Non-Coding RNAs in Lung Tumor Initiation and Progression



Abstract
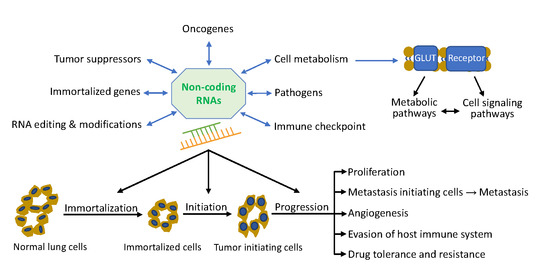
:
1. Introduction
2. Lung Cancer
3. Lung Tumor Initiation
4. Lung Tumor Progression
5. Non-Coding RNA Network in Lung Cancer
5.1. Role of Non-Coding RNAs in Lung Tumor Initiation
5.1.1. Role of Non-Coding RNAs in Lung Tumor Initiating Cells
5.1.2. Role of Non-Coding RNAs in Immortalized Genes
5.1.3. Role of Non-Coding RNAs in Oncogenes and Tumor Suppressors
5.1.4. Role of Non-Coding RNAs in RNA Editing
5.1.5. Role of Non-Coding RNAs in RNA Modifications
5.2. Role of Non-Coding RNAs in Lung Tumor Progression
5.2.1. Role of Non-Coding RNAs in Tumor Cell Proliferation
5.2.2. Role of Non-Coding RNA in Tumor Metastasis
5.2.3. Role of Non-Coding RNA in Angiogenesis
5.2.4. Role of Non-Coding RNA in Evasion of Host Immune System
5.2.5. Role of Non-Coding RNA in Drug Tolerance and Resistance
5.3. Role of Non-Coding RNAs and Metabolism in Lung Cancer
5.3.1. EGFR
5.3.2. MET
5.3.3. PI3K/Akt/mTOR
5.3.4. Ras/Raf/MEK/ERK (MAPK)
5.3.5. Wnt/β-Catenin
5.4. Role of Non-Coding RNAs and Pathogens in Lung Cancer
6. Cell-Free Circulating Non-Coding RNAs in Lung Cancer
7. Pre-Clinical Models for Human Lung Cancer
8. Clinical Trial of Lung Cancer
9. Concluding Remarks and Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, R.T.; Murray, T.; Bolden, S.; Wingo, P.A. Cancer statistics, 2000. CA Cancer J. Clin. 2000, 50, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Mecca, C.; Crinò, L.; Baglivo, S.; Cenci, M.; Metro, G. Non-coding RNAs in lung cancer. Oncoscience 2014, 1, 674–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung Cancer. New Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [Green Version]
- Campling, B.G.; el-Deiry, W.S. Clinical implications of p53 mutations in lung cancer. Methods Mol. Med. 2003, 75, 53–77. [Google Scholar] [CrossRef]
- Remen, T.; Pintos, J.; Abrahamowicz, M.; Siemiatycki, J. Risk of lung cancer in relation to various metrics of smoking history: A case-control study in Montreal. BMC Cancer 2018, 18, 1275. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Miller, K.; Nogueira, L.; Mariotto, A.; Rowland, J.; Yabroff, R.; Alfano, C.; Jemal, A.; Kramer, J.; Siegel, R. Cancer treatment and survivorship statistics, 2019. CA 2019, 69. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Schlessinger, J. Small molecule combats cancer-causing KRAS protein at last. Nature 2019, 575, 294–295. [Google Scholar] [CrossRef]
- Yang, H.; Liang, S.Q.; Schmid, R.A.; Peng, R.W. New Horizons in. Front. Oncol. 2019, 9, 953. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, K.P.; Ou, S.-H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2019, 37, TPS3161. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, B.J.; Morris, J.C.; Steel, J.C. Lung cancer-initiating cells: A novel target for cancer therapy. Target. Oncol. 2013, 8, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Hardavella, G.; George, R.; Sethi, T. Lung cancer stem cells-characteristics, phenotype. Transl. Lung Cancer Res. 2016, 5, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-L.; Patel, A.; Kumar, P.; Chen, Z.-S. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 2012, 31, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Chen, B.; Xu, W.; Zhao, W.; Wu, J. Clinicopathological significance of CD133 in lung cancer: A meta-analysis. Mol. Clin. Oncol. 2014, 2, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, R.D.; Sheridan, S.; Girard, L.; Sato, M.; Kim, Y.; Pollack, J.; Peyton, M.; Zou, Y.; Kurie, J.M.; Dimaio, J.M.; et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004, 64, 9027–9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counter, C.M. The roles of telomeres and telomerase in cell life span. Mutat. Res. 1996, 366, 45–63. [Google Scholar] [CrossRef]
- Smith, J.L.; Lee, L.C.; Read, A.; Li, Q.; Yu, B.; Lee, C.-S.; Luo, J. One-step immortalization of primary human airway epithelial cells capable of oncogenic transformation. Cell Biosci. 2016, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, A.S.; Randell, S.H.; Stewart, S.A.; Elenbaas, B.; Hartwell, K.A.; Brooks, M.W.; Fleming, M.D.; Olsen, J.C.; Miller, S.W.; Weinberg, R.A.; et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 2002, 21, 4577–4586. [Google Scholar] [CrossRef] [Green Version]
- Sussan, T.E.; Pletcher, M.T.; Murakami, Y.; Reeves, R.H. Tumor suppressor in lung cancer 1 (TSLC1) alters tumorigenic growth properties and gene expression. Mol. Cancer 2005, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamura, K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, V.H.; Pipinikas, C.P.; Pennycuick, A.; Lee-Six, H.; Chandrasekharan, D.; Beane, J.; Morris, T.J.; Karpathakis, A.; Feber, A.; Breeze, C.E.; et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med. 2019, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers 2018, 10, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Venugopal, C.; Tokar, T.; McFarlane, N.; Subapanditha, M.K.; Qazi, M.; Bakhshinyan, D.; Vora, P.; Murty, N.K.; Jurisica, I.; et al. Therapeutic Targeting of the Premetastatic Stage in Human Lung-to-Brain Metastasis. Cancer Res. 2018, 78, 5124–5134. [Google Scholar] [CrossRef] [Green Version]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.D.; Le, T.M.; Haggstrom, D.E.; Gentzler, R.D. Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2015, 4, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2016, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Eliasz, S.; Liang, S.; Chen, Y.; De Marco, M.A.; Machek, O.; Skucha, S.; Miele, L.; Bocchetta, M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010, 29, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Chen, H.; Lee, B.H.; Lee, J.Y.H.; Yip, Y.S.; Tan, N.S.; Tan, L.P. Epithelial-mesenchymal transition of cancer cells using bioengineered hybrid scaffold composed of hydrogel/3D-fibrous framework. Sci. Rep. 2019, 9, 8997. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Le, T.; Gerber, D.E. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers 2019, 11, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.C.; Soo, R.A.; Riely, G.J.; Ou, S.I.; Clancy, J.S.; Li, S.; et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.; Goh, B.C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef]
- Passiglia, F.; Van Der Steen, N.; Raez, L.; Pauwels, P.; Gil-Bazo, I.; Santos, E.; Santini, D.; Tesoriere, G.; Russo, A.; Bronte, G.; et al. The role of cMet in non-small cell lung cancer resistant to EGFR-inhibitors: Did we really find the target? Curr. Drug Targets 2014, 15, 1284–1292. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.-Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 63. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- Roman, M.; Baraibar, I.; Lopez, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [Green Version]
- Castro, D.; Moreira, M.; Gouveia, A.M.; Pozza, D.H.; De Mello, R.A. MicroRNAs in lung cancer. Oncotarget 2017, 8, 81679–81685. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lei, T.; Chen, X.; Gu, J.; Huang, J.; Lu, B.; Wang, Z. Long non-coding RNA in lung cancer. Clin. Chim. Acta 2019. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA–LncRNA Interactions. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Springer New York: New York, NY, USA, 2016; pp. 271–286. [Google Scholar]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Di, X.; Jin, X.; Li, R.; Zhao, M.; Wang, K. CircRNAs and lung cancer: Biomarkers and master regulators. Life Sci. 2019, 220, 177–185. [Google Scholar] [CrossRef]
- Zhang, W.C.; Chin, T.M.; Yang, H.; Nga, M.E.; Lunny, D.P.; Lim, E.K.H.; Sun, L.L.; Pang, Y.H.; Leow, Y.N.; Malusay, S.R.Y.; et al. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat. Commun. 2016, 7, 11702. [Google Scholar] [CrossRef]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Faversani, A.; Amatori, S.; Augello, C.; Colombo, F.; Porretti, L.; Fanelli, M.; Ferrero, S.; Palleschi, A.; Pelicci, P.G.; Belloni, E.; et al. miR-494-3p is a novel tumor driver of lung carcinogenesis. Oncotarget 2017, 8, 7231–7247. [Google Scholar] [CrossRef] [Green Version]
- Yin, R.; Zhang, S.; Wu, Y.; Fan, X.; Jiang, F.; Zhang, Z.; Feng, D.; Guo, X.; Xu, L. microRNA-145 suppresses lung adenocarcinoma-initiating cell proliferation by targeting OCT4. Oncol. Rep. 2011, 25, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Xiaotao, X.; Renhua, G.; Yongmei, Y.; Lianke, L.; Wen, G.; Yongqian, S. Reduced miR-31 and let-7 maintain the balance between differentiation and quiescence in lung cancer stem-like side population cells. Biomed. Pharmacother. 2012, 66, 89–97. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, F.; Xu, Y.; Wang, B.; Zhao, Y.; Xu, W.; Shi, L.; Lu, X.; Liu, Q. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol. Appl. Pharmacol. 2015, 282, 9–19. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, J.; Wang, F.; Guan, Z.; Ge, Y.; Yang, X.; Cai, J. LncRNAs and their role in cancer stem cells. Oncotarget 2017, 8, 110685–110692. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Edmonds, M.D.; Boyd, K.L.; Moyo, T.; Mitra, R.; Duszynski, R.; Arrate, M.P.; Chen, X.; Zhao, Z.; Blackwell, T.S.; Andl, T.; et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J. Clin. Invest. 2016, 126, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-H.; Lu, K.-H.; Wang, K.-M.; Sun, M.; Zhang, E.-B.; Yang, J.-S.; Yin, D.-D.; Liu, Z.-L.; Zhou, J.; Liu, Z.-J.; et al. MicroRNA-196a promotes non-small cell lung cancer cell proliferation and invasion through targeting HOXA5. BMC Cancer 2012, 12, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Zhang, X.; Wang, H.M.; Liu, X.M.; Zhang, X.J.; Zheng, B.; Qian, G.R.; Ma, Z.L. MicroRNA-18a-5p functions as an oncogene by directly targeting IRF2 in lung cancer. Cell Death Dis. 2017, 8, e2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Sardo, F.; Forcato, M.; Sacconi, A.; Capaci, V.; Zanconato, F.; Di Agostino, S.; Del Sal, G.; Pandolfi, P.P.; Strano, S.; Bicciato, S.; et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis 2017, 38, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, S.; Yan, W.; Yang, J.; Qin, Y.-J.; Lin, X.-L.; Xie, R.-Y.; Wang, S.-C.; Jin, W.; Gao, F.; et al. MicroRNA-19 triggers epithelial–mesenchymal transition of lung cancer cells accompanied by growth inhibition. Lab. Investig. 2015, 95, 1056–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirono, T.; Jingushi, K.; Nagata, T.; Sato, M.; Minami, K.; Aoki, M.; Takeda, A.H.; Umehara, T.; Egawa, H.; Nakatsuji, Y.; et al. MicroRNA-130b functions as an oncomiRNA in non-small cell lung cancer by targeting tissue inhibitor of metalloproteinase-2. Sci. Rep. 2019, 9, 6956. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Zhou, X.; Yin, Z.; Guo, J.; Hu, T.; Jiang, S.; Liu, L.; Dong, X.; Zhang, S.; Wu, G. Tumor invasion and metastasis regulated by microRNA-184 and microRNA-574-5p in small-cell lung cancer. Oncotarget 2015, 6, 44609–44622. [Google Scholar] [CrossRef]
- Mao, G.; Liu, Y.; Fang, X.; Liu, Y.; Fang, L.; Lin, L.; Liu, X.; Wang, N. Tumor-derived microRNA-494 promotes angiogenesis in non-small cell lung cancer. Angiogenesis 2015, 18, 373–382. [Google Scholar] [CrossRef]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, Y.-J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2011, 18, 74–82. [Google Scholar] [CrossRef]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE 2014, 9, e103305. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wells, J.M.; Chow, K.-H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab. 2019, 1, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Czochor, J.; Steinmetz, A.; Weidhaas, J.B.; Glazer, P.M.; Slack, F.J. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol. Ther. 2011, 12, 908–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhang, Y.; Mao, Y.; Mou, J.; Zhao, J.; Xue, Q.; Wang, D.; Huang, J.; Gao, S.; Gao, Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 2017, 482, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, J.; Yan, M.; Liu, L.; Lin, H.; Zhao, F.; Sun, L.; Zhang, Y.; Cui, Y.; Zhang, F.; et al. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 2015, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Ma, J.; Guillemette, R.; Zhou, M.; Yang, Y.; Yang, Y.; Hock, J.M.; Yu, X. miR-335 inhibits small cell lung cancer bone metastases via IGF-IR and RANKL pathways. Mol. Cancer Res. 2014, 12, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Peng, X.-C.; Zheng, X.-L.; Wang, J.; Qin, Y.-W. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 2009, 66, 169–175. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Li, Y.; Li, C.; Qin, T.; Bai, M.; Zhang, Z.; Jia, R.; Su, Y.; Wang, C. miR-195 suppresses metastasis and angiogenesis of squamous cell lung cancer by inhibiting the expression of VEGF. Mol. Med. Rep. 2019, 20, 2625–2632. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.B.; Liang, L.H.; Wu, K.G.; Wang, L.X.; He, X.; Song, C.; Wang, Y.Q.; Li, Y.H. MiR-140 Expression Regulates Cell Proliferation and Targets PD-L1 in NSCLC. Cell Physiol. Biochem. 2018, 46, 654–663. [Google Scholar] [CrossRef]
- Lv, J.; Qiu, M.; Xia, W.; Liu, C.; Xu, Y.; Wang, J.; Leng, X.; Huang, S.; Zhu, R.; Zhao, M.; et al. High expression of long non-coding RNA SBF2-AS1 promotes proliferation in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2016, 35, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Wang, J.; Shan, B.; Li, B.; Peng, W.; Dong, Y.; Shi, W.; Zhao, W.; He, D.; Duan, M.; et al. The long noncoding RNA LINC00312 induces lung adenocarcinoma migration and vasculogenic mimicry through directly binding YBX1. Mol. Cancer 2018, 17, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashima, M.; Tange, S.; Ishimura, A.; Suzuki, T. MEG3 Long Noncoding RNA Contributes to the Epigenetic Regulation of Epithelial-Mesenchymal Transition in Lung Cancer Cell Lines. J. Biol. Chem. 2017, 292, 82–99. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Zhang, R.; An, X.; Li, Z.; Fang, C.; Pan, B.; Chen, W.; Xu, G.; Han, W. LncRNA HOXA-AS3 confers cisplatin resistance by interacting with HOXA3 in non-small-cell lung carcinoma cells. Oncogenesis 2019, 8, 60. [Google Scholar] [CrossRef]
- Hou, Z.; Zhao, W.; Zhou, J.; Shen, L.; Zhan, P.; Xu, C.; Chang, C.; Bi, H.; Zou, J.; Yao, X.; et al. A long noncoding RNA Sox2ot regulates lung cancer cell proliferation and is a prognostic indicator of poor survival. Int. J. Biochem. Cell Biol. 2014, 53, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Mo, J.; Luo, M.; Yu, Q.; Zhou, S.; Li, T.; Zhang, Y.; Luo, W. c-Myc-activated long non-coding RNA H19 downregulates miR-107 and promotes cell cycle progression of non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12400–12409. [Google Scholar]
- Nie, F.Q.; Sun, M.; Yang, J.S.; Xie, M.; Xu, T.P.; Xia, R.; Liu, Y.W.; Liu, X.H.; Zhang, E.B.; Lu, K.H.; et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 2015, 14, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Li, G.-y.; Jiang, L.; Jia, L.; Zhang, Z.; Li, X.; Wang, R.; Zhou, M.; Zhou, Y.; Zeng, Z.; et al. Long noncoding RNA CAR10 promotes lung adenocarcinoma metastasis via miR-203/30/SNAI axis. Oncogene 2019, 38, 3061–3076. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, J.; Chen, Y.; Li, S.; Jin, M.; Wang, H.; Chen, Z.; Yu, W. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am. J. Cancer Res. 2016, 6, 1099–1107. [Google Scholar]
- Wu, D.; Li, Y.; Zhang, H.; Hu, X. Knockdown of Lncrna PVT1 Enhances Radiosensitivity in Non-Small Cell Lung Cancer by Sponging Mir-195. Cell Physiol. Biochem. 2017, 42, 2453–2466. [Google Scholar] [CrossRef]
- Pan, H.; Jiang, T.; Cheng, N.; Wang, Q.; Ren, S.; Li, X.; Zhao, C.; Zhang, L.; Cai, W.; Zhou, C. Long non-coding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget 2016, 7, 49948–49960. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Yang, X.; Zhang, D.; Luo, J.; Chen, R. Long noncoding RNA LINC01186, regulated by TGF-β/SMAD3, inhibits migration and invasion through Epithelial-Mesenchymal-Transition in lung cancer. Gene 2017, 608, 1–12. [Google Scholar] [CrossRef]
- Wang, H.; Lu, B.; Ren, S.; Wu, F.; Wang, X.; Yan, C.; Wang, Z. Long Noncoding RNA LINC01116 Contributes to Gefitinib Resistance in Non-small Cell Lung Cancer through Regulating IFI44. Mol. Ther. Nucleic Acids 2019, 19, 218–227. [Google Scholar] [CrossRef]
- Castellano, J.J.; Navarro, A.; Vinolas, N.; Marrades, R.M.; Moises, J.; Cordeiro, A.; Saco, A.; Munoz, C.; Fuster, D.; Molins, L.; et al. LincRNA-p21 Impacts Prognosis in Resected Non-Small Cell Lung Cancer Patients through Angiogenesis Regulation. J. Thorac. Oncol. 2016, 11, 2173–2182. [Google Scholar] [CrossRef]
- Tian, X.; Ma, J.; Wang, T.; Tian, J.; Zhang, Y.; Mao, L.; Xu, H.; Wang, S. Long Non-Coding RNA HOXA Transcript Antisense RNA Myeloid-Specific 1-HOXA1 Axis Downregulates the Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in Lung Cancer. Front. Immunol. 2018, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhang, E.b.; Yin, D.d.; Kong, R.; Xu, T.p.; Chen, W.m.; Xia, R.; Shu, Y.q.; De, W. Low expression of long noncoding RNA PANDAR predicts a poor prognosis of non-small cell lung cancer and affects cell apoptosis by regulating Bcl-2. Cell Death Dis. 2015, 6, e1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Feng, S.; Chen, X.; Yang, X.; Mao, R.; Guo, C.; Wang, Z.; Thomas, D.G.; Lin, J.; Reddy, R.M.; et al. Silencing of Long Noncoding RNA MIR22HG Triggers Cell Survival/Death Signaling via Oncogenes YBX1, MET, and p21 in Lung Cancer. Cancer Res. 2018, 78, 3207–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Kong, R.; Chen, F.; Song, Y. A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Mol. Carcinog. 2015, 54, E1–E12. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Qu, X.; Li, W.; Zhong, X.; Li, P.; Yang, S.; Chen, X.; Shao, M.; Zhang, L. The long non-coding RNA, GAS5, enhances gefitinib-induced cell death in innate EGFR tyrosine kinase inhibitor-resistant lung adenocarcinoma cells with wide-type EGFR via downregulation of the IGF-1R expression. J. Hematol. Oncol. 2015, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-F.; Pei, X.; Li, K.-S.; Jin, L.-N.; Wang, F.; Wu, J.; Zhang, X.-M. Circular RNA circFGFR1 promotes progression and anti-PD-1 resistance by sponging miR-381-3p in non-small cell lung cancer cells. Mol. Cancer 2019, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Yin, Z.; Xu, J.; Wu, F.; Huang, Q.; Yang, L.; Jin, Y.; Yang, G. Circulating microRNAs predict the response to anti-PD-1 therapy in non-small cell lung cancer. Genomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Yan, B.; Xin, R.; Ma, T. A novel circular RNA hsa_circ_0020123 exerts oncogenic properties through suppression of miR-144 in non-small cell lung cancer. Am. J. Cancer Res. 2018, 8, 1387–1402. [Google Scholar]
- Cheng, Z.; Yu, C.; Cui, S.; Wang, H.; Jin, H.; Wang, C.; Li, B.; Qin, M.; Yang, C.; He, J.; et al. circTP63 functions as a ceRNA to promote lung squamous cell carcinoma progression by upregulating FOXM1. Nat. Commun. 2019, 10, 3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Xia, W.; Chen, R.; Wang, S.; Xu, Y.; Ma, Z.; Xu, W.; Zhang, E.; Wang, J.; Fang, T.; et al. The Circular RNA circPRKCI Promotes Tumor Growth in Lung Adenocarcinoma. Cancer Res. 2018, 78, 2839–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.H.; Zhu, X.Z.; Huang, K.W.; Zhang, Q.; Fan, Y.X.; Yan, P.W.; Wen, J. Emerging roles of circular RNA hsa_circ_0000064 in the proliferation and metastasis of lung cancer. Biomed. Pharmacother. 2017, 96, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Gou, Q.; Pu, W.; Guo, C.; Yang, Y.; Wu, K.; Liu, Y.; Liu, L.; Wei, Y.-Q.; Peng, Y. Circular RNA F-circEA produced from EML4-ALK fusion gene as a novel liquid biopsy biomarker for non-small cell lung cancer. Cell Res. 2018, 28, 693–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, H. CircRNA circ_0067934 silencing inhibits the proliferation, migration and invasion of NSCLC cells and correlates with unfavorable prognosis in NSCLC. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3053–3060. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, D.; Wei, Y. Overexpressed CDR1as functions as an oncogene to promote the tumor progression via miR-7 in non-small-cell lung cancer. Onco. Targets Ther. 2018, 11, 3979–3987. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ma, W.; Yuan, Y.; Zhang, Y.; Sun, S. Circular RNA hsa_circRNA_103809 promotes lung cancer progression via facilitating ZNF121-dependent MYC expression by sequestering miR-4302. Biochem. Biophys. Res. Commun. 2018, 500, 846–851. [Google Scholar] [CrossRef]
- Wan, L.; Zhang, L.; Fan, K.; Cheng, Z.-X.; Sun, Q.-C.; Wang, J.-J. Circular RNA-ITCH Suppresses Lung Cancer Proliferation via Inhibiting the Wnt/β-Catenin Pathway. Biomed. Res. Int. 2016, 2016, 1579490. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Ma, W.; Ke, Z.; Xie, F. CircRNA hsa_circ_100395 regulates miR-1228/TCF21 pathway to inhibit lung cancer progression. Cell Cycle 2018, 17, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wang, G.; Shen, H.; Fei, X. Hsa_circ_0033155: A potential novel biomarker for non-small cell lung cancer. Exp. Ther. Med. 2018, 16, 3220–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, S.; Mao, Y.; Xu, J.; Yang, S.; Shen, H.; Xu, W.; Fan, W.; Wang, J. CircRNF13 regulates the invasion and metastasis in lung adenocarcinoma by targeting miR-93-5p. Gene 2018, 671, 170–177. [Google Scholar] [CrossRef]
- Wang, Q.; Li, D.C.; Li, Z.F.; Liu, C.X.; Xiao, Y.M.; Zhang, B.; Li, X.D.; Zhao, J.; Chen, L.P.; Xing, X.M.; et al. Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene 2011, 30, 3875–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Wang, Z.; Zhang, X.; Feng, S.; Wang, G.; He, J.; Zhang, B. MicroRNA-545 suppresses cell proliferation by targeting cyclin D1 and CDK4 in lung cancer cells. PLoS ONE 2014, 9, e88022. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Shen, Y.-Q.; Li, Y.-L.; Liang, C.; Zhang, B.-J.; Lu, S.-D.; He, Y.-Y.; Wang, P.; Sun, Q.-L.; Jin, Y.-X.; et al. Direct repression of the oncogene CDK4 by the tumor suppressor miR-486-5p in non-small cell lung cancer. Oncotarget 2016, 7, 34011–34021. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, D.Q.; Liu, D.; Tang, X.J. MiR-613 induces cell cycle arrest by targeting CDK4 in non-small cell lung cancer. Cell Oncol. 2016, 39, 139–147. [Google Scholar] [CrossRef]
- Qin, Y.; Zhou, X.; Huang, C.; Li, L.; Liu, H.; Liang, N.; Chen, Y.; Ma, D.; Han, Z.; Xu, X.; et al. Lower miR-340 expression predicts poor prognosis of non-small cell lung cancer and promotes cell proliferation by targeting CDK4. Gene 2018, 675, 278–284. [Google Scholar] [CrossRef]
- Feng, H.; Ge, F.; Du, L.; Zhang, Z.; Liu, D. MiR-34b-3p represses cell proliferation, cell cycle progression and cell apoptosis in non-small-cell lung cancer (NSCLC) by targeting CDK4. J. Cell Mol. Med. 2019, 23, 5282–5291. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasinski, A.L.; Slack, F.J. miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, S.; Mallick, R.; Kannisto, E.; Sharma, R.; Bshara, W.; Yendamuri, S.; Dhillon, S.S. MiR-205 and MiR-375 microRNA assays to distinguish squamous cell carcinoma from adenocarcinoma in lung cancer biopsies. J. Thorac. Oncol. 2015, 10, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, J.J. The emerging role of RNA editing in plasticity. J. Exp. Biol. 2015, 218, 1812–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Slack, F.J. ADARs Edit MicroRNAs to Promote Leukemic Stem Cell Activity. Cell Stem Cell 2016, 19, 141–142. [Google Scholar] [CrossRef] [Green Version]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Sharpnack, M.F.; Chen, B.; Aran, D.; Kosti, I.; Sharpnack, D.D.; Carbone, D.P.; Mallick, P.; Huang, K. Global Transcriptome Analysis of RNA Abundance Regulation by ADAR in Lung Adenocarcinoma. EBioMedicine 2018, 27, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Anadón, C.; Guil, S.; Simó-Riudalbas, L.; Moutinho, C.; Setien, F.; Martinez-Cardus, A.; Moran, S.; Villanueva, A.; Calaf, M.; Vidal, A.; et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 2015, 35. [Google Scholar] [CrossRef]
- Huang, R.S.; Zheng, Y.L.; Zhao, J.; Chun, X. microRNA-381 suppresses the growth and increases cisplatin sensitivity in non-small cell lung cancer cells through inhibition of nuclear factor-kappaB signaling. Biomed. Pharmacother. 2018, 98, 538–544. [Google Scholar] [CrossRef]
- Nigita, G.; Distefano, R.; Veneziano, D.; Romano, G.; Rahman, M.; Wang, K.; Pass, H.; Croce, C.M.; Acunzo, M.; Nana-Sinkam, P. Tissue and exosomal miRNA editing in Non-Small Cell Lung Cancer. Sci. Rep. 2018, 8, 10222. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Liu, C.; Liu, W.; Xiang, Y.; Diao, L.; Guo, A.-Y.; Han, L. LNCediting: A database for functional effects of RNA editing in lncRNAs. Nucleic Acids Res. 2017, 45, D79–D84. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sang, L.; Gong, Y. N6-methyladenine RNA modification and cancers. Am. J. Cancer Res. 2018, 8, 1957–1966. [Google Scholar]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3–eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.F.; Yang, Q.; Liu, C.X.; Wu, M.; Chen, L.L.; Yang, L. N(6)-Methyladenosines Modulate A-to-I RNA Editing. Mol. Cell 2018, 69, 126–135. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar]
- Zha, W.; Cao, L.; Shen, Y.; Huang, M. Roles of Mir-144-ZFX pathway in growth regulation of non-small-cell lung cancer. PLoS ONE 2013, 8, e74175. [Google Scholar] [CrossRef] [Green Version]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef]
- Singh, M.; Garg, N.; Venugopal, C.; Hallett, R.; Tokar, T.; McFarlane, N.; Mahendram, S.; Bakhshinyan, D.; Manoranjan, B.; Vora, P.; et al. STAT3 pathway regulates lung-derived brain metastasis initiating cell capacity through miR-21 activation. Oncotarget 2015, 6, 27461–27477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, E.; So Youn, K.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of beta-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Veve, R.; Hirsch, F.R.; Franklin, W.A. The E-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer 2002, 36, 115–124. [Google Scholar] [CrossRef]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, X.; Wu, L.; Su, D.; Mao, W.; Fang, B. Immunotherapy for non-small cell lung cancers: Biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer 2018, 18, 1082. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.-J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.-S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Muller-Tidow, C.; Metzger, R.; Kugler, K.; Diederichs, S.; Idos, G.; Thomas, M.; Dockhorn-Dworniczak, B.; Schneider, P.M.; Koeffler, H.P.; Berdel, W.E.; et al. Cyclin E is the only cyclin-dependent kinase 2-associated cyclin that predicts metastasis and survival in early stage non-small cell lung cancer. Cancer Res. 2001, 61, 647–653. [Google Scholar]
- Fujita, Y.; Yagishita, S.; Hagiwara, K.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Tsuta, K.; Nokihara, H.; Tamura, T.; et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol. Ther. 2015, 23, 717–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrini, L.; Giordano, M.; Niccoli, C.; Melfi, F.; Lucchi, M.; Mussi, A.; Fontanini, G. Role of microRNA-33a in regulating the expression of PD-1 in lung adenocarcinoma. Cancer Cell Int. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Chen, Z.; Chen, Y.; Wang, X.; Tang, N. MIR155HG is a prognostic biomarker and associated with immune infiltration and immune checkpoint molecules expression in multiple cancers. Cancer Med. 2019, 8, 7161–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denaro, N.; Merlano, M.C.; Lo Nigro, C. Long noncoding RNAs as regulators of cancer immunity. Mol. Oncol. 2019, 13, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, E.; Gazdar, A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur. Respir. J. 2009, 33, 1485–1497. [Google Scholar] [CrossRef]
- Shtivelman, E.; Hensing, T.; Simon, G.R.; Dennis, P.A.; Otterson, G.A.; Bueno, R.; Salgia, R. Molecular pathways and therapeutic targets in lung cancer. Oncotarget 2014, 5, 1392–1433. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Xiao, T.; Fang, Z.; Sun, Y.; Li, F.; Gao, Y.; Feng, Y.; Li, L.; Wang, Y.; Liu, X.; et al. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J. Biol. Chem. 2012, 287, 23227–23235. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Kang, W.; Nan, Y.; Lei, Z.; Zhongdong, L.; Demin, L.; Lei, S.; Hairong, H. The Downregulation of miR-200c Promotes Lactate Dehydrogenase A Expression and Non-Small Cell Lung Cancer Progression. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 1015–1022. [Google Scholar] [CrossRef]
- Zhai, S.; Zhao, L.; Lin, T.; Wang, W. Downregulation of miR-33b promotes non-small cell lung cancer cell growth through reprogramming glucose metabolism miR-33b regulates non-small cell lung cancer cell growth. J. Cell. Biochem. 2019, 120, 6651–6660. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, H.; Du, L.; Xi, P.; Wang, Q.; Li, Y.; Liu, D. miR-449a Suppresses LDHA-Mediated Glycolysis to Enhance the Sensitivity of Non-Small Cell Lung Cancer Cells to Ionizing Radiation. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 547–556. [Google Scholar] [CrossRef]
- Chen, H.; Pei, H.; Hu, W.; Ma, J.; Zhang, J.; Mao, W.; Nie, J.; Xu, C.; Li, B.; Hei, T.K.; et al. Long non-coding RNA CRYBG3 regulates glycolysis of lung cancer cells by interacting with lactate dehydrogenase A. J. Cancer 2018, 9, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisségur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadász, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Li, M.; Hu, J.; Lei, R.; Xiong, H.; Ji, H.; Yin, H.; Wei, Q.; Hu, G. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene 2017, 36, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef] [PubMed]
- Lima Queiroz, A.; Zhang, B.; Comstock, D.E.; Hao, Y.; Eriksson, M.; Hydbring, P.; Vakifahmetoglu-Norberg, H.; Norberg, E. miR-126-5p targets Malate Dehydrogenase 1 in non-small cell lung carcinomas. Biochem. Biophys. Res. Commun. 2018, 499, 314–320. [Google Scholar] [CrossRef]
- Li, Y.L.; Liu, X.M.; Zhang, C.Y.; Zhou, J.B.; Shao, Y.; Liang, C.; Wang, H.M.; Hua, Z.Y.; Lu, S.D.; Ma, Z.L. MicroRNA-34a/EGFR axis plays pivotal roles in lung tumorigenesis. Oncogenesis 2017, 6, e372. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-C.; Li, S.-J.; Zhang, F.; Pan, J.-Y.; Wang, L.; Yang, C.-L.; Xi, Y.-Y.; Li, D.J. Hsa-miR-329 exerts tumor suppressor function through down-regulation of MET in non-small cell lung cancer. Oncotarget 2016, 7, 21510–21526. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Sang, M.; Li, S.; Sun, X.; Yang, C.; Xi, Y.; Wang, L.; Zhang, F.; Bi, Y.; Fu, Y.; et al. Hsa-miR-139-5p inhibits proliferation and causes apoptosis associated with down-regulation of c-Met. Oncotarget 2015, 6, 39756–39792. [Google Scholar] [CrossRef]
- Sun, C.; Liu, Z.; Li, S.; Yang, C.; Xue, R.; Xi, Y.; Wang, L.; Wang, S.; He, Q.; Huang, J.; et al. Down-regulation of c-Met and Bcl2 by microRNA-206, activates apoptosis, and inhibits tumor cell proliferation, migration and colony formation. Oncotarget 2015, 6, 25533–25574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Yu, Z.; Lu, Y.; Fan, J.; Ni, Y.; Ma, L. microRNA-148a-3p inhibited the proliferation and epithelial-mesenchymal transition progression of non-small-cell lung cancer via modulating Ras/MAPK/Erk signaling. J. Cell. Physiol. 2019, 234, 12786–12799. [Google Scholar] [CrossRef] [PubMed]
- Seviour, E.G.; Sehgal, V.; Mishra, D.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Lee, J.S.; Sood, A.K.; Kim, M.P.; Mills, G.B.; et al. Targeting KRas-dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant miR-193a-3p. Oncogene 2017, 36, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Hu, X.; Zhang, H.; Wang, S.; Zhang, H.; You, C.; Zhang, C.Y.; Liang, H.; Chen, X.; Ba, Y. MiR-193a-3p is an Important Tumour Suppressor in Lung Cancer and Directly Targets KRAS. Cell. Physiol. Biochem. 2017, 44, 1311–1324. [Google Scholar] [CrossRef]
- Ma, Z.; Qiu, X.; Wang, D.; Li, Y.; Zhang, B.; Yuan, T.; Wei, J.; Zhao, B.; Zhao, X.; Lou, J.; et al. MiR-181a-5p inhibits cell proliferation and migration by targeting Kras in non-small cell lung cancer A549 cells. Acta Biochim. Biophys. Sin. 2015, 47, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Middleton, J.; Jeon, Y.-J.; Magee, P.; Veneziano, D.; Laganà, A.; Leong, H.-S.; Sahoo, S.; Fassan, M.; Booton, R.; et al. KRAS induces lung tumorigenesis through microRNAs modulation. Cell Death Dis. 2018, 9, 219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhang, G.; Hu, X.; Wu, L.; Feng, Y.; He, S.; Zhang, Y.; Hu, Z.; Yang, L.; Tian, T.; et al. Oncogenic RAS Regulates Long Noncoding RNA Orilnc1 in Human Cancer. Cancer Res. 2017, 77, 3745–3757. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Lai, S.; Hu, F.; Li, G.; Wang, G.; Luo, X.; Fu, X.; Hu, J. miR-19a contributes to gefitinib resistance and epithelial mesenchymal transition in non-small cell lung cancer cells by targeting c-Met. Sci. Rep. 2017, 7, 2939. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Zhu, L.; Xu, J.; Lu, B.; Yang, Y.; Liu, F.; Wang, Z. MicroRNA-409-3p Functions as a Tumor Suppressor in Human Lung Adenocarcinoma by Targeting c-Met. Cell. Physiol. Biochem. 2014, 34, 1273–1290. [Google Scholar] [CrossRef]
- Shi, H.; Pu, J.; Zhou, X.-L.; Ning, Y.-Y.; Bai, C. Silencing long non-coding RNA ROR improves sensitivity of non-small-cell lung cancer to cisplatin resistance by inhibiting PI3K/Akt/mTOR signaling pathway. Tumour Biol. 2017, 39, 1010428317697568. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhou, X.; Qiao, J.; Bao, A. MiR-142-3p Overexpression Increases Chemo-Sensitivity of NSCLC by Inhibiting HMGB1-Mediated Autophagy. Cell. Physiol. Biochem. 2017, 41, 1370–1382. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, C.; Xu, Y.; Qian, W.; Zheng, M. Negative Regulation of PTEN by MicroRNA-221 and Its Association with Drug Resistance and Cellular Senescence in Lung Cancer Cells. Biomed. Res. Int. 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Lu, C.; Chu, W.; Zhang, B.; Zhen, Q.; Wang, R.; Zhang, Y.; Li, Z.; Lv, B.; Li, H.; et al. MicroRNA-124 suppresses proliferation and glycolysis in non–small cell lung cancer cells by targeting AKT–GLUT1/HKII. Tumor Biol. 2017, 39, 1010428317706215. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, W.; Wang, J.; Zhang, J. MiR-182 promotes glucose metabolism by upregulating hypoxia-inducible factor 1α in NSCLC cells. Biochem. Biophys. Res. Commun. 2018, 504, 400–405. [Google Scholar] [CrossRef]
- Chang, L.; Xu, W.; Zhang, Y.; Gong, F. Long non-coding RNA-NEF targets glucose transportation to inhibit the proliferation of non-small-cell lung cancer cells. Oncol Lett 2019, 17, 2795–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.C.; Zheng, J.Q. Effect of microRNA-145 on proliferation and apoptosis of human non-small cell lung cancer A549 cells by regulating mTOR signaling pathway. J. Cell Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Xu, H.; Shan, J.; Tao, Y.; Hong, J.A.; Inchauste, S.; Zhang, M.; Kunst, T.F.; Mercedes, L.; Schrump, D.S. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Investig. 2013, 123, 1241–1261. [Google Scholar] [CrossRef] [PubMed]
- Mine, M.; Yamaguchi, K.; Sugiura, T.; Chigita, S.; Yoshihama, N.; Yoshihama, R.; Hiyake, N.; Kobayashi, Y.; Mori, Y. miR-203 Inhibits Frizzled-2 Expression via CD82/KAI1 Expression in Human Lung Carcinoma Cells. PLoS ONE 2015, 10, e0131350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xia, H.; Zhuang, Z.; Miao, L.; Chen, X.; Cai, H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. 2014, 5, e1227. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Li, H.; Liu, C.; Hu, B.; Li, T.; Wang, Y. Long noncoding RNA AK126698 inhibits proliferation and migration of non-small cell lung cancer cells by targeting Frizzled-8 and suppressing Wnt/β-catenin signaling pathway. Onco. Targets Ther. 2016, 9, 3815–3827. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.; Zhu, T.; Wu, S.; Liu, S.; Liu, B.; Wu, J.; Cai, J.; Zhu, X.; Zhang, X.; Zeng, M.; et al. Long noncoding RNA LINC00673-v4 promotes aggressiveness of lung adenocarcinoma via activating WNT/β-catenin signaling. Proc. Natl. Acad. Sci. 2019, 116, 14019–14028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunato, O.; Boeri, M.; Moro, M.; Verri, C.; Mensah, M.; Conte, D.; Caleca, L.; Roz, L.; Pastorino, U.; Sozzi, G. Mir-660 is downregulated in lung cancer patients and its replacement inhibits lung tumorigenesis by targeting MDM2-p53 interaction. Cell Death Dis. 2014, 5, e1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, C.; Li, Y.; Wang, P.; Yue, Z.; Xie, S. miR-98 regulates cisplatin-induced A549 cell death by inhibiting TP53 pathway. Biomed. Pharmacother. 2011, 65, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, M.; Jiang, N.; Shi, K.; Qian, R. A regulatory circuit of circ-MTO1/miR-17/QKI-5 inhibits the proliferation of lung adenocarcinoma. Cancer Biol. Ther. 2019, 20, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.C.; Kajino, T.; Nakatochi, M.; Arima, C.; Shimada, Y.; Suzuki, M.; Miyoshi, H.; Yatabe, Y.; Yanagisawa, K.; Takahashi, T. miR-342-3p regulates MYC transcriptional activity via direct repression of E2F1 in human lung cancer. Carcinogenesis 2015, 36, 1464–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Shu-Ling, W.; Jing-Bo, H.; Ying, Z.; Rong, H.; Xiang-Qun, L.; Wen-Jie, C.; Lin-Fu, Z. MiR-451a attenuates doxorubicin resistance in lung cancer via suppressing epithelialmesenchymal transition (EMT) through targeting c-Myc. Biomed. Pharmacother. 2020, 125, 109962. [Google Scholar] [CrossRef]
- Zhu, D.; Yu, Y.; Wang, W.; Wu, K.; Liu, D.; Yang, Y.; Zhang, C.; Qi, Y.; Zhao, S. Long noncoding RNA PART1 promotes progression of non-small cell lung cancer cells via JAK-STAT signaling pathway. Cancer Med. 2019, 8, 6064–6081. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Zhang, T. Downregulation of MicroRNA-135 Promotes Sensitivity of Non-Small Cell Lung Cancer to Gefitinib by Targeting TRIM16. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 1005–1014. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.-T.; Lin, H.-H.; Lien, Y.-C.; Wang, Y.-H.; Hong, C.-F.; Kao, Y.-R.; Lin, S.-C.; Chang, Y.-C.; Lin, S.-Y.; Chen, S.-J.; et al. EGFR Promotes Lung Tumorigenesis by Activating miR-7 through a Ras/ERK/Myc Pathway That Targets the Ets2 Transcriptional Repressor ERF. Cancer Res. 2010, 70, 8822–8831. [Google Scholar] [CrossRef] [Green Version]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliore, C.; Morando, E.; Ghiso, E.; Anastasi, S.; Leoni, V.P.; Apicella, M.; Cora, D.; Sapino, A.; Pietrantonio, F.; De Braud, F.; et al. miR-205 mediates adaptive resistance to MET inhibition via ERRFI1 targeting and raised EGFR signaling. EMBO Mol. Med. 2018, 10, e8746. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Nam, B.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Kim, W.S.; Choi, C.-M.; Chang, E.-J.; Koh, J.S.; et al. Enhanced Glycolysis Supports Cell Survival in EGFR-Mutant Lung Adenocarcinoma by Inhibiting Autophagy-Mediated EGFR Degradation. Cancer Res. 2018, 78, 4482–4496. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual MET–EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef] [Green Version]
- Vanhove, K.; Graulus, G.-J.; Mesotten, L.; Thomeer, M.; Derveaux, E.; Noben, J.-P.; Guedens, W.; Adriaensens, P. The Metabolic Landscape of Lung Cancer: New Insights in a Disturbed Glucose Metabolism. Front. Oncol. 2019, 9, 1215. [Google Scholar] [CrossRef] [Green Version]
- Fumarola, C.; Bonelli, M.A.; Petronini, P.G.; Alfieri, R.R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 2014, 90, 197–207. [Google Scholar] [CrossRef]
- Wang, L.; Chen, R.; Zhang, Y. miR-296-3p targets APEX1 to suppress cell migration and invasion of non-small-cell lung cancer. Oncol. Lett. 2019, 18, 2612–2618. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Lin, Y.; Meng, S.; Guo, Y.; Zhang, J.; Zhang, W. miRNA-296-3p modulates chemosensitivity of lung cancer cells by targeting CX3CR1. Am. J. Transl. Res. 2016, 8, 1848–1856. [Google Scholar] [PubMed]
- Xu, T.; Jiang, L.; Wang, Z. The progression of HMGB1-induced autophagy in cancer biology. Onco. Targets Ther. 2018, 12, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Cai, J.; Fang, L.; Huang, Y.; Li, R.; Yuan, J.; Yang, Y.; Zhu, X.; Chen, B.; Wu, J.; Li, M. miR-205 Targets PTEN and PHLPP2 to Augment AKT Signaling and Drive Malignant Phenotypes in Non–Small Cell Lung Cancer. Cancer Res. 2013, 73, 5402–5415. [Google Scholar] [CrossRef] [Green Version]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Gao, H.; Xu, R.; Wang, H.; Mei, J.; Liu, C. The interplay between HIF-1α and noncoding RNAs in cancer. J. Exp. Clin. Cancer Res. 2020, 39, 27. [Google Scholar] [CrossRef]
- Zhou, C.; Ye, L.; Jiang, C.; Bai, J.; Chi, Y.; Zhang, H. Long noncoding RNA HOTAIR, a hypoxia-inducible factor-1α activated driver of malignancy, enhances hypoxic cancer cell proliferation, migration, and invasion in non-small cell lung cancer. Tumour Biol. 2015, 36, 9179–9188. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wilson, M.J. miR-520c and miR-373 upregulate MMP9 expression by targeting mTOR and SIRT1, and activate the Ras/Raf/MEK/Erk signaling pathway and NF-κB factor in human fibrosarcoma cells. J. Cell. Physiol. 2012, 227, 867–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [Green Version]
- R Babu, K.; Tay, Y. The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. Int. J. Mol. Sci. 2019, 20, 5335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C. microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. Int. J. Mol. Sci. 2019, 20, 6094. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D. Wnt Signaling Pathway in Non-Small Cell Lung Cancer. JNCI 2013, 106. [Google Scholar] [CrossRef]
- Xia, Y.; He, Z.; Liu, B.; Wang, P.; Chen, Y. Downregulation of Meg3 enhances cisplatin resistance of lung cancer cells through activation of the WNT/β-catenin signaling pathway. Mol. Med. Rep. 2015, 12, 4530–4537. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gao, L.; Zhang, H.; Wang, D.; Wang, M.; Zhu, J.; Pang, C.; Wang, C. Succinate dehydrogenase 5 (SDH5) regulates glycogen synthase kinase 3β-β-catenin-mediated lung cancer metastasis. J. Biol. Chem. 2013, 288, 29965–29973. [Google Scholar] [CrossRef] [Green Version]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt–β-catenin signalling through Dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Rharass, T.; Lemcke, H.; Lantow, M.; Kuznetsov, S.A.; Weiss, D.G.; Panáková, D. Ca2+-mediated mitochondrial reactive oxygen species metabolism augments Wnt/β-catenin pathway activation to facilitate cell differentiation. J. Biol. Chem. 2014, 289, 27937–27951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chocarro-Calvo, A.; García-Martínez, J.M.; Ardila-González, S.; De la Vieja, A.; García-Jiménez, C. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol. Cell 2013, 49, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-t.; Li, X.; Liu, Z.-l.; Xu, Z.; Dai, W.; Zhang, K.; Wu, J.-s.; Arshad, B.; Wu, K.-n.; Kong, L.-q. Hepatitis B virus reactivation and antiviral prophylaxis during lung cancer chemotherapy: A systematic review and meta-analysis. PLOS ONE 2017, 12, e0179680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.-H.; Liao, W.-Y.; Ho, C.-C.; Chen, K.-Y.; Shih, J.-Y.; Chen, J.-S.; Lin, Z.-Z.; Lin, C.-C.; Yang, J.C.-H.; Yu, C.-J. Incidence of hepatitis B reactivation during epidermal growth factor receptor tyrosine kinase inhibitor treatment in non–small-cell lung cancer patients. Eur. J. Cancer 2019, 117, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Dundar, H.Z.; Aksoy, F.; Aksoy, S.A.; Tasar, P.; Ugras, N.; Tunca, B.; Egeli, U.; Cecener, G.; Yerci, O.; Kaya, E. Overexpression of miR-21 Is Associated With Recurrence in Patients With Hepatitis B Virus–Mediated Hepatocellular Carcinoma Undergoing Liver Transplantation. Transplant. Proc. 2019, 51, 1157–1161. [Google Scholar] [CrossRef]
- Jin, J.; Tang, S.; Xia, L.; Du, R.; Xie, H.; Song, J.; Fan, R.; Bi, Q.; Chen, Z.; Yang, G.; et al. MicroRNA-501 promotes HBV replication by targeting HBXIP. Biochem. Biophys. Res. Commun. 2013, 430, 1228–1233. [Google Scholar] [CrossRef]
- Feng, J.; Yang, G.; Liu, Y.; Gao, Y.; Zhao, M.; Bu, Y.; Yuan, H.; Yuan, Y.; Yun, H.; Sun, M.; et al. LncRNA PCNAP1 modulates hepatitis B virus replication and enhances tumor growth of liver cancer. Theranostics 2019, 9, 5227–5245. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G1-modulated P53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef]
- Yang, X.; Li, H.; Sun, H.; Fan, H.; Hu, Y.; Liu, M.; Li, X.; Tang, H. Hepatitis B Virus-Encoded MicroRNA Controls Viral Replication. J. Virol. 2017, 91, e01919. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Wang, X.; Ding, X.; Li, Y.; Zhang, X.; Xie, P.; Yang, J.; Wang, S. MicroRNA-141 represses HBV replication by targeting PPARA. PLoS ONE 2012, 7, e34165. [Google Scholar] [CrossRef] [Green Version]
- Oura, K.; Fujita, K.; Morishita, A.; Iwama, H.; Nakahara, M.; Tadokoro, T.; Sakamoto, T.; Nomura, T.; Yoneyama, H.; Mimura, S.; et al. Serum microRNA-125a-5p as a potential biomarker of HCV-associated hepatocellular carcinoma. Oncol. Lett. 2019, 18, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Mao, W.; Zheng, S.; Ye, J. Epidermal growth factor receptor-regulated miR-125a-5p--a metastatic inhibitor of lung cancer. FEBS J. 2009, 276, 5571–5578. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, J.; Wang, H.; Shi, J.; Wu, K.; Liu, S.; Liu, Y.; Wu, J. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013, 9, e1003248. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnow, P.; Sagan, S.M. Unraveling the Mysterious Interactions Between Hepatitis C Virus RNA and Liver-Specific MicroRNA-122. Annu. Rev. Virol. 2016, 3, 309–332. [Google Scholar] [CrossRef]
- Chen, F.-F.; Yan, J.-J.; Lai, W.-W.; Jin, Y.-T.; Su, I.-J. Epstein-barr virus-associated nonsmall cell lung carcinoma. Cancer 1998, 82, 2334–2342. [Google Scholar] [CrossRef]
- Han, A.J.; Xiong, M.; Zong, Y.S. Association of Epstein-Barr Virus With Lymphoepithelioma-Like Carcinoma of the Lung in Southern China. Am. J. Clin. Pathol. 2000, 114, 220–226. [Google Scholar] [CrossRef]
- Gómez-Román, J.J.; Martínez, M.N.; Fernández, S.L.; Val-Bernal, J.F. Epstein–Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod. Pathol. 2009, 22, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xiong, H.; Yan, S.; Wu, N.; Lu, Z. Identification and Characterization of Epstein-Barr Virus Genomes in Lung Carcinoma Biopsy Samples by Next-Generation Sequencing Technology. Sci. Rep. 2016, 6, 26156. [Google Scholar] [CrossRef] [Green Version]
- Kuzembayeva, M.; Hayes, M.; Sugden, B. Multiple functions are mediated by the miRNAs of Epstein-Barr virus. Curr Opin Virol 2014, 7, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr Top. Microbiol Immunol 2015, 391, 181–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movassagh, M.; Oduor, C.; Forconi, C.; Moormann, A.M.; Bailey, J.A. Sensitive detection of EBV microRNAs across cancer spectrum reveals association with decreased survival in adult acute myelocytic leukemia. Sci. Rep. 2019, 9, 20321. [Google Scholar] [CrossRef]
- Koshiol, J.; Gulley, M.L.; Zhao, Y.; Rubagotti, M.; Marincola, F.M.; Rotunno, M.; Tang, W.; Bergen, A.W.; Bertazzi, P.A.; Roy, D.; et al. Epstein-Barr virus microRNAs and lung cancer. Br. J. Cancer 2011, 105, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.C.-m.; Leung, C.-C. Management of co-existent tuberculosis and lung cancer. Lung Cancer 2018, 122, 83–87. [Google Scholar] [CrossRef]
- Abd-El-Fattah, A.A.; Sadik, N.A.H.; Shaker, O.G.; Aboulftouh, M.L. Differential MicroRNAs Expression in Serum of Patients with Lung Cancer, Pulmonary Tuberculosis, and Pneumonia. Cell Biochem. Biophys. 2013, 67, 875–884. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, D.; Yao, R.; Wang, M.; Wang, Y.; Yao, Y.; Li, X.; Zhang, H. microRNA expression profiles associated with survival, disease progression, and response to gefitinib in completely resected non-small-cell lung cancer with EGFR mutation. Med. Oncol. 2013, 30, 750. [Google Scholar] [CrossRef]
- Shen, H.; Shen, J.; Wang, L.; Shi, Z.; Wang, M.; Jiang, B.-h.; Shu, Y. Low miR-145 expression level is associated with poor pathological differentiation and poor prognosis in non-small cell lung cancer. Biomed. Pharmacother. 2015, 69, 301–305. [Google Scholar] [CrossRef]
- Yuxia, M.; Zhennan, T.; Wei, Z. Circulating miR-125b is a novel biomarker for screening non-small-cell lung cancer and predicts poor prognosis. J. Cancer Res. Clin. Oncol. 2012, 138, 2045–2050. [Google Scholar] [CrossRef]
- Stenvold, H.; Donnem, T.; Andersen, S.; Al-Saad, S.; Busund, L.-T.; Bremnes, R.M. Stage and tissue-specific prognostic impact of miR-182 in NSCLC. BMC Cancer 2014, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Zaporozhchenko, I.A.; Morozkin, E.S.; Skvortsova, T.E.; Ponomaryova, A.A.; Rykova, E.Y.; Cherdyntseva, N.V.; Polovnikov, E.S.; Pashkovskaya, O.A.; Pokushalov, E.A.; Vlassov, V.V.; et al. Plasma miR-19b and miR-183 as Potential Biomarkers of Lung Cancer. PLoS ONE 2016, 11, e0165261. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Liu, X.; Luo, P.; Jing, W.; Zhu, M.; Tu, J. Identification of Circulating Long Noncoding RNA HOTAIR as a Novel Biomarker for Diagnosis and Monitoring of Non–Small Cell Lung Cancer. Technol. Cancer Res. Treat. 2017, 16, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Feng, X.B.; Tan, Q.; Luo, P.; Jing, W.; Zhu, M.; Liang, C.; Tu, J.; Ning, Y. Identification of Circulating Long Noncoding RNA Linc00152 as a Novel Biomarker for Diagnosis and Monitoring of Non-Small-Cell Lung Cancer. Dis. Markers 2017, 2017, 7439698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Lv, T.; Shi, X.; Liu, H.; Zhu, Q.; Zeng, J.; Yang, W.; Yin, J.; Song, Y. Circulating long noncoding RNA GAS5 is a novel biomarker for the diagnosis of nonsmall cell lung cancer. Medicine 2016, 95, e4608. [Google Scholar] [CrossRef] [PubMed]
- Kamel, L.M.; Atef, D.M.; Mackawy, A.M.H.; Shalaby, S.M.; Abdelraheim, N. Circulating long non-coding RNA GAS5 and SOX2OT as potential biomarkers for diagnosis and prognosis of non-small cell lung cancer. Biotechnol. Appl. Biochem. 2019, 66, 634–642. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, E.J.; Lee, S.; Tan, X.; Liu, X.; Park, S.; Kang, K.; Yoon, J.-S.; Ko, Y.H.; Kurie, J.M.; et al. MiR-34a and miR-34b/c have distinct effects on the suppression of lung adenocarcinomas. Exp. Mol. Med. 2019, 51, 9. [Google Scholar] [CrossRef] [Green Version]
- Ekert, J.E.; Johnson, K.; Strake, B.; Pardinas, J.; Jarantow, S.; Perkinson, R.; Colter, D.C. Three-Dimensional Lung Tumor Microenvironment Modulates Therapeutic Compound Responsiveness In Vitro – Implication for Drug Development. PLoS ONE 2014, 9, e92248. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.-J.; Chun, S.-M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Movia, D.; Bazou, D.; Volkov, Y.; Prina-Mello, A. Multilayered Cultures of NSCLC cells grown at the Air-Liquid Interface allow the efficacy testing of inhaled anti-cancer drugs. Sci. Rep. 2018, 8, 12920. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Berghmans, T.; Ameye, L.; Lafitte, J.-J.; Colinet, B.; Cortot, A.; CsToth, I.; Holbrechts, S.; Lecomte, J.; Mascaux, C.; Meert, A.-P.; et al. Prospective Validation Obtained in a Similar Group of Patients and with Similar High Throughput Biological Tests Failed to Confirm Signatures for Prediction of Response to Chemotherapy and Survival in Advanced NSCLC: A Prospective Study from the European Lung Cancer Working Party. Front. Oncol. 2015, 4, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghmans, T.; Ameye, L.; Willems, L.; Paesmans, M.; Mascaux, C.; Lafitte, J.J.; Meert, A.P.; Scherpereel, A.; Cortot, A.B.; Cstoth, I.; et al. Identification of microRNA-based signatures for response and survival for non-small cell lung cancer treated with cisplatin-vinorelbine A ELCWP prospective study. Lung Cancer 2013, 82, 340–345. [Google Scholar] [CrossRef]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—an update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
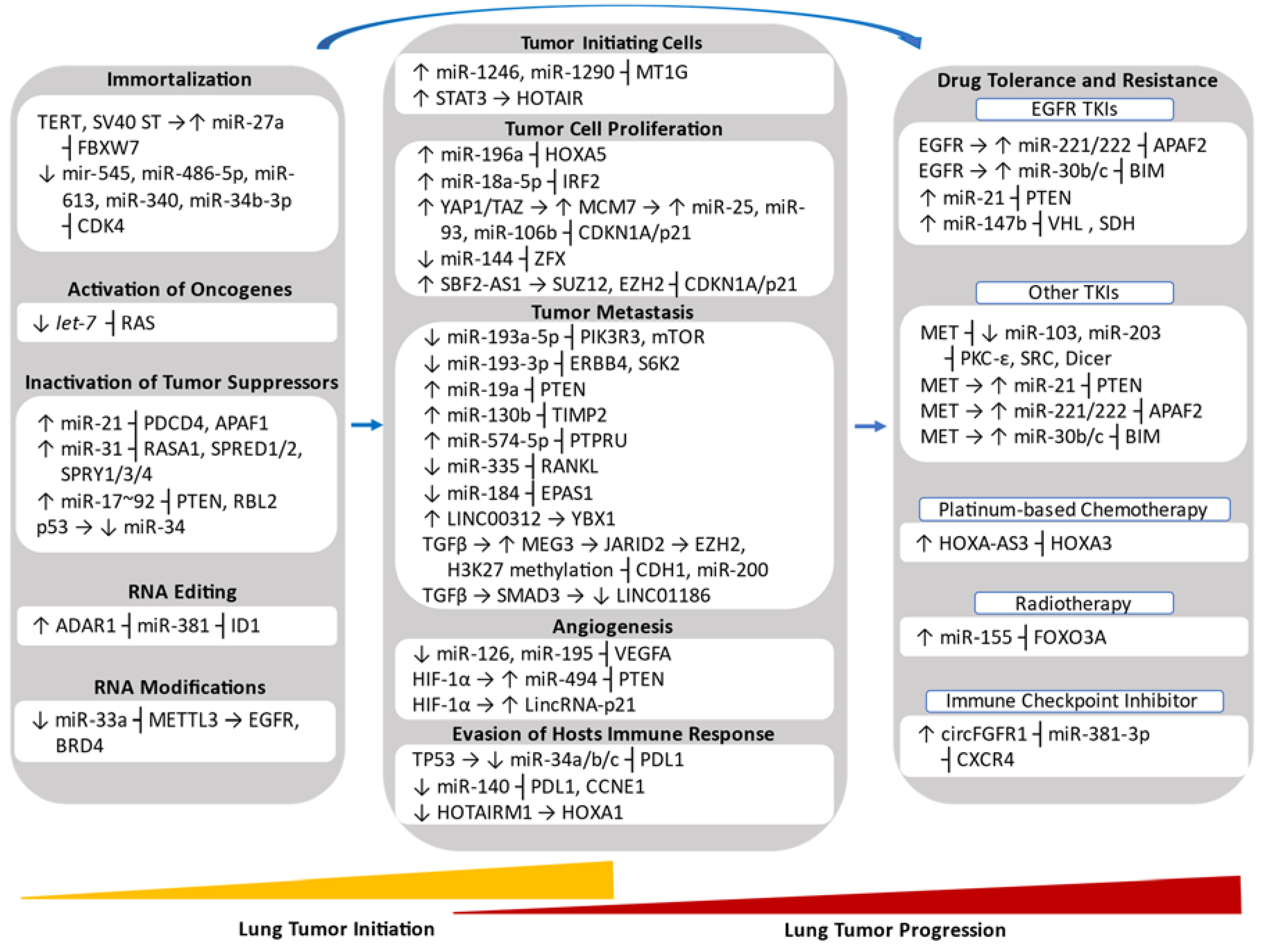



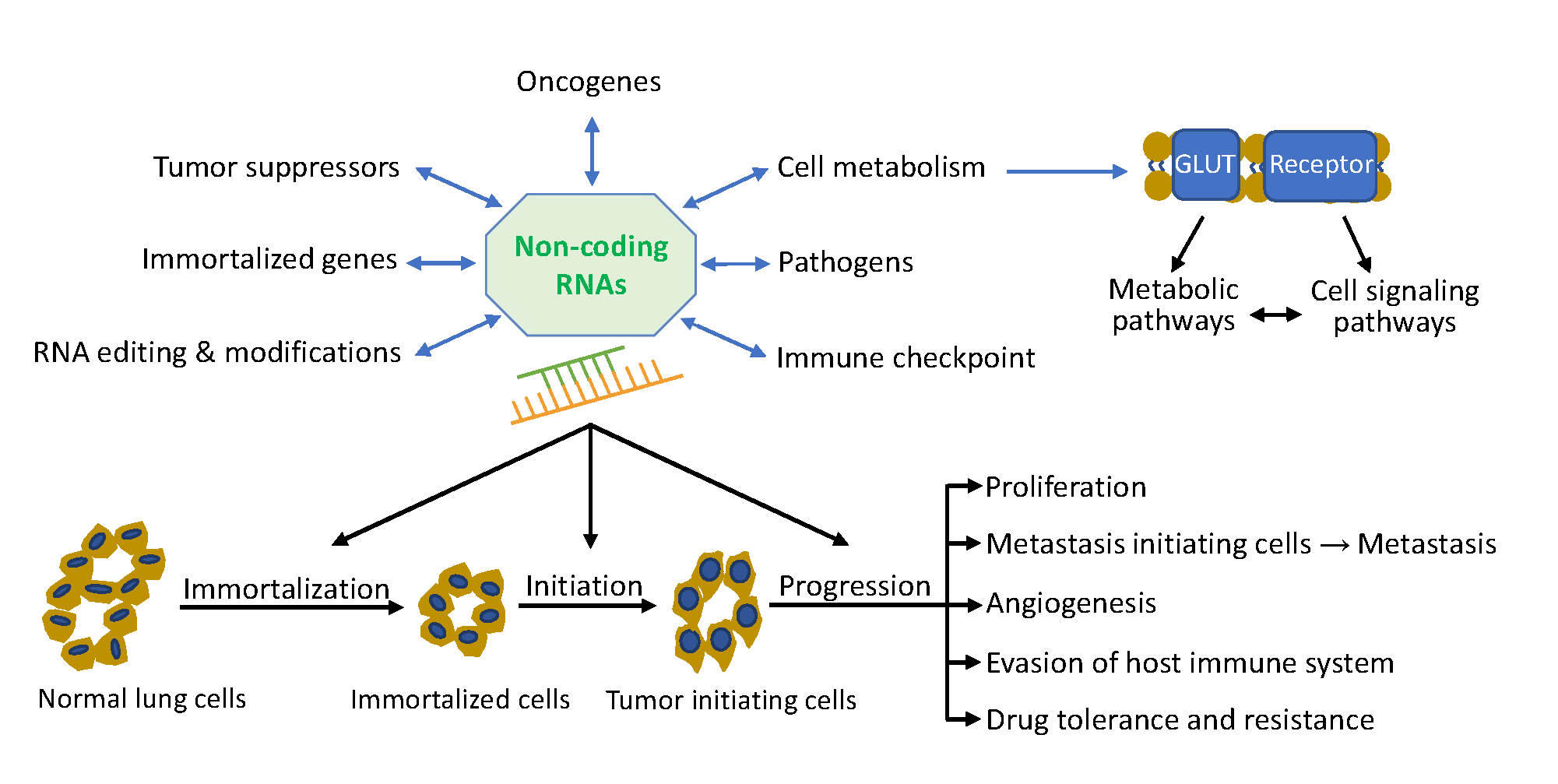{kind=link}
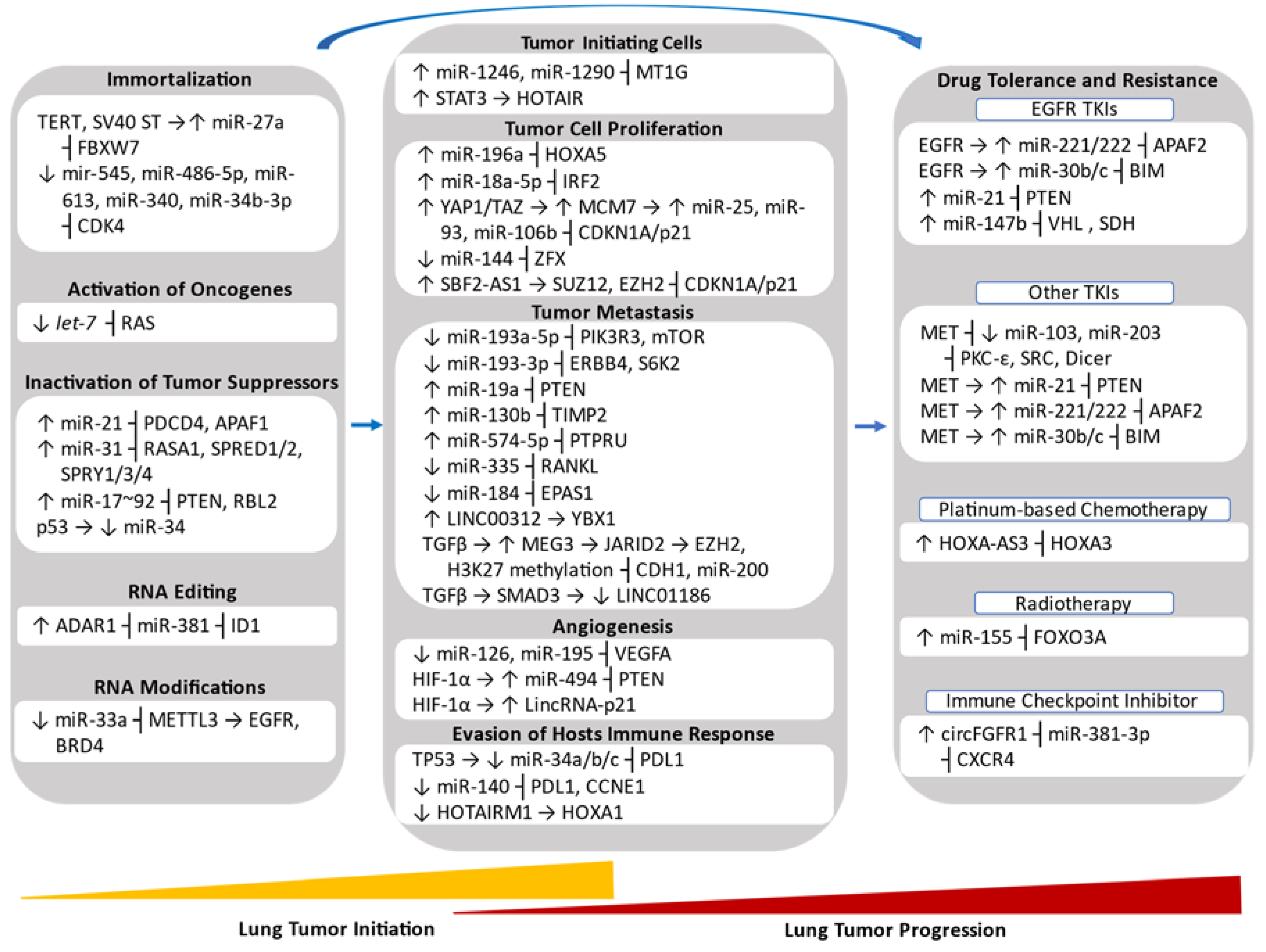{kind=link}
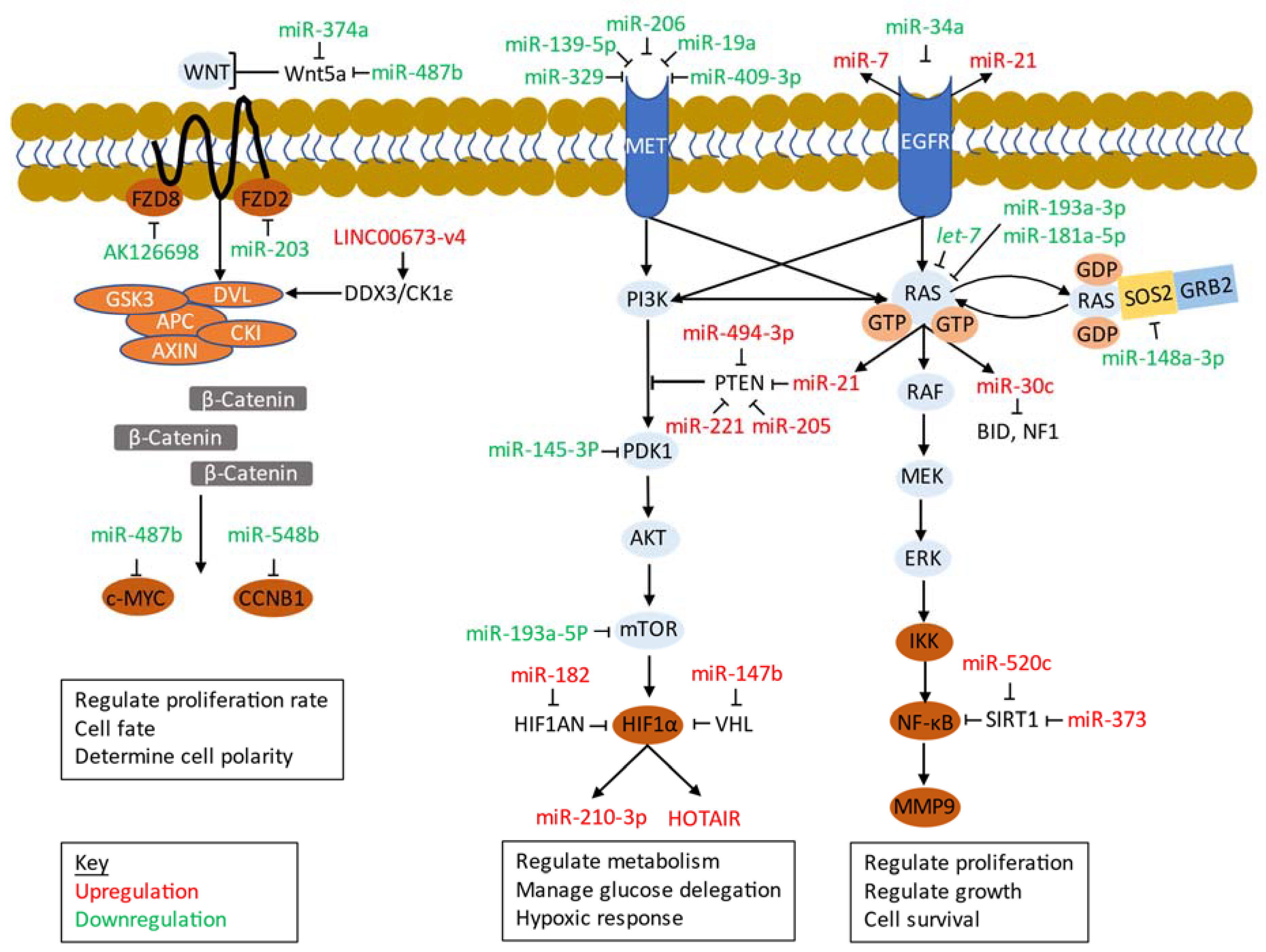{kind=link}
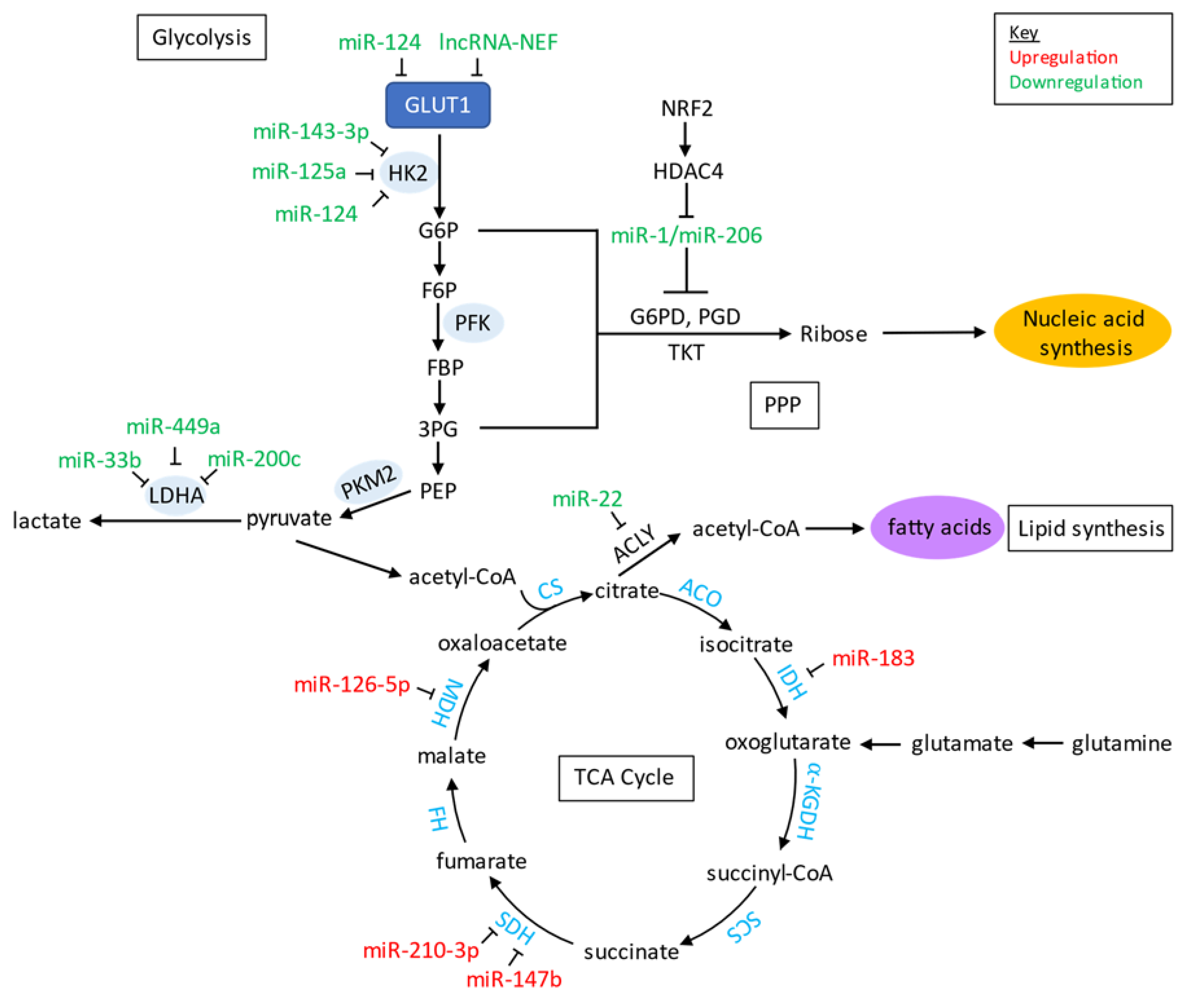{kind=link}
| miRNA | Lung Cancer Subtype | Downstream Direct Targets | Hallmark | Initiation (I)/Progression (P) | References |
|---|---|---|---|---|---|
| Upregulated miRNAs | |||||
| miR-1246, miR-1290 | LUAD | MT1G | Enrichment of TICs | I and P | [83] |
| miR-21 | NSCLC | PDCD4, APAF1 | Inactivation of Tumor Suppressor | I | [90] |
| miR-31 | NSCLC | RASA1, SPRED1/2, SPRY1/3/4 | Inactivation of Tumor Suppressor | I | [91] |
| miR-17~92 | SCLC | PTEN, RBL2 | Inactivation of Tumor Suppressor | I | [92] |
| miR-196a | NSCLC | HOXA5 | Promotion of Metastasis | P | [93] |
| miR-18a-5p | NSCLC | IRF2 | Promotion of Proliferation and Inhibition of Apoptosis | I | [94] |
| miR-25, miR-93, miR-106b | NSCLC | p21 | Promotion of Proliferation | P | [95] |
| miR-19a | NSCLC | PTEN | Promotion of EMT and Metastasis | P | [96] |
| miR-130b | NSCLC | TIMP2 | Promotion of Metastasis | P | [97] |
| miR-574-5p | SCLC | PTPRU | Promotion of Metastasis | P | [98] |
| miR-494 | NSCLC | PTEN | Promotion of Angiogenesis | P | [99] |
| miR-221/222 | NSCLC | APAF1 | Promotion of Drug Resistance to EGFR TKI gefitinib | P | [100] |
| miR-30b/c | NSCLC | BIM | Promotion of Drug Resistance to EGFR TKI gefitinib | P | [100] |
| miR-21 | NSCLC | PTEN | Promotion of Drug Resistance to EGFR TKI gefitinib | P | [101] |
| miR-147b | LUAD | VHL, SDH | Promotion of Drug Tolerance to EGFR TKI osimertinib | P | [102] |
| miR-155 | NSCLC | FOXO3A | Promotion of Resistance to Radiotherapy | I | [103] |
| Downregulated miRNAs | |||||
| miR-184 | SCLC | EPAS1/HIF-2α | Promotion of Metastasis | P | [98] |
| miR-103, miR-203 | NSCLC | PKC-ε, SRC and Dicer | Promotion of Drug Resistance to EGFR TKI gefitinib | P | [100] |
| let-7 | NSCLC | RAS | Activation of Oncogene | I | [104] |
| miR-33 | NSCLC | METTL3 | Promotion of RNA Modifications | I and P | [105] |
| miR-193a-5p | NSCLC | PIK3R3, mTOR | Promotion of Metastasis | P | [106] |
| miR-193a-3p | NSCLC | ERBB4, S6K2 | Promotion of Metastasis | P | [106] |
| miR-335 | SCLC | RANKL | Promotion of Metastasis | P | [107] |
| miR-126 | LUAD | VEGFA | Promotion of Angiogenesis | P | [108] |
| miR-195 | LUSC | VEGFA | Promotion of Angiogenesis | P | [109] |
| miR-34a/b/c | NSCLC | PDL1 | Evasion of Host Immune Response | P | [110] |
| miR-140 | NSCLC | PDL1, CCNE1 | Evasion of Host Immune Response | P | [111] |
| LncRNA and circRNA | Lung Cancer Subtype | Downstream Direct Targets | Hallmark | Initiation (I)/Progression (P) | References |
|---|---|---|---|---|---|
| Upregulated LncRNAs | |||||
| HOTAIR | NSCLC | N.A. | Enrichment of TICs | I | [88] |
| SBF2-AS1 | NSCLC/SCLC | SUZ12, EZH2 | Promotion of Tumor Cell Proliferation | P | [112] |
| LINC00312 | LUAD | YBX1 | Promotion of Vasculogenic Mimicry and Metastasis | P | [113] |
| MEG3 | NSCLC | JARID2 | Promotion of TGFβ-induced EMT | I and P | [114] |
| HOXA-AS3 | LUAD, LUSC | HOXA3 | Promotion of Drug Resistance to Platinum-based Chemotherapy | P | [115] |
| SOX2OT | LUSC | N.A. | Promotion of Tumor Cell Proliferation | P | [116] |
| H19 | NSCLC | miR-107 | Promotion of Tumor Cell Proliferation | P | [117] |
| ANRIL | NSCLC | EZH2 | Promotion of Tumor Cell Proliferation and Metastasis | P | [118] |
| CAR10 | LUAD | miR-30, miR-203 | Promotion of Metastasis | P | [119] |
| MALAT1 | NSCLC | miR-204 | Promotion of Metastasis | P | [120] |
| PVT1 | NSCLC | miR-195 | Promotion of Resistance to Radiotherapy | P | [121] |
| BC087858 | NSCLC | N.A. | Promotion of Drug Resistance to EGFR TKI gefitinib | P | [122] |
| Downregulated LncRNAs | |||||
| LINC01186 | NSCLC | N.A. | Inhibition of TGFβ-induced EMT | P | [123,124] |
| LincRNA-p21 | LUAD | N.A. | Promotion of Hypoxia-induced Angiogenesis | P | [125] |
| HOTAIRM1 | LUAD | HOXA1 | Inhibition of Evasion of Hosts Immune Response | P | [126] |
| PANDAR | NSCLC | NF-YA | Inhibition of Tumor Cell Proliferation | P | [127] |
| MIR22HG | NSCLC | YBX1 | Inhibition of Tumor Cell Proliferation | P | [128] |
| GAS5 | NSCLC | N.A. | Inhibition of Tumor Cell Proliferation, Activation of Tumor Suppressors | I/P | [129] |
| GAS5 | NSCLC | IGF-1R | Promotion of Apoptosis, Inhibition of Drug Resistance to EGFR TKI gefitinib | P | [130] |
| Upregulated circRNAs | |||||
| circFGFR1 | NSCLC | miR-381-3p | Promotion of Progression and Resistance to Immune Checkpoint Inhibitors | P | [131,132] |
| circ_0020123 | NSCLC | miR-144 | Promotion of Tumor Cell Proliferation | P | [133] |
| circTP63 | LUSC | miR-873-3p | Promotion of Tumor Cell Proliferation | P | [134] |
| circPRKCI | LUAD | miR-545, miR-589 | Promotion of Tumor Cell Proliferation, Promotion of Drug Resistance to EGFR TKI gefitinib | P | [135] |
| circ_0000064 | NSCLC | N.A. | Promotion of Tumor Cell Proliferation and Metastasis, Inhibition of Cell Apoptosis | P | [136] |
| F-circEA | NSCLC | N.A. | Promotion of Metastasis | P | [137] |
| circ_0067934 | NSCLC | N.A. | Promotion of EMT and Metastasis | P | [138] |
| CDR1as | NSCLC | miR-7 | Activation of Oncogene, Inhibition of Apoptosis | P | [139] |
| circ_103809 | NSCLC | miR-4302 | Activation of Oncogene | P | [140] |
| Downregulated circRNAs | |||||
| cir-ITCH | NSCLC | miR-7, miR-214 | Inhibition of Tumor Cell Proliferation | P | [141] |
| circ_100395 | NSCLC | miR-1228 | Inhibition of Tumor Cell Proliferation and Metastasis | P | [142] |
| circ_0033155 | NSCLC | PTEN | Inhibition of Tumor Cell Proliferation and Metastasis | P | [143] |
| circRNF13 | NSCLC | miR-93-5p | Inhibition of Metastasis | P | [144] |
| Name of ncRNA | Type of ncRNA | Targets | Signaling Pathway | Metabolic Pathway | References |
|---|---|---|---|---|---|
| miR-21 | miRNA | PTEN | Ras/Raf/MEK/ERK, PI3K/Akt/mTOR | Glycolysis, Glucose-mediated TCA Cycle | [101] |
| miR-147b | miRNA | VHL, SDH | EGFR | ETC, TCA Cycle | [102] |
| miR-34a | miRNA | EGFR | Ras/Raf/MEK/ERK, PI3K/Akt/mTOR | Glycolysis | [199] |
| miR-329 | miRNA | MET | Ras/Raf/MEK/ERK, PI3K/Akt/mTOR | N.A. | [200] |
| miR-139-5p | miRNA | MET | Ras/Raf/MEK/ERK, PI3K/Akt/mTOR | N.A. | [201] |
| miR-206 | miRNA | MET | Ras/Raf/MEK/ERK, PI3K/Akt/mTOR | N.A. | [202] |
| miR-148a-3p | miRNA | SOS2 | Ras/Raf/MEK/ERK | N.A. | [203] |
| miR-193a-3p | miRNA | KRAS | Ras/Raf/MEK/ERK | N.A. | [204,205] |
| miR-181a-5p | miRNA | KRAS | Ras/Raf/MEK/ERK | N.A. | [206] |
| miR-30c | miRNA | BID, NF1 | Ras/Raf/MEK/ERK | Glucose-mediated TCA Cycle | [207] |
| Orilnc1 | lncRNA | N.A. | Ras/Raf/MEK/ERK | N.A. | [208] |
| miR-494-3p | miRNA | PTEN | PI3K/Akt/mTOR | Glycolysis | [85] |
| miR-19a | miRNA | MET | PI3K/Akt/mTOR | N.A. | [209] |
| miR-409-3p | miRNA | MET | PI3K/Akt/mTOR | N.A. | [210] |
| ROR | lncRNA | N.A. | PI3K/Akt/mTOR | Glycolysis | [211] |
| miR-142-3p | miRNA | HMGB1 | PI3K/Akt/mTOR | Glycolysis | [212] |
| miR-221 | miRNA | PTEN | PI3K/Akt/mTOR | Glycolysis | [213] |
| miR-124 | miRNA | GLUT1, HK2 | PI3K/Akt/mTOR | Glycolysis | [214] |
| miR-182 | miRNA | HIF1AN | PI3K/Akt/mTOR | ETC, TCA Cycle | [215] |
| lncRNA-NEF | lncRNA | GLUT1 | PI3K/Akt/mTOR | Glycolysis | [216] |
| miR-145-3p | miRNA | PDK1 | PI3K/Akt/mTOR | N.A. | [217] |
| miR-487b | miRNA | KRAS, Wnt5a, MYC | Wnt/β-Catenin | N.A. | [218] |
| miR-203 | miRNA | FZD2 | Wnt/β-Catenin | N.A. | [219] |
| miR-548b | miRNA | CCNB1 | Wnt/β-Catenin | N.A. | [220] |
| miR-374a | miRNA | Wnt5a | Wnt/β-Catenin | N.A. | [220] |
| AK126698 | lncRNA | FZD8 | Wnt/β-Catenin | ETC | [221] |
| LINC00673-v4 | lncRNA | DDX3 | Wnt/β-Catenin | N.A. | [222] |
| miR-660 | miRNA | MDM2 | p53 | N.A. | [223] |
| miR-98miR-453 | miRNA | TP53 | p53 | N.A. | [224] |
| circ-MTO1 | circRNA | miR-17 ┤QKI-5 | Notch | N.A. | [225] |
| circRNA_103809 | circRNA | miR-4302 ┤ZN121 | MYC | N.A. | [140] |
| miR-342-3p | miRNA | E2F1 | MYC | N.A. | [226] |
| miR-451a | miRNA | MYC | MYC | N.A. | [227] |
| PART1 | lncRNA | miR-635 | JAK/STAT | N.A. | [228] |
| miR-135 | miRNA | TRIM16 | JAK/STAT | N.A. | [229] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, R.M.; Moreno, C.; Zhang, W.C. Non-Coding RNAs in Lung Tumor Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2774. https://doi.org/10.3390/ijms21082774
Santos RM, Moreno C, Zhang WC. Non-Coding RNAs in Lung Tumor Initiation and Progression. International Journal of Molecular Sciences. 2020; 21(8):2774. https://doi.org/10.3390/ijms21082774
Chicago/Turabian StyleSantos, Ruben Mercado, Cerena Moreno, and Wen Cai Zhang. 2020. "Non-Coding RNAs in Lung Tumor Initiation and Progression" International Journal of Molecular Sciences 21, no. 8: 2774. https://doi.org/10.3390/ijms21082774
APA StyleSantos, R. M., Moreno, C., & Zhang, W. C. (2020). Non-Coding RNAs in Lung Tumor Initiation and Progression. International Journal of Molecular Sciences, 21(8), 2774. https://doi.org/10.3390/ijms21082774