Traumatic Brain Injury and Blood–Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition


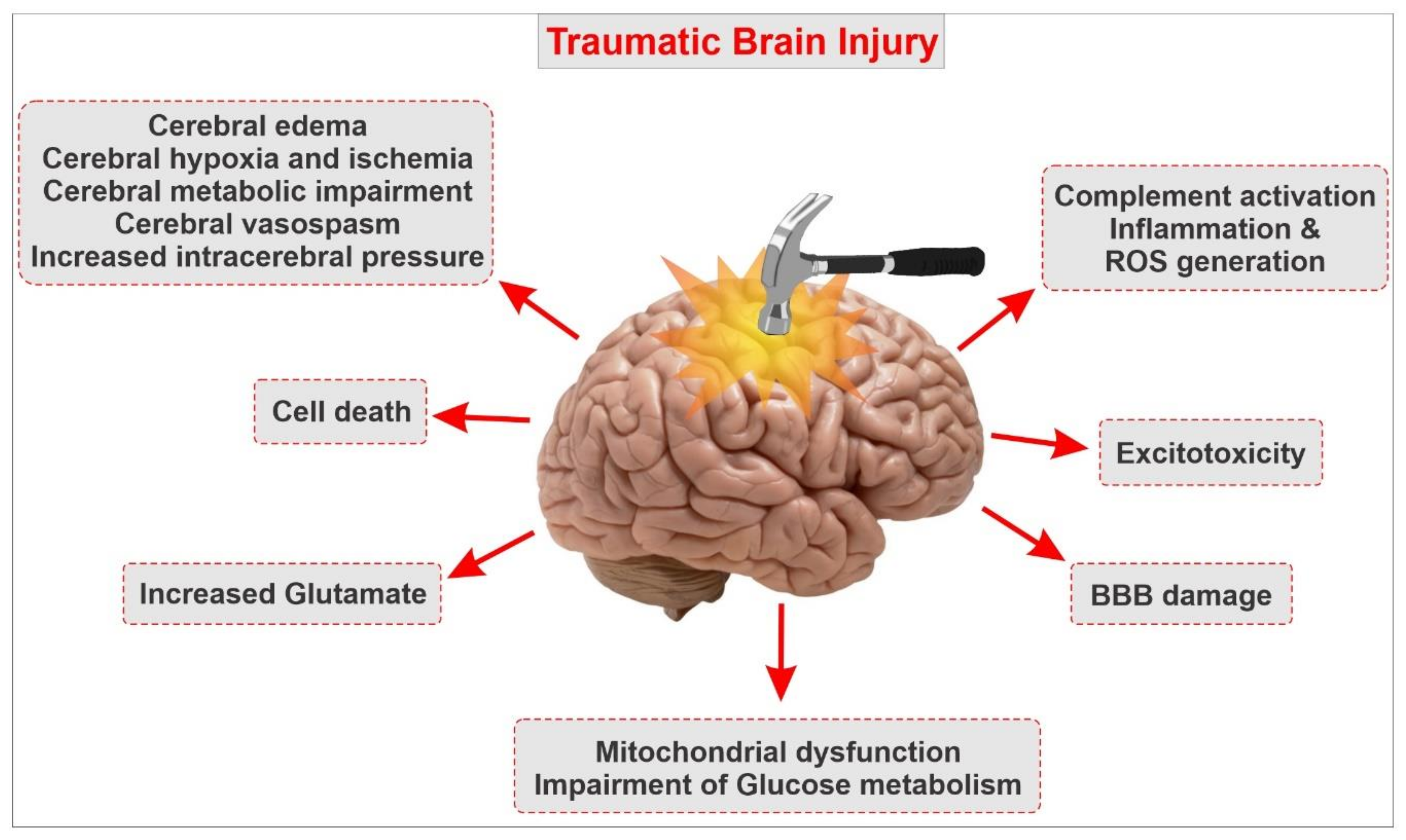
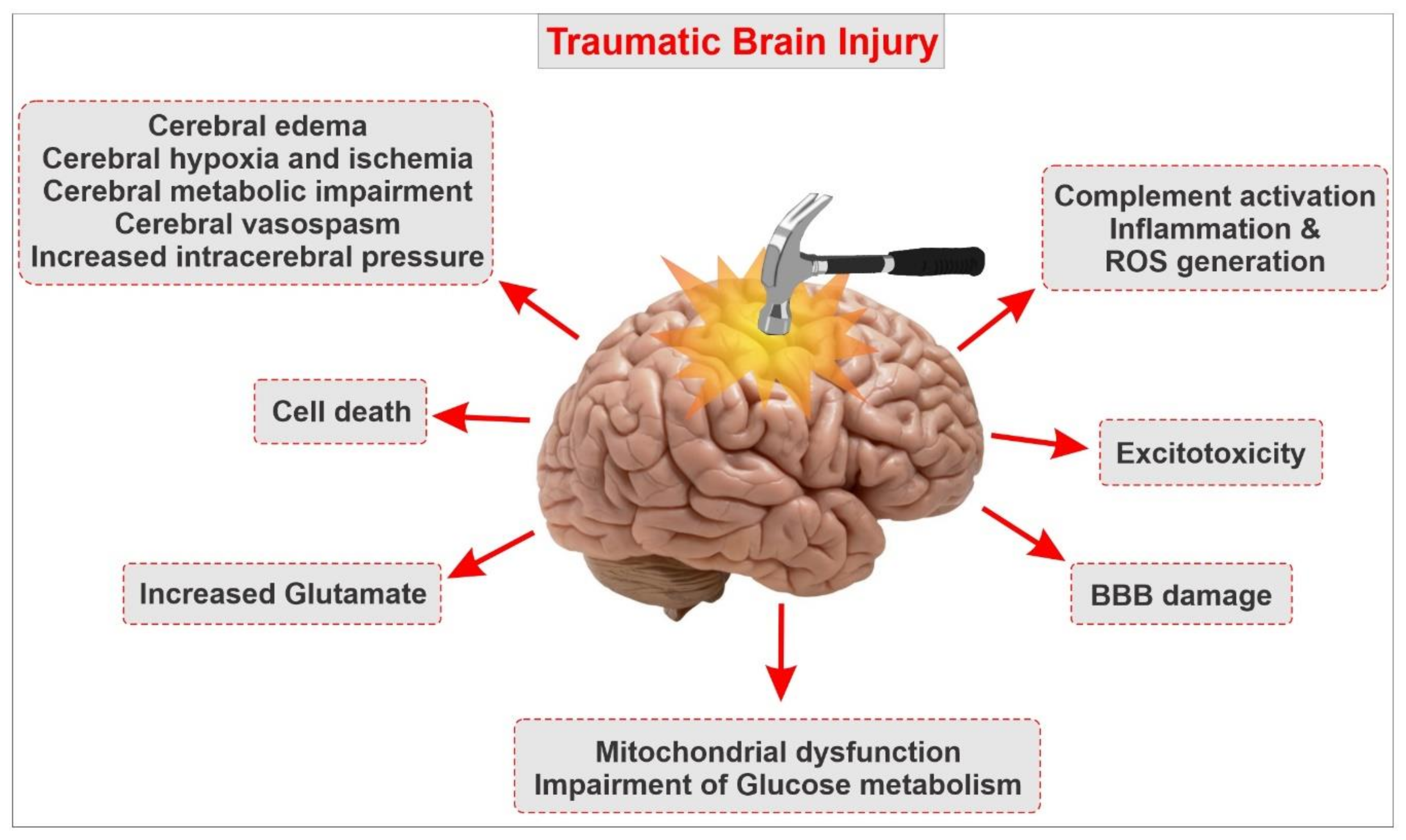{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Blood–Brain Barrier Interface
3. Chronic Smoking: A Major Comorbid Factor for BBB Dysfunction and Significant Neurological Disorders
4. Pathophysiology and Underlying Causes of TBI
5. TBI and Breakdown of the BBB
6. Post-TBI Cell Death Mechanisms
7. Excitotoxicity
8. Neuroinflammation
9. Cerebral Edema Formation
10. Oxidative Stress and Influence of Cigarette Smoking on the Pathophysiology of TBI
11. Current Treatments of TBI
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| AJ | Adherens junctions |
| AMPA | α-amino-3-hydroxy5-methyl-4-isoxazole-propionic acid |
| ApoE | Apo-lipoprotein E |
| BBB | Blood–Brain Barrier |
| BCRP | Breast-cancer-resistance protein |
| CNS | Central nervous system |
| CDC | lefts for Disease Control and Prevention |
| FTC | Federal Trade Control |
| ICAM1 | Intercellular adhesion molecule 1 |
| IL-6 | Interleukin-6 |
| MF | Metformin |
| MMP-2 | Matrix metalloproteinase-2 |
| MRP | Multidrug-resistant protein |
| NAD(P)H | Quinone reductase I |
| NF-ĸB | Nuclear factor kappa-light chain-enhancer of activated B cells |
| NMDA | N-methyl-D-aspartic acid |
| NRF2 | Nuclear factor erythroid 2-related factor |
| OS | Oxidative stress |
| PARP1 | Poly(ADP-Ribose) polymerase 1 |
| PECAM-1 | Platelet Endothelial Cell Adhesion Molecule-1 |
| Pgp | P-glycoprotein |
| ROS | Reactive oxygen species |
| SAA1 | Serum Amyloid A1 |
| SCSM | Single cigarette smoking machine |
| STAT3 | Signal Transducer and Activator Of Transcription-3 |
| SFN | Sulforaphane |
| TBI | Traumatic Brain Injury |
| TGF-β | transforming growth factor β |
| TJ | Tight Junction |
| TNF-α | tumor necrosis factor-alpha |
| TS | Tobacco smoke |
| VCAM1 | Vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
| ZO-1 | Zonula occludens-1 |
References
- Sharma, R.; Shultz, S.R.; Robinson, M.; Belli, A.; Hibbs, M.L.; O’Brien, T.J.; Semple, B.D. Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav. Immun. 2019, 79, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Price, L.; Wilson, C.; Grant, A.G. Blood–brain barrier pathophysiology following traumatic brain injury. In Translational Research in Traumatic Brain Injury; CRC Press: Boca Raton, FL, USA, 2015; pp. 85–96. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. In-vitro blood–brain barrier modeling: A review of modern and fast-advancing technologies. Br. J. Pharmacol. 2018, 38, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Deeb, G.; Rahal, R.; Atwi, K.; Mondello, S.; Mady, H.E.S.M.; Gali, A.; Sleiman, E.; Marei, H.E. Mesenchymal stem cells in the treatment of traumatic brain injury. Front. Neurol. 2017, 8, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, B.D.; Zamani, A.; Rayner, G.; Shultz, S.R.; Jones, N.C. Affective, neurocognitive and psychosocial disorders associated with traumatic brain injury and post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 27–41. [Google Scholar] [CrossRef]
- Laker, S.R. Epidemiology of concussion and mild traumatic brain injury. PM R 2011, 3, S354–S358. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [Green Version]
- De La Tremblaye, P.B.; O’Neil, D.A.; Laporte, M.J.; Cheng, J.P.; Beitchman, J.A.; Thomas, T.C.; Bondi, C.O.; Kline, A.E. Elucidating opportunities and pitfalls in the treatment of experimental traumatic brain injury to optimize and facilitate clinical translation. Neurosci. Biobehav. Rev. 2018, 85, 160–175. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Durazzo, T.C.; Abadjian, L.; Kincaid, A.; Bilovsky-Muniz, T.; Boreta, L.; Gauger, G.E. The influence of chronic cigarette smoking on neurocognitive recovery after mild traumatic brain injury. J. Neurotrauma 2013, 30, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Benady, A.; Freidin, D.; Pick, C.G.; Rubovitch, V. GM1 ganglioside prevents axonal regeneration inhibition and cognitive deficits in a mouse model of traumatic brain injury. Sci. Rep. 2018, 8, 13340. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.W. Neurobiological consequences of traumatic brain injury. Dialog- Clin. Neurosci. 2011, 13, 287–300. [Google Scholar]
- Wanner, I.-B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Englander, J.; Cifu, D.X.; Diaz-Arrastia, R.; Center, M.S.K.T. Seizures after traumatic brain injury. Arch. Phys. Med. Rehabil. 2014, 95, 1223–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, S.; Neuhaus, W. The Blood–Brain Barrier as a Target in Traumatic Brain Injury Treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Amoo, M.; O’Halloran, P.J.; Leo, A.-M.; O’Loughlin, A.; Mahon, P.; Lim, C. Outcomes of emergency neurosurgical intervention in neuro-critical care patients with traumatic brain injury at Cork University Hospital. Br. J. Neurosurg. 2018, 32, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Risacher, S.L.; McAllister, T.W.; Saykin, A.J. Traumatic brain injury and age at onset of cognitive impairment in older adults. J. Neurol. 2016, 263, 1280–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, J.V.; Maas, A.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Sahyouni, R.; Gutierrez, P.; Gold, E.; Robertson, R.T.; Cummings, B.J. Effects of concussion on the blood–brain barrier in humans and rodents. J. Concussion 2017, 1, 2059700216684518. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Boil. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D.; et al. Curcumin plays neuroprotective roles against traumatic brain injury partly via Nrf2 signaling. Toxicol. Appl. Pharmacol. 2018, 346, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Prata, C.; Sega, F.V.D.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: A deep relationship. Oxidative Med. Cell. Longev. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, J.L. Blood–brain barrier and traumatic brain injury. J. Neurosci. Res. 2013, 92, 141–147. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. Anti-diabetic countermeasures against tobacco smoke-dependent cerebrovascular toxicity: Use and effect of rosiglitazone. Int. J. Mol. Sci. 2019, 20, 4225. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.R.; Bruce-Keller, A.J. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta 2011, 1822, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P.; Martinez, J.; Miller, B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 2011, 13, 13. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [Green Version]
- Erdő, F.; Denes, L.; De Lange, E. Age-associated physiological and pathological changes at the blood–brain barrier: A review. Br. J. Pharmacol. 2016, 37, 4–24. [Google Scholar] [CrossRef] [Green Version]
- Broux, B.; Gowing, E.; Prat, A. Glial regulation of the blood-brain barrier in health and disease. Semin. Immunopathol. 2015, 37, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Assessing the protective effect of rosiglitazone against electronic cigarette/tobacco smoke-induced blood–brain barrier impairment. BMC Neurosci. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, P.; Ramasundarahettige, C.; Landsman, V.; Rostron, B.; Thun, M.; Anderson, R.N.; McAfee, T.; Peto, R. 21st-century hazards of smoking and benefits of cessation in the United States. N. Engl. J. Med. 2013, 368, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Naik, P.; Cucullo, L. Differential cerebrovascular toxicity of various tobacco products: A regulatory perspective. J. Pharmacovigil. 2015, 3, 1–2. [Google Scholar] [CrossRef]
- Sajja, R.K.; Rahman, S.; Cucullo, L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. Br. J. Pharmacol. 2015, 36, 539–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrissobolis, S. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733. [Google Scholar] [CrossRef]
- Hossain, M.; Sathe, T.; Fazio, V.; Mazzone, P.; Weksler, B.; Janigro, D.; Rapp, E.; Cucullo, L. Tobacco smoke: A critical etiological factor for vascular impairment at the blood–brain barrier. Brain Res. 2009, 1287, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Aseervatham, S.B.; Choi, S.; Krishnan, J.; Kandasamy, R. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86. [Google Scholar] [CrossRef]
- Geraghty, P.P.; Wyman, A.E.M.; Garcia-Arcos, I.P.; Dabo, A.J.M.; Gadhvi, S.M.; Foronjy, R.M. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front. Physiol. 2013, 4, 267. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Muromoto, R.; Sekine, Y.; Togi, S.; Kitai, Y.; Kon, S.; Oritani, K. Signal transducer and activator of transcription 3 regulation by novel binding partners. World J. Boil. Chem. 2015, 6, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Kryndushkin, D.; Balarezo, M.G.; Campbell, A.M.; Saavedra, J.M.; Shewmaker, F.P.; Symes, A.J. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am. J. Pathol. 2015, 185, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klünemann, H.H.; Schuierer, G.; et al. Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2011, 43, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubovitch, V.; Ten-Bosch, M.; Zohar, O.; Harrison, C.R.; Tempel-Brami, C.; Stein, E.; Hoffer, B.J.; Balaban, C.; Schreiber, S.; Chiu, W.-T.; et al. A mouse model of blast-induced mild traumatic brain injury. Exp. Neurol. 2011, 232, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A Review of the Molecular Mechanisms of Traumatic Brain Injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef]
- Quintard, H.; Patet, C.; Suys, T.; Marques-Vidal, P.; Oddo, M. Normobaric Hyperoxia is Associated with Increased Cerebral Excitotoxicity After Severe Traumatic Brain Injury. Neurocrit. Care 2014, 22, 243–250. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of Oxidative Stress and Neurovascular Inflammation in the Pathogenesis of Traumatic Brain Injury. Mol. Neurobiol. 2014, 51, 966–979. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2–ARE signaling pathway as a potential mechanism. Free. Radic. Boil. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef]
- Pop, V.; Badaut, J. A Neurovascular Perspective for Long-Term Changes After Brain Trauma. Transl. Stroke Res. 2011, 2, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Tagge, C.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.; Zhang, X.-L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 2018, 141, 422–458. [Google Scholar] [CrossRef] [PubMed]
- Dulla, C.G.; Coulter, U.A.; Žiburkus, J. From Molecular Circuit Dysfunction to Disease: Case Studies in Epilepsy, Traumatic Brain Injury and Alzheimer’s Disease. Neuroscientist 2015, 22, 295–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muradashvili, N.; Lominadze, D. Role of fibrinogen in cerebrovascular dysfunction after traumatic brain injury. Brain Inj. 2013, 27, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.; Hanrahan, F.; Gobbo, O.; Kelly, M.E.; Kiang, A.-S.; Humphries, M.M.; Nguyen, A.T.; Ozaki, E.; Keaney, J.; Blau, C.W.; et al. Targeted suppression of claudin-5 decreases cerebral oedema and improves cognitive outcome following traumatic brain injury. Nat. Commun. 2012, 3, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borutaite, V.; Tolkovsky, A.M.; Fricker, M.; Brown, G.C.; Coleman, M. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar]
- Sun, G.-Z.; Gao, F.-F.; Zhao, Z.-M.; Sun, H.; Xu, W.; Wu, L.-W.; He, Y.-C. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen. Res. 2016, 11, 1260–1266. [Google Scholar] [CrossRef]
- Liu, G.; Zou, H.; Luo, T.; Long, M.; Bian, J.; Liu, X.; Gu, J.; Yuan, Y.; Song, R.; Wang, Y.; et al. Caspase-dependent and caspase-independent pathways are involved in cadmium-induced apoptosis in primary rat proximal tubular cell culture. PLoS ONE 2016, 11, e0166823. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, S.; Poksay, K.S.; Schilling, B.; Crippen, D.; Gibson, B.W.; Bredesen, D.E. Identification of new modulators and protein alterations in non-apoptotic programmed cell death. J. Cell. Biochem. 2010, 111, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, X.; Wang, L.; Ding, P.; Zhang, Y.; Han, W.; Ma, D. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J. Cell Sci. 2004, 117, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Fujikawa, D. The role of excitotoxic programmed necrosis in acute brain injury. Comput. Struct. Biotechnol. J. 2015, 13, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Stoica, B.A.; Faden, A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-C.; Hickey, R.W.; Chen, Y.; Bayir, H.; Sullivan, M.L.; Chu, C.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W.; Graham, S.; et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant γ-glutamylcysteinyl ethyl ester. Br. J. Pharmacol. 2007, 28, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, B.; Du Toit, A.; Hofmeyr, J.-H.S. Defining and measuring autophagosome flux—concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Chen, S.; Huang, K.-X.; Le, W. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H. Autophagy in traumatic brain injury: A new target for therapeutic intervention. Front. Mol. Neurosci. 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gao, J.; Zhao, M.; Jing, X.; Cui, Y.; Xu, X.; Wang, K.; Zhang, W.; Cui, J. The effects of BMSCs transplantation on autophagy by CX43 in the hippocampus following traumatic brain injury in rats. Neurol. Sci. 2013, 35, 677–682. [Google Scholar] [CrossRef]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed head injury induces upregulation of beclin 1 at the cortical site of injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef]
- Zhang, M.-H.; Zhou, X.-M.; Gao, J.-L.; Wang, K.-J.; Cui, J.-Z. PI3K/Akt/mTOR pathway participates in neuroprotection by dexmedetomidine inhibits neuronic autophagy following traumatic brain injury in rats. Int. J. Res. Med. Sci. 2014, 2, 1569–1575. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef]
- Kapoor, S.; Kim, S.-M.; Farook, J.M.; Mir, S.; Saha, R.; Sen, N. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J. Neurosci. 2013, 33, 17398–17403. [Google Scholar] [CrossRef]
- Song, Y.; Li, T.; Liu, Z.; Xu, Z.; Zhang, Z.; Chi, L.; Liu, Y. Inhibition of Drp1 after traumatic brain injury provides brain protection and improves behavioral performance in rats. Chem. Biol. Interact. 2019, 304, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Acosta, S.; Pena, I.D.; Tajiri, N.; Kaneko, Y.; Lozano, D.; Gonzales-Portillo, G.S. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L. Fbsbioscience.Org Traumatic brain injury a review of characteristics molecular basis and management. Front. Biosci. 2016, 21, 890–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasker, R.C. Spreading depolarisations and traumatic brain injury: Time course and mechanisms. Lancet Neurol. 2012, 11, 389–390. [Google Scholar] [CrossRef]
- Park, J.-Y.; Amarsanaa, K.; Cui, Y.; Lee, J.-H.; Wu, J.; Yang, Y.-S.; Eun, S.-Y.; Jung, S.-C. Methyl lucidone exhibits neuroprotective effects on glutamate-induced oxidative stress in HT-22 cells via Nrf-2/HO-1 signaling. Appl. Biol. Chem. 2019, 62, 1–9. [Google Scholar] [CrossRef]
- Xin, H.; Cui, Y.; An, Z.; Yang, Q.; Zou, X.; Yu, N. Attenuated glutamate induced ROS production by antioxidative compounds in neural cell lines. RSC Adv. 2019, 9, 34735–34743. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Manias, J.; Eubanks, J.H.; Stewart, D.J. Increased expression of vascular endothelial growth factor-D following brain injury. Int. J. Mol. Sci. 2019, 20, 1594. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Nagai, N.; Umemura, K. A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front. Cell. Neurosci. 2016, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Li, S.; Chung, S.H.; Zhu, L.; Stayt, J.; Su, T.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Gillies, M.C. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-β1-induced permeability of centrally derived vascular endothelium. Eur. J. Cell Boil. 2011, 90, 323–332. [Google Scholar] [CrossRef]
- Li, F.; Lan, Y.; Wang, Y.; Wang, J.; Yang, G.; Meng, F.; Han, H.; Meng, A.; Wang, Y.; Yang, X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 2011, 20, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Acosta, S.A.; Tajiri, N.; de la Pena, I.; Bastawrous, M.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J. Cell. Physiol. 2015, 230, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Fluiter, K.; Opperhuizen, A.L.; Morgan, B.P.; Baas, F.; Ramaglia, V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J. Immunol. 2014, 192, 2339–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Mahmood, A.; Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin. J. Traumatol. 2018, 21, 137–151. [Google Scholar] [CrossRef]
- Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A.; Leker, R. Subacute treatment with vascular endothelial growth factor after traumatic brain injury increases angiogenesis and gliogenesis. Neuroscience 2012, 202, 334–341. [Google Scholar] [CrossRef]
- Weston, N.M.; Sun, D. The Potential of stem cells in treatment of traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2018, 18, 1. [Google Scholar] [CrossRef]
- Sun, D. The potential of endogenous neurogenesis for brain repair and regeneration following traumatic brain injury. Neural Regen. Res. 2014, 9, 688–692. [Google Scholar] [CrossRef]
- Sun, D.; McGinn, M.; Hankins, J.E.; Mays, K.M.; Rolfe, A.; Colello, R.J. Aging- and injury-related differential apoptotic response in the dentate gyrus of the hippocampus in rats following brain trauma. Front. Aging Neurosci. 2013, 5, 95. [Google Scholar] [CrossRef]
- Xu, H.-M. Th1 cytokine-based immunotherapy for cancer. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 482–494. [Google Scholar] [CrossRef]
- Ye, L.; Huang, Y.; Zhao, L.; Li, Y.; Sun, L.; Zhou, Y.; Qian, G.; Zheng, J. IL-1β and TNF-α induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J. Neurochem. 2013, 125, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.; Ghosh, S. NF-κB, Inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Mettang, M.; Reichel, S.N.; Lattke, M.; Palmer, A.; Abaei, A.; Rasche, V.; Huber-Lang, M.; Baumann, B.; Wirth, T. IKK2/NF-κB signaling protects neurons after traumatic brain injury. FASEB J. 2018, 32, 1916–1932. [Google Scholar] [CrossRef] [Green Version]
- Badaut, J.; Ashwal, S.; Obenaus, A. Aquaporins in cerebrovascular disease: A target for treatment of brain edema? Cerebrovasc. Dis. 2011, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2–ARE pathway. Free. Radic. Boil. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef]
- Mutinati, M.; Pantaleo, M.; Roncetti, M.; Piccinno, M.; Rizzo, A.; Sciorsci, R. Oxidative stress in neonatology. A review. Reprod. Domest. Anim. 2013, 49, 7–16. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Cong, Z.-X.; Zhou, X.-M.; Xu, J.-G.; Jia, Y.; Ding, Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J. Surg. Res. 2014, 186, 328–337. [Google Scholar] [CrossRef]
- Arent, A.M.; De Souza, L.; Walz, R.; Dafre, A.L. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. BioMed Res. Int. 2014, 2014, 1–18. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.P.O.; Rodrigues, F.; Della-Pace, I.D.; Mota, B.C.; Oliveira, S.M.; Gewehr, C.D.C.V.; Bobinski, F.; De Oliveira, C.V.; Brum, J.S.; Oliveira, M.S.; et al. The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: Role of inflammatory and oxidative brain damage. Neurochem. Int. 2013, 63, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Álvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/macrophage polarization dynamics following traumatic brain injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yan, H.; Ni, H.; Liang, W.; Jin, W. Expression of nuclear factor erythroid 2-related factor 2 following traumatic brain injury in the human brain. NeuroReport 2019, 30, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, M.; Wang, H.; Gao, C.-C.; Zhu, L.; Lin, Y.-X.; Fang, J.; Ding, K. Activation of the Nrf2-ARE signal pathway after blast induced traumatic brain injury in mice. Int. J. Neurosci. 2019, 1–7. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Wang, H.; Xu, J.-G.; Ding, K.; Li, T. Deletion of Nrf2 exacerbates oxidative stress after traumatic brain injury in mice. Cell. Mol. Neurobiol. 2015, 35, 713–721. [Google Scholar] [CrossRef]
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Mahmood, A.; Chopp, M. Emerging treatments for traumatic brain injury. Expert Opin. Emerg. Drugs 2009, 14, 67–84. [Google Scholar] [CrossRef]
- Langham, J.; Goldfrad, C.; Teasdale, G.; Shaw, D.; Rowan, K. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst. Rev. 2003, 2003, CD000565. [Google Scholar] [CrossRef]
- Berman, R.F.; Verweij, B.H.; Muizelaar, J.P. Neurobehavioral protection by the neuronal calcium channel blocker Ziconotide in a model of traumatic diffuse brain injury in rats. J. Neurosurg. 2000, 93, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Verweij, B.H.; Muizelaar, J.P.; Vinas, F.C.; Peterson, P.L.; Xiong, Y.; Lee, C.P. Improvement in mitochondrial dysfunction as a new surrogate efficiency measure for preclinical trials: Dose—Response and time-window profiles for administration of the calcium channel blocker Ziconotide in experimental brain injury. J. Neurosurg. 2000, 93, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Sorani, M.D.; Morabito, D.; Rosenthal, G.; Giacomini, K.M.; Manley, G.T. Characterizing the dose-response relationship between mannitol and intracranial pressure in traumatic brain injury patients using a high-frequency physiological data collection system. J. Neurotrauma 2008, 25, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.; Schierhout, G. Mannitol for acute traumatic brain injury. Cochrane Database Syst. Rev. 2005, CD001049. [Google Scholar] [CrossRef] [Green Version]
- Giacino, J.T.; Whyte, J.; Bagiella, E.; Kalmar, K.; Childs, N.; Khademi, A.; Eifert, B.; Long, D.; Katz, D.; Cho, S.; et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. New Engl. J. Med. 2012, 366, 819–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, E.; Maura, L.S.; Ohlinger, M.J. Amantadine enhancement of arousal and cognition after traumatic brain injury. Ann. Pharmacother. 2008, 42, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Liu, Z. Nuclear factor erythroid 2-related factor 2 (Nrf2) mediates neuroprotection in traumatic brain injury at least in part by inactivating microglia. Med. Sci. Monit. 2016, 22, 2161–2166. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, V.; Kaur, I.P. Evaluating potential of curcumin loaded solid lipid nanoparticles in aluminium induced behavioural, biochemical and histopathological alterations in mice brain. Food Chem. Toxicol. 2011, 49, 2906–2913. [Google Scholar] [CrossRef]
- Agarwal, N.B.; Jain, S.; Agarwal, N.K.; Mediratta, P.K.; Sharma, K.K. Modulation of pentylenetetrazole-induced kindling and oxidative stress by curcumin in mice. Phytomedicine 2011, 18, 756–759. [Google Scholar] [CrossRef]
- Carmona-Ramírez, I.; Santamaría, A.; Tobon-Velasco, J.C.; Orozco-Ibarra, M.; Gonzalez-Herrera, I.G.; Pedraza-Chaverri, J.; Maldonado, P.D. RETRACTED: Curcumin restores Nrf2 levels and prevents quinolinic acid-induced neurotoxicity. J. Nutr. Biochem. 2013, 24, 14–24. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; Vasto, S.; Sonya, V.; Abraham, N.G.; Nader, A.G.; Caruso, C.; Calogero, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: A nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Alcântara, G.F.T.; Simões-Neto, E.; Da Cruz, G.M.P.; Nobre, M.E.P.; Neves, K.; Andrade, G.; Brito, G.A.D.C.; Viana, G.S.D.B. Curcumin reverses neurochemical, histological and immuno-histochemical alterations in the model of global brain ischemia. J. Tradit. Complement. Med. 2016, 7, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, P.; Hall, T.C.; Paladugu, L.; Kolli, N.; Learman, C.; Rossignol, J.; Dunbar, G.L. A comparative study of dietary curcumin, nanocurcumin, and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5×-familial Alzheimer’s disease mice. Histochem. Cell Biol. 2016, 146, 609–625. [Google Scholar] [CrossRef]
- Wang, B.-F.; Cui, Z.-W.; Zhong, Z.-H.; Sun, Y.-H.; Sun, Q.-F.; Yang, G.-Y.; Bian, L.-G. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharmacol. Sin. 2015, 36, 939–948. [Google Scholar] [CrossRef]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef]
- Tu, Z.-S.; Wang, Q.; Sun, D.-D.; Dai, F.; Zhou, B. Design, synthesis, and evaluation of curcumin derivatives as Nrf2 activators and cytoprotectors against oxidative death. Eur. J. Med. Chem. 2017, 134, 72–85. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Schubert, D.; Gomez-Pinilla, F. Brain and spinal cord interaction. Neurorehabilit. Neural Repair 2011, 25, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.-T.; Bian, C.; Yuan, J.-C.; Chu, W.-H.; Xiang, X.; Chen, F.; Wang, C.-S.; Feng, H.; Lin, J.-K. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway in experimental traumatic brain injury. J. Neuroinflamm. 2014, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. Br. J. Pharmacol. 2018, 39, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Soane, L.; Dai, W.L.; Fiskum, G.; Bambrick, L.L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010, 88, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gillespie, S.; Becker, F.; Vital, S.A.; Nguyen, V.; Alexander, J.S.; Evans, P.C.; Gavins, F.N. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc. Pharmacol. 2016, 85, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Dinkova-Kostova, A.T. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef]
- Zhao, X.; Wen, L.; Dong, M.; Lu, X. Sulforaphane activates the cerebral vascular Nrf2–ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 2016, 1653, 1–7. [Google Scholar] [CrossRef]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free. Radic. Boil. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Moon, D.-O.; Kim, M.-O.; Kang, S.-H.; Choi, Y.H.; Kim, G.-Y. Sulforaphane suppresses TNF-α-mediated activation of NF-κB and induces apoptosis through activation of reactive oxygen species-dependent caspase-3. Cancer Lett. 2009, 274, 132–142. [Google Scholar] [CrossRef]
- Checker, R.; Gambhir, L.; Thoh, M.; Sharma, D.; Sandur, S.K. Sulforaphane, a naturally occurring isothiocyanate, exhibits anti-inflammatory effects by targeting GSK3β/Nrf-2 and NF-κB pathways in T cells. J. Funct. Foods 2015, 19, 426–438. [Google Scholar] [CrossRef]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2014, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J. Neuroinflamm. 2014, 11, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.J.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Boil. 2017, 12, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K.; Young, J.L.; Zirlik, A.; Macfarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin Inhibits Proinflammatory Responses and Nuclear Factor-κB in Human Vascular Wall Cells. Arter. Thromb. Vasc. Boil. 2006, 26, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin Inhibits Cytokine-Induced Nuclear Factor κB Activation Via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Li, S.N.; Wang, X.; Zeng, Q.T.; Feng, Y.B.; Cheng, X.; Mao, X.B.; Wang, T.H.; Deng, H.P. Metformin inhibits nuclear factor kappaB activation and decreases serum high-sensitivity C-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels 2009, 24, 446–453. [Google Scholar] [CrossRef]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.I.; Kim, B.H.; Kim, S.K.; Song, G.Y.; et al. Metformin inhibits P-glycoprotein expression via the NF-kappaB pathway and CRE transcriptional activity through AMPK activation. Br. J. Pharmacol. 2011, 162, 1096–1108. [Google Scholar] [CrossRef] [Green Version]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Boil. 2017, 13, 353–362. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Cheng, J.; Liu, Y.; Wu, J.; Wang, X.; Wei, S.; Zhou, X.; Qin, Z.; Jia, J.; Zhen, X.-C. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav. Immun. 2014, 40, 131–142. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Tanokashira, D.; Kurata, E.; Fukuokaya, W.; Kawabe, K.; Kashiwada, M.; Takeuchi, H.; Nakazato, M.; Taguchi, A. Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Bio 2018, 8, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X.; et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic. Boil. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Lequerica, A.; Jasey, N.; Tremont, J.N.P.; Chiaravalloti, N.D. Pilot study on the effect of ramelteon on sleep disturbance after traumatic brain injury: Preliminary evidence from a clinical trial. Arch. Phys. Med. Rehabilitation 2015, 96, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Bergold, P. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp. Neurol. 2015, 275, 367–380. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivandzade, F.; Alqahtani, F.; Cucullo, L. Traumatic Brain Injury and Blood–Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition. Int. J. Mol. Sci. 2020, 21, 2721. https://doi.org/10.3390/ijms21082721
Sivandzade F, Alqahtani F, Cucullo L. Traumatic Brain Injury and Blood–Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition. International Journal of Molecular Sciences. 2020; 21(8):2721. https://doi.org/10.3390/ijms21082721
Chicago/Turabian StyleSivandzade, Farzane, Faleh Alqahtani, and Luca Cucullo. 2020. "Traumatic Brain Injury and Blood–Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition" International Journal of Molecular Sciences 21, no. 8: 2721. https://doi.org/10.3390/ijms21082721