Roles of Nrf2 in Protecting the Kidney from Oxidative Damage


Abstract
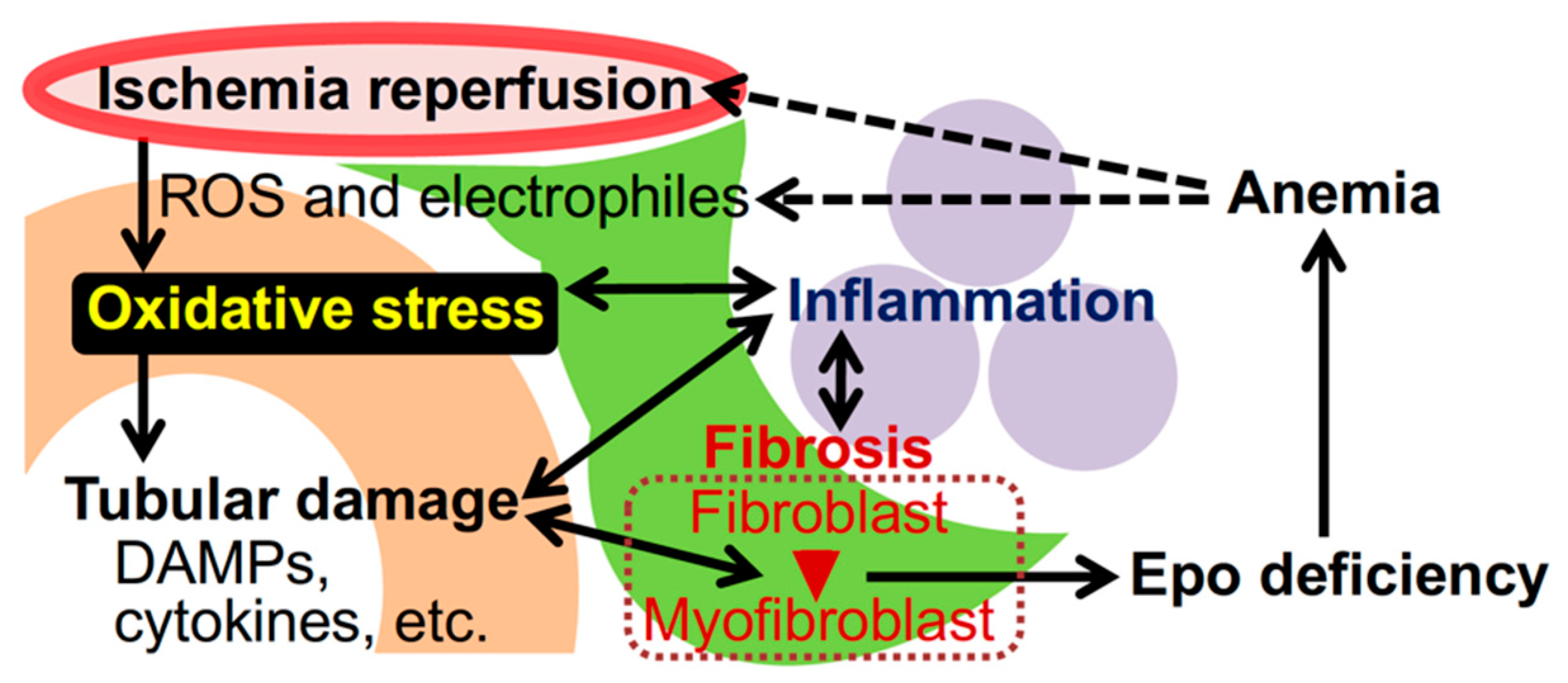
:1. Introduction
2. Oxidative Stress in Kidney
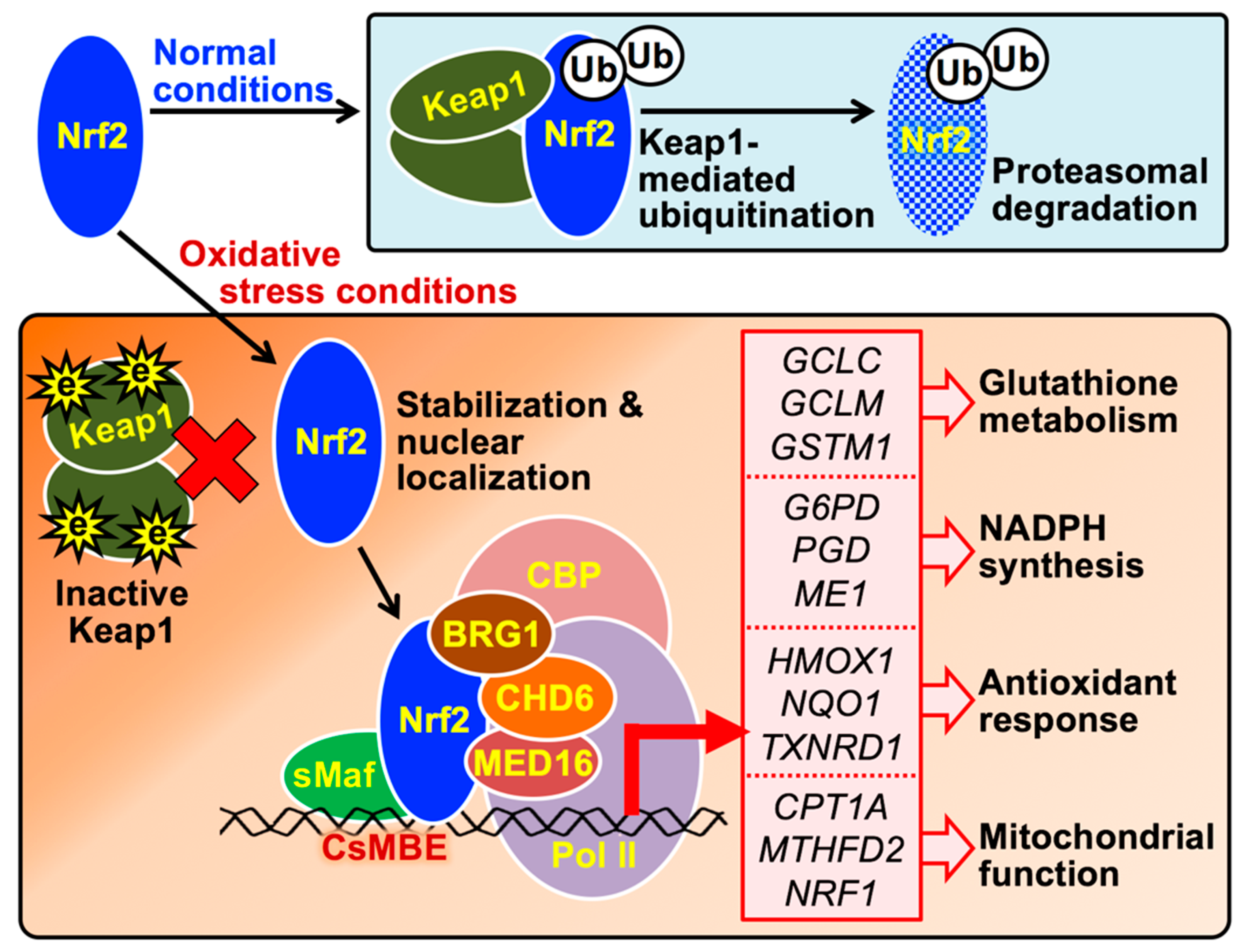
3. Role of the Keap1-Nrf2 System in Cytoprotection against Oxidative Stress
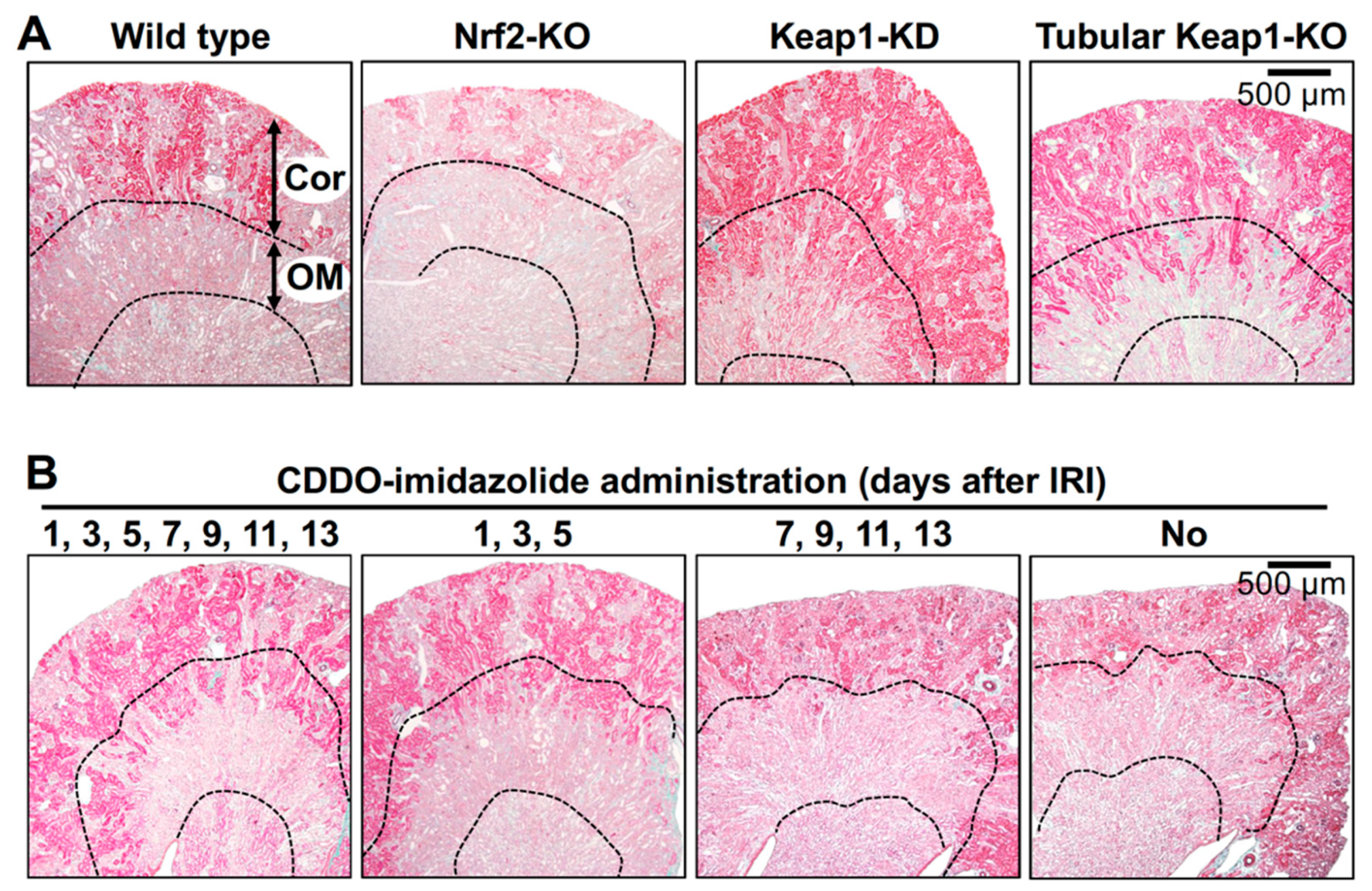
4. Mouse Genetic Models Used to Investigate the Roles of Nrf2 in the Kidney
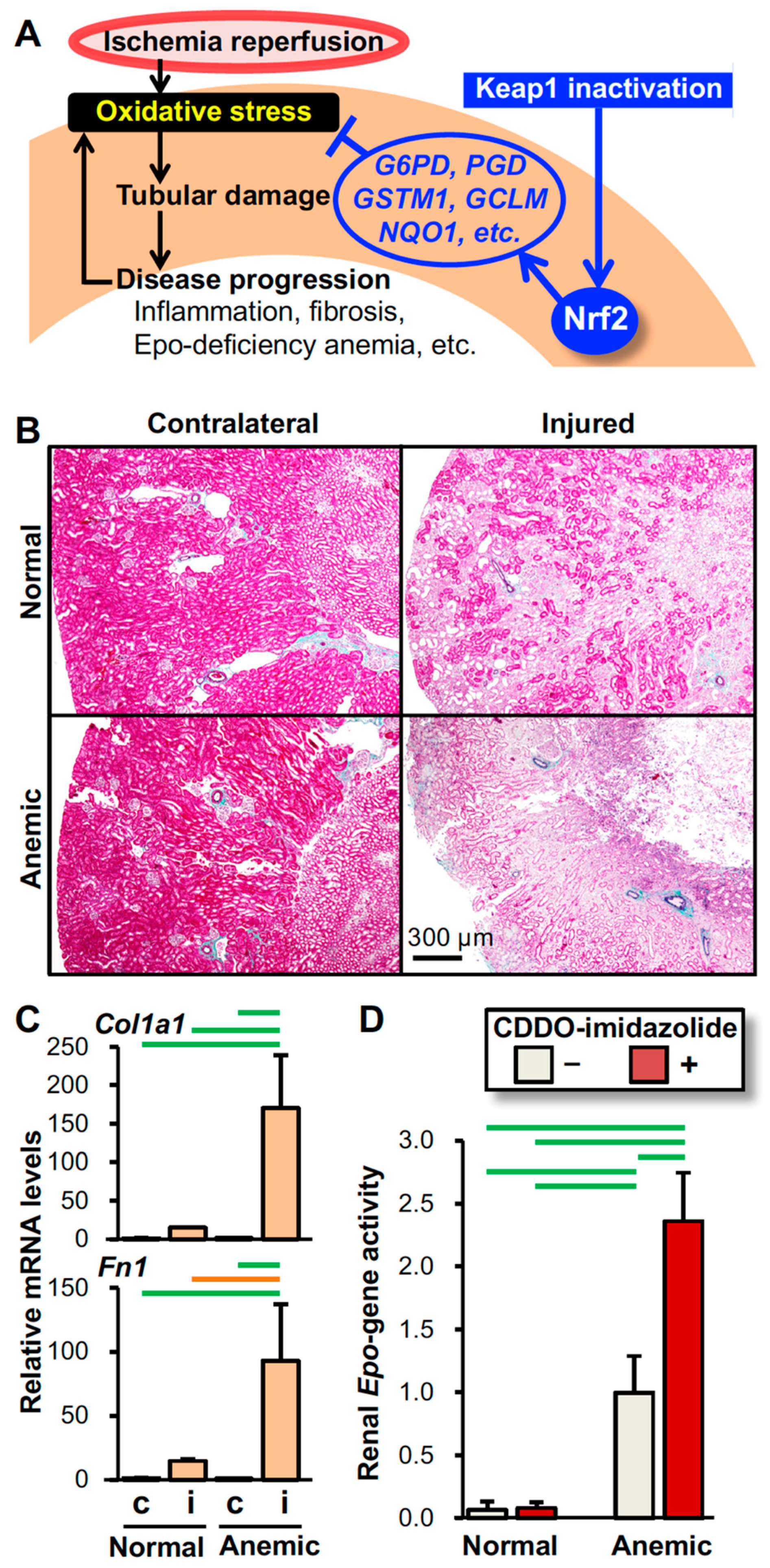
5. Mechanism of Renoprotection by Nrf2
6. Beneficial Effects of Nrf2 Activation for Kidney Disease Treatment
7. Clinical Impact of Pharmacological Activation of Nrf2 on Kidney Injury
8. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxydeoxyguanosine |
| AKI | acute kidney injury |
| BRG1 | brahma-related gene 1 |
| CBP | cyclic AMP-response element binding protein (CREB)-binding protein |
| CDDO | 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl] |
| CHD6 | chromodomain helicase DNA binding protein 6 |
| CKD | chronic kidney disease |
| CNC | Cap ‘n’ Collar |
| CPT1A | carnitine palmitoyltransferase 1A |
| CsMBE | CNC and small Maf binding element |
| DAMPs | damage-associated molecular patterns |
| eGFR | estimated glomerular filtration rate |
| Epo | erythropoietin |
| ESRD | end-stage renal disease |
| G6PD | glucose-6-phosphate dehydrogenase 6 |
| GCLC | glutamate cysteine ligase catalytic subunit |
| GCLM | glutamate cysteine ligase modifier subunit |
| GFP | green fluorescent protein |
| GSTM1 | glutathione S-transferase µ1 |
| HMOX1 | heme oxygenase 1 |
| IRI | ischemia-reperfusion injury |
| Keap1 | Kelch-like ECH-associated protein 1 |
| ME1 | malic enzyme 1 |
| MED16 | mediator subunit 16 |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase 2 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NQO1 | NADPH quinone oxidoreductase 1 |
| NRF1 | nuclear respiratory factor 1 |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PGD | 6-phosphogluconate dehydrogenase |
| PPP | pentose phosphate pathway |
| REP cell | renal Epo-producing cell |
| ROS | reactive oxygen species |
| sMaf | small musculoaponeurotic fibrosarcoma |
| TXNRD1 | thioredoxin reductase 1 |
References
- Sato, Y.; Yanagita, M. Renal anemia: From incurable to curable. Am. J. Physiol. Renal Physiol. 2013, 305, F1239–F1248. [Google Scholar] [CrossRef] [Green Version]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef]
- Ohsaki, Y.; O’Connor, P.; Mori, T.; Ryan, R.P.; Dickinson, B.C.; Chang, C.J.; Lu, Y.; Ito, S.; Cowley, A.W., Jr. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am. J. Physiol. Renal Physiol. 2012, 302, F95–F102. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Eckardt, K.U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; De Zeeuw, D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from kidney disease: Improving global outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summary of Recommendation Statements. Kidney Int. Suppl. 2012, 2, 8–12. [CrossRef] [PubMed] [Green Version]
- Chapter 1: Definition and classification of CKD. Kidney Int. Suppl. 2013, 3, 19–62. [CrossRef] [PubMed] [Green Version]
- Humphreys, B.D.; Cantaluppi, V.; Portilla, D.; Singbartl, K.; Yang, L.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Acute Dialysis Quality Initiative, X.W.G. Targeting endogenous repair pathways after AKI. J. Am. Soc. Nephrol. 2016, 27, 990–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. Acute Kidney Injury Advisory Group of the American Society of, N. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Murtagh, F.E.; Burns, A.; Moranne, O.; Morton, R.L.; Naicker, S. Supportive care: Comprehensive conservative care in end-stage kidney disease. Clin. J. Am. Soc. Nephrol. 2016, 11, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Lo, L.J.; Go, A.S.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordonez, J.D.; Hsu, C.Y. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009, 76, 893–899. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007, 72, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [Green Version]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Tanaka, T.; Nangaku, M. Nuclear factor erythroid 2-related factor 2 as a treatment target of kidney diseases. Curr. Opin. Nephrol. Hypertens. 2020, 29, 128–135. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 system to prevent kidney disease progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [Green Version]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. Antioxid. Redox. Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Layton, A.T. Recent advances in renal hypoxia: Insights from bench experiments and computer simulations. Am. J. Physiol. Renal Physiol. 2016, 311, F162–F165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijano, C.; Castro, L.; Peluffo, G.; Valez, V.; Radi, R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: Direct measurements and formation of hydrogen peroxide and peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3404–H3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, T.; Maruyama, A.; Kang, M.I.; Kawatani, Y.; Shibata, T.; Uchida, K.; Warabi, E.; Noguchi, N.; Itoh, K.; Yamamoto, M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 2005, 280, 27244–27250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.D.; Santos, S.S.; Meireles, T.; Romero, N.; Glorieux, G.; Pecoits-Filho, R.; Zhang, D.D.; Nakao, L.S. Uremic toxins promote accumulation of oxidized protein and increased sensitivity to hydrogen peroxide in endothelial cells by impairing the autophagic flux. Biochem. Biophys. Res. Commun. 2020, 523, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Popkov, V.A.; Silachev, D.N.; Zalevsky, A.O.; Zorov, D.B.; Plotnikov, E.Y. Mitochondria as a source and a target for uremic toxins. Int. J. Mol. Sci. 2019, 20, 3094. [Google Scholar] [CrossRef] [Green Version]
- Otsuki, A.; Suzuki, M.; Katsuoka, F.; Tsuchida, K.; Suda, H.; Morita, M.; Shimizu, R.; Yamamoto, M. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic. Biol. Med. 2016, 91, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Zhang, J.; Hosoya, T.; Maruyama, A.; Nishikawa, K.; Maher, J.M.; Ohta, T.; Motohashi, H.; Fukamizu, A.; Shibahara, S.; Itoh, K.; et al. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem. J. 2007, 404, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell. Biol. 2015, 36, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.Y.; Gladwell, W.; Yamamoto, M.; Kleeberger, S.R. Exacerbated airway toxicity of environmental oxidant ozone in mice deficient in Nrf2. Oxid. Med. Cell. Longev. 2013, 2013, 254069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L27–L36. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, R.; Kosai, H.; Masuo, M.; Izumiyama, K.; Noshikawaji, T.; Morimoto, M.; Kumaki, S.; Miyazaki, Y.; Motohashi, H.; Yamamoto, M.; et al. Nrf2 suppresses allergic lung inflammation by attenuating the type 2 innate lymphoid cell response. J. Immunol. 2019, 202, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Eba, S.; Hoshikawa, Y.; Moriguchi, T.; Mitsuishi, Y.; Satoh, H.; Ishida, K.; Watanabe, T.; Shimizu, T.; Shimokawa, H.; Okada, Y.; et al. The nuclear factor erythroid 2-related factor 2 activator oltipraz attenuates chronic hypoxia-induced cardiopulmonary alterations in mice. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Katsumata, Y.; Shinmura, K.; Sugiura, Y.; Tohyama, S.; Matsuhashi, T.; Ito, H.; Yan, X.; Ito, K.; Yuasa, S.; Ieda, M.; et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension 2014, 63, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 suppresses oxidative stress and inflammation in App knock-in Alzheimer’s disease model mice. Mol. Cell. Biol. 2020, 40, e00467-19. [Google Scholar] [CrossRef]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic beta-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagishita, Y.; Uruno, A.; Chartoumpekis, D.V.; Kensler, T.W.; Yamamoto, M. Nrf2 represses the onset of type 1 diabetes in non-obese diabetic mice. J. Endocrinol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keleku-Lukwete, N.; Suzuki, M.; Otsuki, A.; Tsuchida, K.; Katayama, S.; Hayashi, M.; Naganuma, E.; Moriguchi, T.; Tanabe, O.; Engel, J.D.; et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 12169–12174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maicas, N.; Ferrandiz, M.L.; Brines, R.; Ibanez, L.; Cuadrado, A.; Koenders, M.I.; van den Berg, W.B.; Alcaraz, M.J. Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid. Redox. Signal. 2011, 15, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Itoh, K.; Nagayoshi, E.; Haruta, J.; Kimura, T.; O’Connor, T.; Harada, T.; Yamamoto, M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 2001, 59, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Masui, S.; Itoh, T.; Miyajima, A.; Yamamoto, M. Nrf2 activation ameliorates hepatotoxicity induced by a heme synthesis inhibitor. Toxicol. Sci. 2019, 167, 227–238. [Google Scholar] [CrossRef]
- Skoko, J.J.; Wakabayashi, N.; Noda, K.; Kimura, S.; Tobita, K.; Shigemura, N.; Tsujita, T.; Yamamoto, M.; Kensler, T.W. Loss of Nrf2 in mice evokes a congenital intrahepatic shunt that alters hepatic oxygen and protein expression gradients and toxicity. Toxicol. Sci. 2014, 141, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Warabi, E.; Sugimoto, H.; Horie, M.; Gotoh, N.; Tokushige, K.; Hashimoto, E.; Utsunomiya, H.; Takahashi, H.; Ishii, T.; et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J. Gastroenterol. 2013, 48, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Knatko, E.V.; Ibbotson, S.H.; Zhang, Y.; Higgins, M.; Fahey, J.W.; Talalay, P.; Dawe, R.S.; Ferguson, J.; Huang, J.T.; Clarke, R.; et al. Nrf2 activation protects against solar-simulated ultraviolet radiation in mice and humans. Cancer Prev. Res (Phila.) 2015, 8, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honkura, Y.; Matsuo, H.; Murakami, S.; Sakiyama, M.; Mizutari, K.; Shiotani, A.; Yamamoto, M.; Morita, I.; Shinomiya, N.; Kawase, T.; et al. NRF2 is a key target for prevention of noise-induced hearing loss by reducing oxidative damage of cochlea. Sci. Rep. 2016, 6, 19329. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [Green Version]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Aleksunes, L.M.; Goedken, M.J.; Chen, C.; Reisman, S.A.; Manautou, J.E.; Klaassen, C.D. Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicol. Appl. Pharmacol. 2008, 231, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, R.J.; Chartoumpekis, D.V.; Rush, B.M.; Zhou, D.; Fu, H.; Kensler, T.W.; Liu, Y. Keap1 hypomorphism protects against ischemic and obstructive kidney disease. Sci. Rep. 2016, 6, 36185. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Shimizu, A.; Pastan, I.; Taguchi, K.; Naganuma, E.; Suzuki, T.; Hosoya, T.; Yokoo, T.; Saito, A.; Miyata, T.; et al. Keap1 inhibition attenuates glomerulosclerosis. Nephrol. Dial. Transpl. 2014, 29, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Reddy, N.M.; Higbee, E.M.; Potteti, H.R.; Noel, S.; Racusen, L.; Kensler, T.W.; Sporn, M.B.; Reddy, S.P.; Rabb, H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int. 2014, 85, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.; Reddy, S.P.; Rabb, H. T Lymphocyte-Specific Activation of Nrf2 Protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef] [Green Version]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Suzuki, T.; Seki, S.; Hiramoto, K.; Naganuma, E.; Kobayashi, E.H.; Yamaoka, A.; Baird, L.; Takahashi, N.; Sato, H.; Yamamoto, M. Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat. Commun. 2017, 8, 14577. [Google Scholar] [CrossRef] [Green Version]
- Noel, S.; Arend, L.J.; Bandapalle, S.; Reddy, S.P.; Rabb, H. Kidney epithelium specific deletion of kelch-like ECH-associated protein 1 (Keap1) causes hydronephrosis in mice. BMC Nephrol. 2016, 17, 110. [Google Scholar] [CrossRef] [Green Version]
- Morito, N.; Yoh, K.; Hirayama, A.; Itoh, K.; Nose, M.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 deficiency improves autoimmune nephritis caused by the fas mutation lpr. Kidney Int. 2004, 65, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Ghosh, A.; Lo, C.S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Nrf2 deficiency upregulates intrarenal angiotensin-converting enzyme-2 and angiotensin 1-7 receptor expression and attenuates hypertension and nephropathy in diabetic mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.A.; Nicholas, S.B.; Norris, K.C.; Vaziri, N.D. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol. Dial. Transpl. 2013, 28, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: Potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef]
- Zoja, C.; Corna, D.; Nava, V.; Locatelli, M.; Abbate, M.; Gaspari, F.; Carrara, F.; Sangalli, F.; Remuzzi, G.; Benigni, A. Analogs of bardoxolone methyl worsen diabetic nephropathy in rats with additional adverse effects. Am. J. Physiol. Renal Physiol. 2013, 304, F808–F819. [Google Scholar] [CrossRef] [Green Version]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011, 20, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011, 20, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Navas, C.; Horswell, S.; Salm, M.; Bardella, C.; Rowan, A.; Stares, M.; Castro-Giner, F.; Fisher, R.; de Bruin, E.C.; et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat. Commun. 2015, 6, 6336. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am. J. Physiol. Renal Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, X.; Fan, J.; Chen, W.; Wang, J.; Zeng, Y.; Feng, X.; Yu, X.; Yang, X. Bardoxolone methyl (BARD) ameliorates aristolochic acid (AA)-induced acute kidney injury through Nrf2 pathway. Toxicology 2014, 318, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Nagasu, H.; Sogawa, Y.; Kidokoro, K.; Itano, S.; Yamamoto, T.; Satoh, M.; Sasaki, T.; Suzuki, T.; Yamamoto, M.; Wigley, W.C.; et al. Bardoxolone methyl analog attenuates proteinuria-induced tubular damage by modulating mitochondrial function. FASEB J. 2019, 33, 12253–12263. [Google Scholar] [CrossRef] [Green Version]
- Molina-Jijón, E.; Tapia, E.; Zazueta, C.; El Hafidi, M.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Medina-Campos, O.N.; Zarco-Márquez, G.; Torres, I.; Pedraza-Chaverri, J. Curcumin prevents Cr(VI)-induced renal oxidant damage by a mitochondrial pathway. Free Radic. Biol. Med. 2011, 51, 1543–1557. [Google Scholar] [CrossRef]
- Tapia, E.; Soto, V.; Ortiz-Vega, K.M.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Santamaría, J.; García-Niño, W.R.; Correa, F.; Zazueta, C.; et al. Curcumin induces Nrf2 nuclear translocation and prevents glomerular hypertension, hyperfiltration, oxidant stress, and the decrease in antioxidant enzymes in 5/6 nephrectomized rats. Oxid. Med. Cell. Longev. 2012, 2012, 269039. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Pan, X.; Fu, H.; Zheng, Y.; Dai, Y.; Yin, Y.; Chen, Q.; Hao, Q.; Bao, D.; Hou, D. Effect of curcumin on glycerol-induced acute kidney injury in rats. Sci. Rep. 2017, 7, 10114. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, J.; Liu, H.; Yuan, J.; Yin, Y.; Wang, T.; Cheng, B.; Sun, S.; Guo, Z. Curcumin ameliorates glyoxylate-induced calcium oxalate deposition and renal injuries in mice. Phytomedicine 2019, 61, 152861. [Google Scholar] [CrossRef]
- Jin, F.; Chen, X.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Bisdemethoxycurcumin attenuates cisplatin-induced renal injury through anti-apoptosis, anti-oxidant and anti-inflammatory. Eur. J. Pharmacol. 2020, 874, 173026. [Google Scholar] [CrossRef]
- Feng, X.; Guan, W.; Zhao, Y.; Wang, C.; Song, M.; Yao, Y.; Yang, T.; Fan, H. Dexmedetomidine ameliorates lipopolysaccharide-induced acute kidney injury in rats by inhibiting inflammation and oxidative stress via the GSK-3β/Nrf2 signaling pathway. J. Cell Physiol. 2019, 234, 18994–19009. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Q.; Li, C.; Lu, Y.; Hu, H.; Qin, B.; Xun, Y.; Zhu, Y.; Wu, Y.; Zhang, J.; et al. Inhibiting inflammation and modulating oxidative stress in oxalate-induced nephrolithiasis with the Nrf2 activator dimethyl fumarate. Free Radic. Biol. Med. 2019, 134, 9–22. [Google Scholar] [CrossRef]
- Oh, C.J.; Kim, J.Y.; Choi, Y.K.; Kim, H.J.; Jeong, J.Y.; Bae, K.H.; Park, K.G.; Lee, I.K. Dimethylfumarate attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. PLoS ONE 2012, 7, e45870. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Wei, W.; Fan, X.; Ci, X. Farrerol attenuates cisplatin-induced nephrotoxicity by inhibiting the reactive oxygen species-mediated oxidation, inflammation, and apoptotic signaling pathways. Front. Physiol. 2019, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Qin, Z.; Tang, J.; Xu, Z.; Li, R.; Jiang, X.; Yang, C.; Xing, Q.; Qi, X.; Tang, M.; et al. RTA-408 protects kidney from ischemia-reperfusion injury in mice via activating Nrf2 and Downstream GSH Biosynthesis gene. Oxid. Med. Cell. Longev. 2017, 2017, 7612182. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, L.; Sun, C.; Zhang, B.; Li, J.; Sun, J.; Zhang, Y.; Sun, D. Paeonol alleviates epirubicin-induced renal injury in mice by regulating Nrf2 and NF-κB pathways. Eur. J. Pharmacol. 2017, 795, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, L.; Wang, S.; Zhang, C.; Zheng, L.; Jia, Y.; Xu, M.; Zhu, T.; Zhang, Y.; Rong, R. Resveratrol alleviates inflammatory responses and oxidative stress in rat kidney ischemia-reperfusion injury and H2O2-induced NRK-52E cells via the Nrf2/TLR4/NF-κB pathway. Cell Physiol. Biochem. 2018, 45, 1677–1689. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging (Albany NY) 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zou, Y.; Xing, J.; Fu, Y.Y.; Wang, K.Y.; Wan, P.Z.; Zhai, X.Y. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3β/Nrf2 pathway. Oxid. Med. Cell. Longev. 2020, 2020, 6286984. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wang, P.; Qiao, Y.; Ge, Y.; Wang, Y.; Quan, S.; Yao, R.; Zhuang, S.; Wang, L.J.; Du, Y.; et al. Genetic and pharmacologic targeting of glycogen synthase kinase 3β reinforces the Nrf2 antioxidant defense against podocytopathy. J. Am. Soc Nephrol. 2016, 27, 2289–2308. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liao, G.; Zhou, Q.; Lv, D.; Holthfer, H.; Zou, H. Sulforaphane attenuates contrast-induced nephropathy in rats via Nrf2/HO-1 pathway. Oxid. Med. Cell. Longev. 2016, 2016, 9825623. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Kang, N.I.; Lee, H.K.; Jang, K.Y.; Park, J.W.; Park, B.H. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem. Pharmacol. 2008, 75, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Yamamoto, M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflugers Arch. 2016, 468, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Yamazaki, S.; Moriguchi, T.; Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Abe, M.; Kiyomoto, H.; Ito, S.; et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1599–1616. [Google Scholar] [CrossRef] [Green Version]
- Souma, T.; Nezu, M.; Nakano, D.; Yamazaki, S.; Hirano, I.; Sekine, H.; Dan, T.; Takeda, K.; Fong, G.H.; Nishiyama, A.; et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J. Am. Soc. Nephrol. 2016, 27, 428–438. [Google Scholar] [CrossRef]
- Gouva, C.; Nikolopoulos, P.; Ioannidis, J.P.; Siamopoulos, K.C. Treating anemia early in renal failure patients slows the decline of renal function: A randomized controlled trial. Kidney Int. 2004, 66, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.E.; Sanches, T.R.; Volpini, R.A.; Shimizu, M.H.; Kuriki, P.S.; Camara, N.O.; Seguro, A.C.; Andrade, L. Effects of continuous erythropoietin receptor activator in sepsis-induced acute kidney injury and multi-organ dysfunction. PLoS ONE 2012, 7, e29893. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Souma, T.; Hirano, I.; Pan, X.; Minegishi, N.; Suzuki, N.; Yamamoto, M. A mouse model of adult-onset anaemia due to erythropoietin deficiency. Nat. Commun. 2013, 4, 1950. [Google Scholar] [CrossRef] [Green Version]
- Westenfelder, C.; Biddle, D.L.; Baranowski, R.L. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. 1999, 55, 808–820. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, L.; Dey, S.; Alnaeeli, M.; Suresh, S.; Rogers, H.; Teng, R.; Noguchi, C.T. Erythropoietin action in stress response, tissue maintenance and metabolism. Int. J. Mol. Sci. 2014, 15, 10296–10333. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Sasaki, Y.; Kato, K.; Yamazaki, S.; Kurasawa, M.; Yorozu, K.; Shimonaka, Y.; Yamamoto, M. Efficacy estimation of erythropoiesis-stimulating agents using erythropoietin-deficient anemic mice. Haematologica 2016, 101, e356–e360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [Green Version]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; de Boer, I.H. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. 2013, 24, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Matsushita, K.; Woodward, M.; Bilo, H.J.; Chalmers, J.; Heerspink, H.J.; Lee, B.J.; Perkins, R.M.; Rossing, P.; Sairenchi, T.; et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: A meta-analysis. Lancet 2012, 380, 1662–1673. [Google Scholar] [CrossRef] [Green Version]
- de Zeeuw, D.; Akizawa, T.; Agarwal, R.; Audhya, P.; Bakris, G.L.; Chin, M.; Krauth, M.; Lambers Heerspink, H.J.; Meyer, C.J.; McMurray, J.J.; et al. Rationale and trial design of bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes: The occurrence of renal events (BEACON). Am. J. Nephrol. 2013, 37, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Kashtan, C.E.; Ding, J.; Garosi, G.; Heidet, L.; Massella, L.; Nakanishi, K.; Nozu, K.; Renieri, A.; Rheault, M.; Wang, F.; et al. Alport syndrome: A unified classification of genetic disorders of collagen IV alpha345: A position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018, 93, 1045–1051. [Google Scholar] [CrossRef]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/βTrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse | Phenotype in Kidney Disease | References |
|---|---|---|
| Nrf2-KO (systemic) | Vulnerable to kidney disease models (autoimmune nephritis, diabetic nephropathy, toxic injury, ureteral obstruction, podocyte injury, and IRI) | [56,57,58,59,60,61,62,63,64,65,66,67] |
| Resistant to kidney disease models (autoimmune nephritis and diabetic nephropathy) | [71,72] | |
| Keap1-KD (systemic) | Resistant to kidney disease models (ureteral obstruction, podocyte injury, and IRI) | [63,64,67] |
| Keap1-KO (myeloid cell) | No remarkable effect against renal IRI. | [67] |
| (T cell) | Resistant to IRI | [66] |
| (developing tubule) | Hydronephrosis in neonates | [69,70] |
| (adult tubule) | Resistant to IRI | [67] |
| Compound | Kidney Disease Model | Outcome | Reference |
|---|---|---|---|
| CDDO-imidazolide | IRI (m) | Improved | [65,67] |
| CDDO-ME | IRI (m), toxic injury (m), and proteinuria-induced tubular damage (m) | Improved | [82,83,84] |
| Curcumin | Toxic injury (r, m) and 5/6 nephrectomy (r) | Improved | [85,86,87,88,89] |
| Dexmedetomidine | Lipopolysaccharide-induced injury (r) | Improved | [90] |
| Dimethyl fumarate | Toxic injury (r) and ureteral obstruction (m) | Improved | [91,92] |
| Farrerol | Toxic injury (r) | Improved | [93] |
| Omaveloxolone | IRI (m) | Improved | [94] |
| Paeonol | Toxic injury (m) | Improved | [95] |
| Resveratrol | IRI (r) and aging-related injury (m) | Improved | [96,97] |
| Roxadustat | Toxic injury (m) | Improved | [98] |
| SB216763 | Toxic injury (m) | Improved | [99] |
| Sulforaphane | IRI (m), pristane-induced Lupus nephritis (m), and contrast-induced nephropathy (r) | Improved | [100,101,102] |
| Phase | Period | CKD Stage | Registration ID | |
|---|---|---|---|---|
| BEAM | II | 2009–2010 | 3–4 | NCT00811889 |
| BEACON | III | 2011–2012 † | 4 | NCT01351675 |
| TSUBAKI | II | 2015–2017 | 3–4 | NCT02316821 |
| AYAME | III | 2018–2022 | 3–4 | NCT03550443 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. https://doi.org/10.3390/ijms21082951
Nezu M, Suzuki N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. International Journal of Molecular Sciences. 2020; 21(8):2951. https://doi.org/10.3390/ijms21082951
Chicago/Turabian StyleNezu, Masahiro, and Norio Suzuki. 2020. "Roles of Nrf2 in Protecting the Kidney from Oxidative Damage" International Journal of Molecular Sciences 21, no. 8: 2951. https://doi.org/10.3390/ijms21082951
APA StyleNezu, M., & Suzuki, N. (2020). Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. International Journal of Molecular Sciences, 21(8), 2951. https://doi.org/10.3390/ijms21082951