dorrie, A.; Hoffmann, E.; Resch, K.; Kracht, M. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J. Biol. Chem. 2003, 278, 40213–40223. [Google Scholar] [CrossRef] [Green Version]
- Colombo, R.; Caldarelli, M.; Mennecozzi, M.; Giorgini, M.L.; Sola, F.; Cappella, P.; Perrera, C.; Depaolini, S.R.; Rusconi, L.; Cucchi, U.; et al. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010, 70, 10255–10264. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Davis, R.J. c-Jun NH2-terminal kinase is required for lineage-specific differentiation but not stem cell self-renewal. Mol. Cell Biol. 2010, 30, 1329–1340. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.V.; Dixon, G.; Verma, N.; Rosen, B.P.; Gordillo, M.; Luo, R.; Xu, C.; Wang, Q.; Soh, C.L.; Yang, D.; et al. Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat. Genet. 2019, 51, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.; Ki, M.O.; Chen, H.; Cho, Y.Y.; Kim, S.H.; Yu, D.H.; Lee, S.Y.; Lee, K.Y.; Bae, K.; Peng, C.; et al. JNK1 and 2 play a negative role in reprogramming to pluripotent stem cells by suppressing Klf4 activity. Stem Cell Res. 2014, 12, 139–152. [Google Scholar] [CrossRef]
- Neganova, I.; Shmeleva, E.; Munkley, J.; Chichagova, V.; Anyfantis, G.; Anderson, R.; Passos, J.; Elliott, D.J.; Armstrong, L.; Lako, M. JNK/SAPK signaling is essential for efficient reprogramming of human fibroblasts to induced pluripotent stem cells. Stem Cells 2016, 34, 1198–1212. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Okada, S.; Fukuda, T.; Inada, K.; Tokuhisa, T. Prolonged expression of c-fos suppresses cell cycle entry of dormant hematopoietic stem cells. Blood 1999, 93, 816–825. [Google Scholar] [CrossRef]
- Xiao, X.; Lai, W.; Xie, H.; Liu, Y.; Guo, W.; Liu, Y.; Li, Y.; Li, Y.; Zhang, J.; Chen, W.; et al. Targeting JNK pathway promotes human hematopoietic stem cell expansion. Cell Discov. 2019, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Tian, A.; Jiang, J. Intestinal stem cell response to injury: Lessons from Drosophila. Cell. Mol. Life Sci. 2016, 73, 3337–3349. [Google Scholar] [CrossRef]
- Bond, A.M.; Ming, G.L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Bengoa-Vergniory, N.; Gorrono-Etxebarria, I.; Gonzalez-Salazar, I.; Kypta, R.M. A switch from canonical to noncanonical Wnt signaling mediates early differentiation of human neural stem cells. Stem Cells 2014, 32, 3196–3208. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.Z.; Yu, S.P.; Lee, J.H.; Chen, D.; Taylor, T.M.; Deveau, T.C.; Yu, A.C.; Wei, L. Regulatory role of the JNK-STAT1/3 signaling in neuronal differentiation of cultured mouse embryonic stem cells. Cell. Mol. Neurobiol. 2014, 34, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmarsh, A.J.; Davis, R.J. Role of mitogen-activated protein kinase kinase 4 in cancer. Oncogene 2007, 26, 3172–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramek, D.; Kotsinas, A.; Meixner, A.; Wada, T.; Elling, U.; Pospisilik, J.A.; Neely, G.G.; Zwick, R.H.; Sigl, V.; Forni, G.; et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat. Genet. 2011, 43, 212–219. [Google Scholar] [CrossRef]
- Lee, J.J.; Lee, J.H.; Ko, Y.G.; Hong, S.I.; Lee, J.S. Prevention of premature senescence requires JNK regulation of Bcl-2 and reactive oxygen species. Oncogene 2010, 29, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Girnius, N.; Edwards, Y.J.; Garlick, D.S.; Davis, R.J. The cJUN NH2-terminal kinase (JNK) signaling pathway promotes genome stability and prevents tumor initiation. eLife 2018, 7, e36389. [Google Scholar] [CrossRef]
- Ohta, K.; Haraguchi, N.; Kano, Y.; Kagawa, Y.; Konno, M.; Nishikawa, S.; Hamabe, A.; Hasegawa, S.; Ogawa, H.; Fukusumi, T.; et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int. J. Oncol. 2013, 42, 121–1218. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Kaoud, T.S.; Edupuganti, R.; Zhang, T.; Kogawa, T.; Zhao, Y.; Chauhan, G.B.; Giannoukos, D.N.; Qi, Y.; Tripathy, D.; et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene 2017, 36, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Nasrazadani, A.; Van Den Berg, C.L. c-Jun N-terminal kinase 2 regulates multiple receptor tyrosine kinase pathways in mouse mammary tumor growth and metastasis. Genes. Cancer 2011, 2, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Wang, H.; May, S.; Song, X.; Fueyo, J.; Fuller, G.N.; Wang, H. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J. Neurooncol. 2008, 88, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Kim, M.J.; Kim, R.K.; Lim, E.J.; Choi, K.S.; An, S.; Hwang, S.G.; Kang, S.G.; Suh, Y.; Park, M.J.; et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 2012, 31, 4655–4666. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Han, S.Y.; Wang, C.; Su, W.; Harshyne, L.; Holgado-Madruga, M.; Wong, A.J. c-Jun NH(2)-terminal kinase 2alpha2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 2006, 66, 10024–10031. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Park, C.S.; Suppipat, K.; Mistretta, T.A.; Puppi, M.; Horton, T.M.; Rabin, K.; Gray, N.S.; Meijerink, J.P.P.; Lacorazza, H.D. Inactivation of KLF4 promotes T-cell acute lymphoblastic leukemia and activates the MAP2K7 pathway. Leukemia 2017, 31, 1314–1324. [Google Scholar] [CrossRef]
- Okada, M.; Shibuya, K.; Sato, A.; Seino, S.; Suzuki, S.; Seino, M.; Kitanaka, C. Targeting the K-Ras—JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget 2014, 5, 5100–5112. [Google Scholar] [CrossRef] [Green Version]
- Seino, M.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, S.; Ohta, T.; Kurachi, H.; Kitanaka, C. Requirement of JNK signaling for self-renewal and tumor-initiating capacity of ovarian cancer stem cells. Anticancer Res. 2014, 34, 4723–4731. [Google Scholar]
- Hess, P.; Pihan, G.; Sawyers, C.L.; Flavell, R.A.; Davis, R.J. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nat. Genet. 2002, 32, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, F.; Celso, C.L.; Scadden, D. Advances in Experimental Medicine and Biology; Meshorer, E., Plath, K., Eds.; Springer Science+Business Media: Heidelberg, Germany, 2010; pp. 155–168. [Google Scholar]
- Li, M.; Sun, L.; Luo, Y.; Xie, C.; Pang, Y.; Li, Y. High-mobility group box 1 released from astrocytes promotes the proliferation of cultured neural stem/progenitor cells. Int. J. Mol. Med. 2014, 34, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Y.; Yin, H.M.; Wang, H.; Su, D.; Xia, Y.; Yan, L.F.; Fang, B.; Liu, W.; Wang, Y.M.; Gu, A.H.; et al. Transforming growth factor-beta1 regulates the nascent hematopoietic stem cell niche by promoting gluconeogenesis. Leukemia 2018, 32, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Laboureau, J.; Gabison, E.; Verrecchia, F.; Mauviel, A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J. Biol. Chem. 2003, 278, 24624–24628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mong, P.Y.; Petrulio, C.; Kaufman, H.L.; Wang, Q. Activation of Rho kinase by TNF-alpha is required for JNK activation in human pulmonary microvascular endothelial cells. J. Immunol. 2008, 180, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.; Celia-Terrassa, T.; Kumar, S.; Hang, X.; Wei, Y.; Choudhury, A.; Hwang, J.; Peng, J.; Nixon, B.; Grady, J.J.; et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 2018, 360, eaan4153. [Google Scholar] [CrossRef] [Green Version]
- Himes, S.R.; Sester, D.P.; Ravasi, T.; Cronau, S.L.; Sasmono, T.; Hume, D.A. The JNK are important for development and survival of macrophages. J. Immunol. 2006, 176, 2219–2228. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Borovski, T.; De Sousa, E.; Melo, F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, S.C. Type I collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br. J. Cancer 2009, 101, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation mediated by jnk in myeloid cells promotes the development of hepatitis and hepatocellular carcinoma. Cell Rep. 2016, 15, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insua-Rodriguez, J.; Pein, M.; Hongu, T.; Meier, J.; Descot, A.; Lowy, C.M.; De Braekeleer, E.; Sinn, H.P.; Spaich, S.; Sutterlin, M.; et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 2018, 10, e9003. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Wang, L.; Zhang, C. Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol. Lett. 2018, 16, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Cai, X.; Cao, C.; Li, J.; Chen, F.; Zhang, S.; Liu, B.; Zhang, W.; Zhang, X.; Ye, L. Inflammatory factor TNF-alpha promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-kappaB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget 2017, 8, 58338–58352. [Google Scholar] [PubMed] [Green Version]
- Mahla, R.S. Stem Cells Applications in Regenerative Medicine and Disease Therapeutics. Int. J. Cell Biol. 2016, 2016, 6940283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaoud, T.S.; Mitra, S.; Lee, S.; Taliaferro, J.; Cantrell, M.; Linse, K.D.; Van Den Berg, C.L.; Dalby, K.N. Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chem. Biol. 2011, 6, 658–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Anderson, A.; Nguyen, K.; Ojeda, S.S.; Ortiz-Rivera, I.; Nguyen, T.N.; Zhang, T.; Kaoud, T.S.; Gray, N.S.; Dalby, K.N.; et al. JNK2 Is Required for the Tumorigenic Properties of Melanoma Cells. ACS Chem. Biol. 2019, 14, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting cancer stemness in the clinic: From hype to hope. Cell Stem Cell 2019, 21, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008, 68, 6932–6941. [Google Scholar] [CrossRef] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semba, T.; Sammons, R.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. https://doi.org/10.3390/ijms21072613
Semba T, Sammons R, Wang X, Xie X, Dalby KN, Ueno NT. JNK Signaling in Stem Cell Self-Renewal and Differentiation. International Journal of Molecular Sciences. 2020; 21(7):2613. https://doi.org/10.3390/ijms21072613
Chicago/Turabian StyleSemba, Takashi, Rachel Sammons, Xiaoping Wang, Xuemei Xie, Kevin N. Dalby, and Naoto T. Ueno. 2020. "JNK Signaling in Stem Cell Self-Renewal and Differentiation" International Journal of Molecular Sciences 21, no. 7: 2613. https://doi.org/10.3390/ijms21072613
APA StyleSemba, T., Sammons, R., Wang, X., Xie, X., Dalby, K. N., & Ueno, N. T. (2020). JNK Signaling in Stem Cell Self-Renewal and Differentiation. International Journal of Molecular Sciences, 21(7), 2613. https://doi.org/10.3390/ijms21072613