Non-Apoptotic Cell Death Signaling Pathways in Melanoma


Abstract
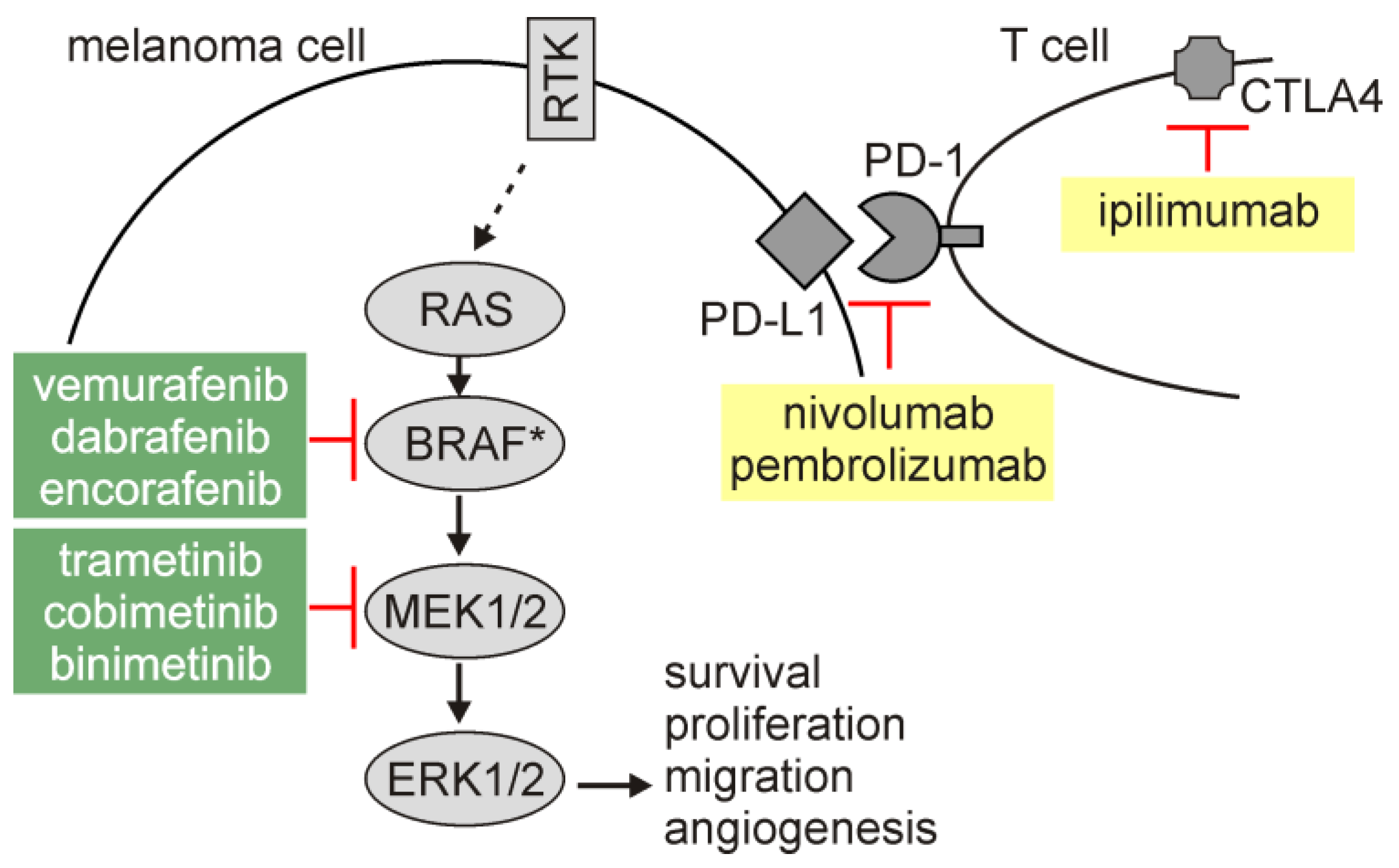
1. Introduction
2. Autophagy
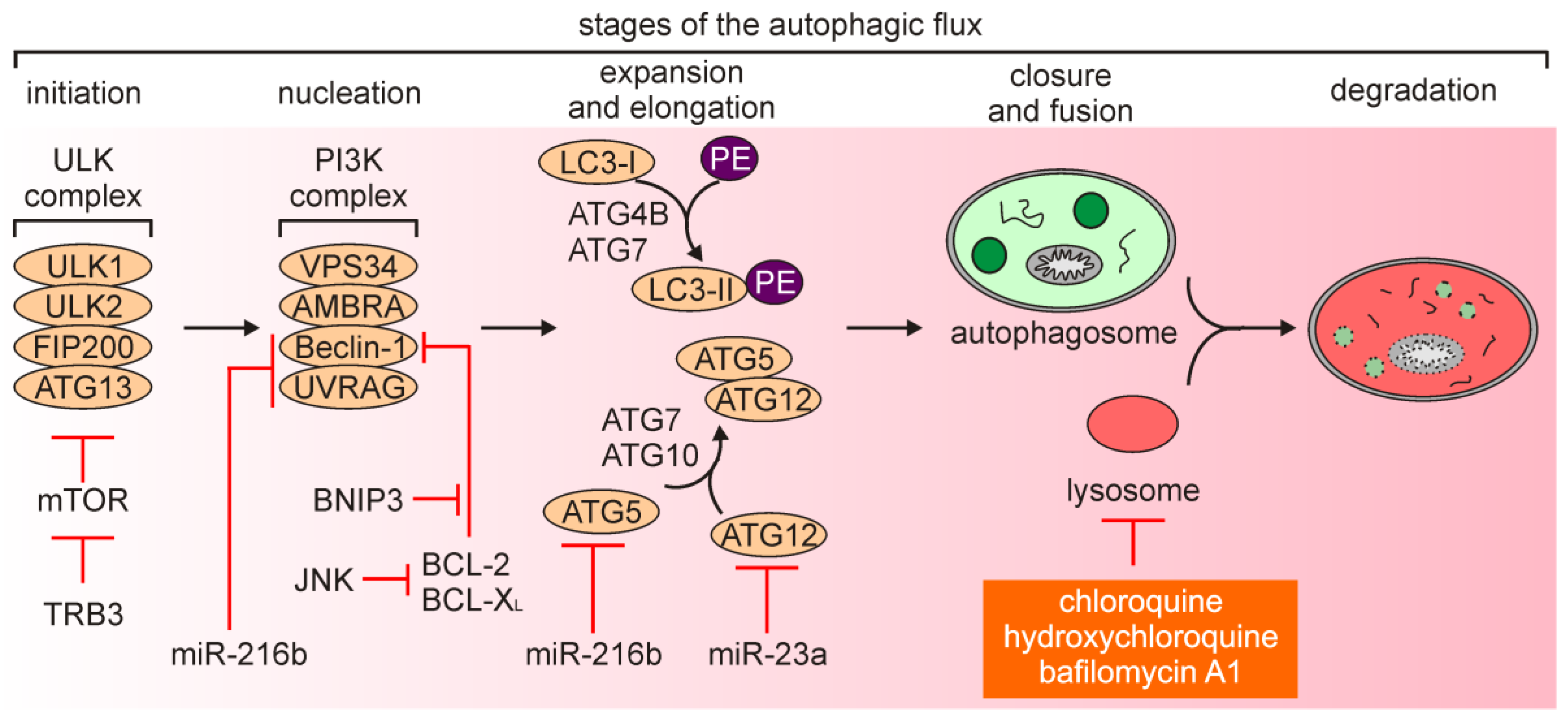
2.1. An Overview of Autophagy and Autophagy-Dependent Cell Death
2.2. Autophagy in Melanoma
3. Necroptosis
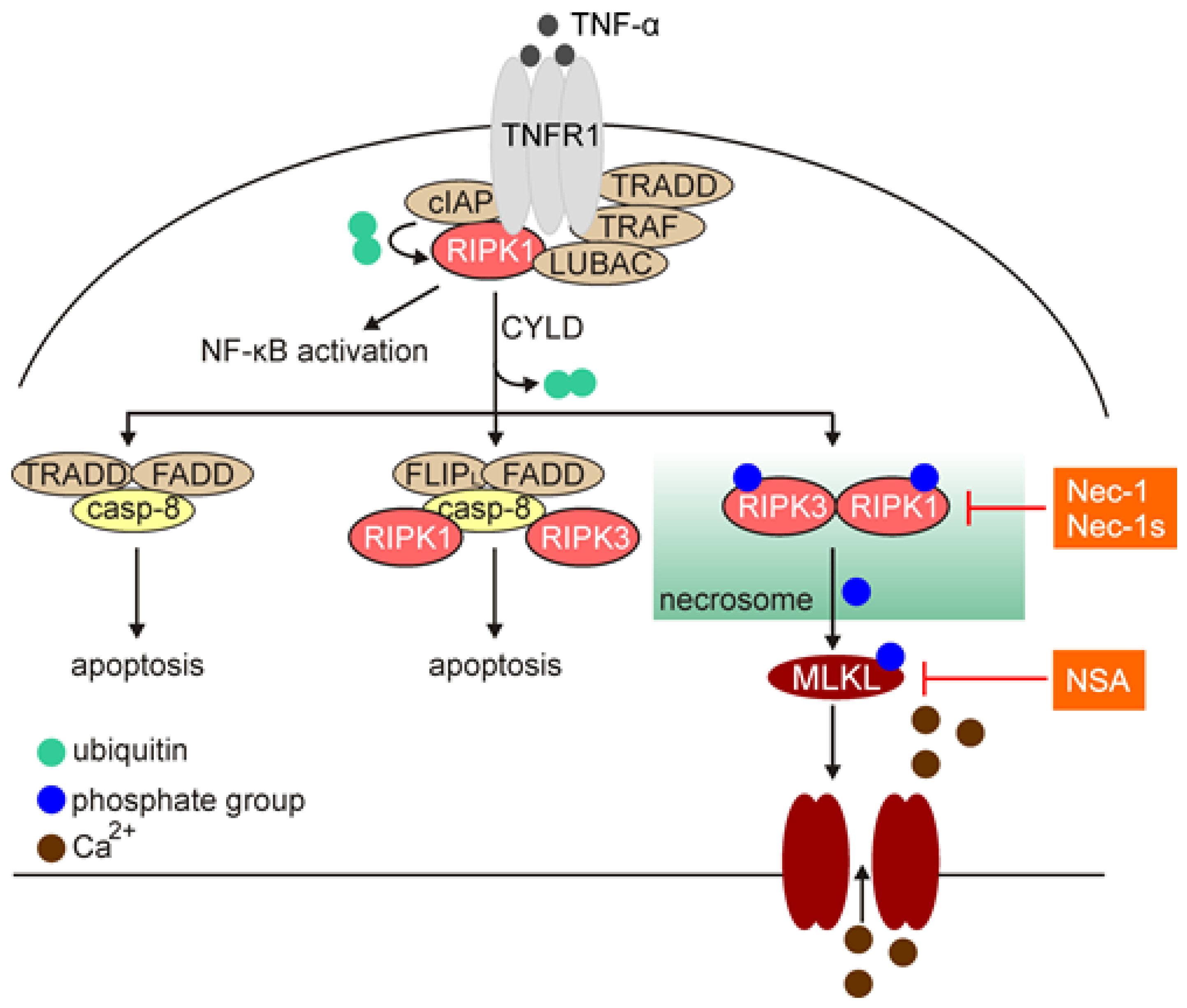
3.1. An Overview of Necroptotic Signaling Pathway
3.2. Necroptosis in Melanoma
4. Ferroptosis
4.1. An Overview of Ferroptosis
4.2. Ferroptosis in Melanoma
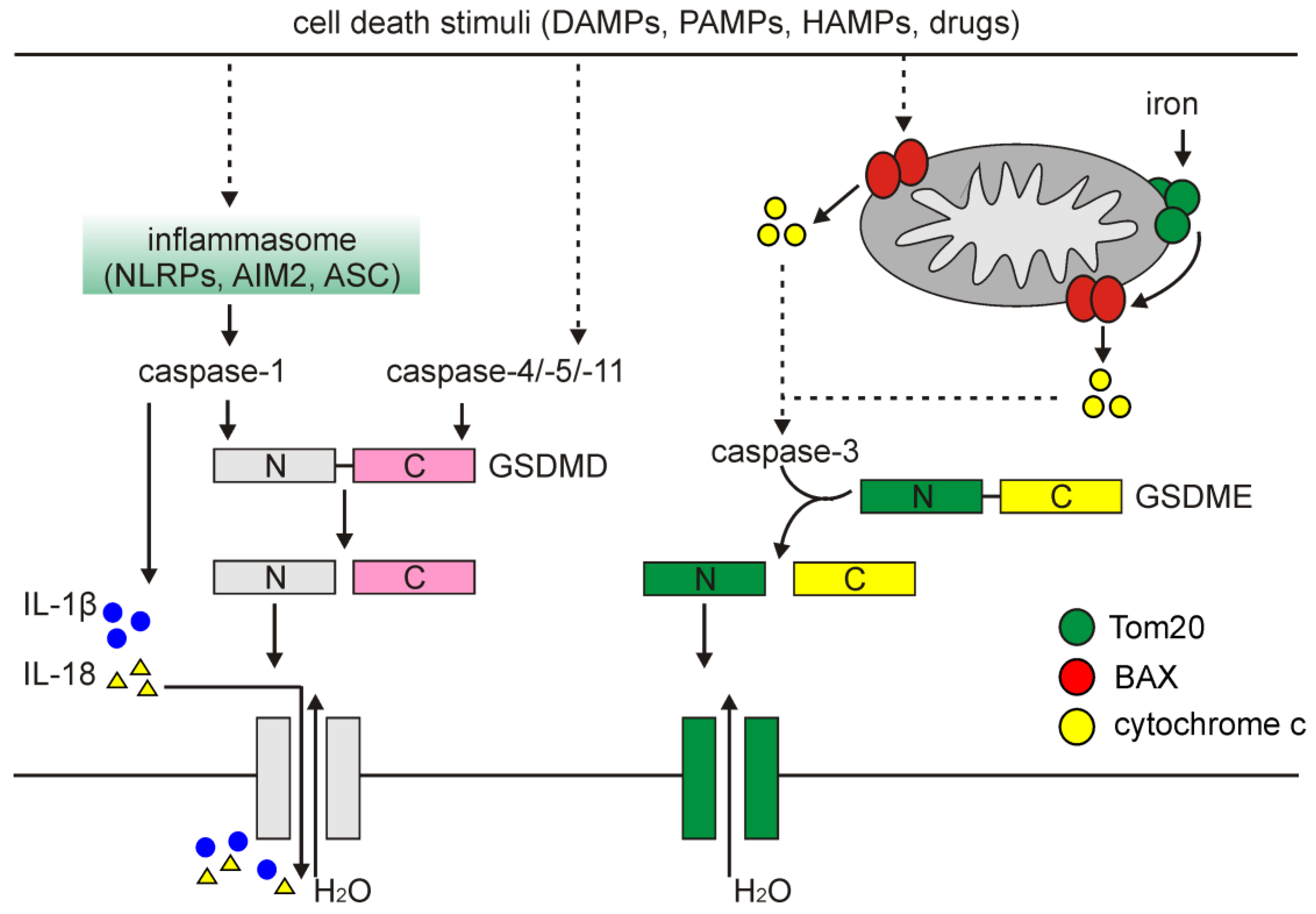
5. Pyroptosis
5.1. An Overview of Pyroptosis
5.2. Pyroptosis in Melanoma
6. Conclusions and Future Directions
Funding
Conflicts of Interest
Abbreviations
| ATGs | autophagy-related genes |
| BCL-2 | B-cell leukemia/lymphoma 2 |
| BNIP3 | BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| BRAF | B-Raf proto-oncogene serine/threonine kinase |
| BRAFi | inhibitors of mutated BRAF |
| CDR1as | cerebellar degeneration-related 1 antisense |
| DAMPs | damage-associated molecular patterns |
| DC | dendritic cell |
| GPX4 | glutathione peroxidase 4 |
| GSDM | gasdermin |
| GSH | glutathione (reduced) |
| HMGB1 | high mobility group box 1 |
| HSP90 | heat shock protein 90 |
| IFN | interferon |
| IL-1β | interleukin-1β |
| LC3 | microtubule-associated protein light chain 3 |
| MEKi | inhibitors of MEK1/2 |
| MITF | microphthalmia-associated transcription factor |
| MLKL | mixed lineage kinase domain-like |
| NF-κB | nuclear factor-kappa B |
| NGFR | nerve growth factor receptor |
| NK | natural killer |
| NRF-2 | nuclear factor erythroid 2-related factor 2 |
| PD-L1 | programmed death-ligand 1 |
| OXPHOS | oxidative phosphorylation |
| RCD | regulated cell death |
| RIPK | receptor-interacting protein kinase |
| ROS | reactive oxygen species |
| SQSTM1/p62 | sequestosome 1 |
| TNF-α | tumor necrosis factor alpha |
| UV | ultraviolet |
| WNT | Wingless-type |
References
- Vaux, D.L.; Korsmeyer, S.J. Cell death in development. Cell 1999, 96, 245–254. [Google Scholar] [CrossRef]
- Opferman, J.T.; Korsmeyer, S.J. Apoptosis in the development and maintenance of the immune system. Nat. Immunol. 2003, 4, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88. [Google Scholar] [CrossRef]
- Fulda, S. Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Semin. Cell Dev. Biol. 2015, 39, 20–25. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. Pro-apoptotic activity of BH3-only proteins and BH3 mimetics: From theory to potential cancer therapy. Anticancer Agents Med. Chem. 2012, 12, 966–981. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Hartman, M.; Czyz, M. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Klöditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, G. Mechanisms and therapeutic regulation of pyroptosis in inflammatory diseases and cancer. Int. J. Mol. Sci. 2020, 21, 1456. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. C.A. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Kontogianni, G.; Piroti, G.; Maglogiannis, I.; Chatziioannou, A.; Papadodima, O. Dissecting the mutational landscape of cutaneous melanoma: An omic analysis based on patients from Greece. Cancers (Basel) 2018, 10, 96. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Cartlidge, R.A.; Thomas, G.R.; Cagnol, S.; Jong, K.A.; Molton, S.A.; Finch, A.J.; McMahon, M. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 2008, 21, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. Anti-apoptotic proteins on guard of melanoma cell survival. Cancer Lett. 2013, 331, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Mielczarek-Lewandowska, A.; Hartman, M.L.; Czyz, M. Inhibitors of HSP90 in melanoma. Apoptosis 2020, 25, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. Pro-survival role of MITF in melanoma. J. Investig. Dermatol. 2015, 135, 352–358. [Google Scholar] [CrossRef]
- Bennett, P.E.; Bemis, L.; Norris, D.A.; Shellman, Y.G. miR in melanoma development: miRNAs and acquired hallmarks of cancer in melanoma. Physiol. Genom. 2013, 45, 1049–1059. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Gajos-Michniewicz, A.; Czyz, M. MCL-1, BCL-XL and MITF are diversely employed in adaptive response of melanoma cells to changes in microenvironment. PLoS ONE 2015, 10, e0128796. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Noman, M.Z.; Gajos-Michniewicz, A.; Chouaib, S.; Czyz, M. Gene expression profiling identifies microphthalmia-associated transcription factor (MITF) and Dickkopf-1 (DKK1) as regulators of microenvironment-driven alterations in melanoma phenotype. PLoS ONE 2014, 9, e95157. [Google Scholar] [CrossRef]
- Calance, D.N.; Steixner, C.; Gross, S.; Schuler-Thurner, B.; Knoll, G.; Ehrenschwender, M. Hypertonicity primes malignant melanoma cells for apoptosis. Apoptosis 2018, 23, 201–209. [Google Scholar] [CrossRef]
- Rohrbeck, L.; Gong, J.N.; Lee, E.F.; Kueh, A.J.; Behren, A.; Tai, L.; Lessene, G.; Huang, D.C.; Fairlie, W.D.; Strasser, A.; et al. Hepatocyte growth factor renders BRAF mutant human melanoma cell lines resistant to PLX4032 by downregulating the pro-apoptotic BH3-only proteins PUMA and BIM. Cell Death Differ. 2016, 23, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, J.N.; Nazarian, R.; Wang, Q.; Guo, D.; Hsueh, T.; Mok, S.; Sazegar, H.; MacConaill, L.E.; Barretina, J.G.; Kehoe, S.M.; et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J. Transl. Med. 2010, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Missert, D.J.; Asciolla, J.J.; Podgrabinska, S.; Wieder, S.Y.; Izadmehr, S.; Belbin, G.; Skobe, M.; Chipuk, J.E. Anti-apoptotic BCL-2 proteins govern cellular outcome following B-RAF(V600E) inhibition and can be targeted to reduce resistance. Oncogene 2015, 34, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Pedini, F.; De Luca, G.; Felicetti, F.; Puglisi, R.; Boe, A.; Arasi, M.B.; Fratini, F.; Mattia, G.; Spada, M.; Caporali, S.; et al. Joint action of miR-126 and MAPK/PI3K inhibitors against metastatic melanoma. Mol. Oncol. 2019, 13, 1836–1854. [Google Scholar] [CrossRef] [PubMed]
- Zalesna, I.; Osrodek, M.; Hartman, M.L.; Rozanski, M.; Sztiller-Sikorska, M.; Niewinna, K.; Nejc, D.; Czyz, M. Exogenous growth factors bFGF, EGF and HGF do not influence viability and phenotype of V600EBRAF melanoma cells and their response to vemurafenib and trametinib in vitro. PLoS ONE 2017, 12, e0183498. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Rozanski, M.; Osrodek, M.; Zalesna, I.; Czyz, M. Vemurafenib and trametinib reduce expression of CTGF and IL-8 in V600EBRAF melanoma cells. Lab. Investig. 2017, 97, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Gajos-Michniewicz, A.; Talaj, J.; Mielczarek-Lewandowska, A.; Czyz, M. Selective BH3 mimetics potentiate encorafenib activity in inducing apoptosis in BRAFV600E melanoma cells. Cell Death Dis. 2020, in press. [Google Scholar]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Carè, A.; Matarrese, P. Cell death-based treatments of melanoma: Conventional treatments and new therapeutic strategies. Cell Death Dis. 2018, 9, 112. [Google Scholar] [CrossRef]
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef]
- Santana-Magal, N.; Farhat-Younis, L.; Gutwillig, A.; Gleiberman, A.; Rasoulouniriana, D.; Tal, L.; Netanely, D.; Shamir, R.; Blau, R.; Feinmesser, M.; et al. Melanoma-secreted lysosomes trigger monocyte-derived dendritic cell apoptosis and limit cancer immunotherapy. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell. Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Soengas, M.S. Mitophagy or how to control the Jekyll and Hyde embedded in mitochondrial metabolism: Implications for melanoma progression and drug resistance. Pigment Cell Melanoma Res. 2012, 25, 721–731. [Google Scholar] [CrossRef]
- Germain, K.; Kim, P.K. Pexophagy: A model for selective autophagy. Int. J. Mol. Sci. 2020, 16, 578. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Therapeutic targeting of autophagy. EBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Guan, J.L. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol. Cancer Res. 2011, 9, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Wallot-Hieke, N.; Verma, N.; Schlütermann, D.; Berleth, N.; Deitersen, J.; Böhler, P.; Stuhldreier, F.; Wu, W.; Seggewiß, S.; Peter, C.; et al. Systematic analysis of ATG13 domain requirements for autophagy induction. Autophagy 2018, 14, 743–763. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, S.; Martin, S.; Maes, H.; Agostinis, P. Adapt, recycle, and move on: Proteostasis and trafficking mechanisms in melanoma. Front. Oncol. 2016, 6, 240. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Agostinis, P. Autophagy and mitophagy interplay in melanoma progression. Mitochondrion 2014, 19, 58–68. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic regulation of p62 is critical for cancer therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef]
- Lalaoui, N.; Lindqvist, L.M.; Sandow, J.J.; Ekert, P.G. The molecular relationships between apoptosis, autophagy and necroptosis. Semin. Cell Dev. Biol. 2015, 39, 63–69. [Google Scholar] [CrossRef]
- Bialik, S.; Vasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell. Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Tsujioka, M.; Yoshida, T.; Tajima-Sakurai, H.; Nishida, Y.; Matsuoka, Y.; Yoshino, I.; Tsujimoto, Y.; Shimizu, S. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017, 24, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/ceramide pathways and autophagy in the onset and progression of melanoma: Novel therapeutic targets and opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef]
- Di Leo, L.; Bodemeyer, V.; De Zio, D. The complex role of autophagy in melanoma evolution: New perspectives from mouse models. Front. Oncol. 2020, 9, 1506. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, Z.; Simon, H.U. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014, 10, 372–373. [Google Scholar] [CrossRef]
- Sample, A.; Zhao, B.; Wu, C.; Qian, S.; Shi, X.; Aplin, A.; He, Y.Y. The autophagy receptor adaptor p62 is up-regulated by UVA radiation in melanocytes and in melanoma cells. Photochem. Photobiol. 2018, 94, 432–437. [Google Scholar] [CrossRef]
- Zanchetta, L.M.; Garcia, A.; Lyng, F.; Walsh, J.; Murphy, J.E. Mitophagy and mitochondrial morphology in human melanoma-derived cells post exposure to simulated sunlight. Int. J. Radiat. Biol. 2011, 87, 506–517. [Google Scholar] [CrossRef]
- Konstantakou, E.G.; Velentzas, A.D.; Anagnostopoulos, A.K.; Giannopoulou, A.F.; Anastasiadou, E.; Papassideri, I.S.; Voutsinas, G.; Tsangaris, G.T.; Stravopodis, D.J. Unraveling the human protein atlas of metastatic melanoma in the course of ultraviolet radiation-derived photo-therapy. J. Proteom. 2018, 188, 119–138. [Google Scholar] [CrossRef]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Maddodi, N.; Huang, W.; Havighurst, T.; Kim, K.; Longley, B.J.; Setaluri, V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J. Investig. Dermatol. 2010, 130, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Maida, I.; Guida, S.; Azzariti, A.; Porcelli, L.; Tommasi, S.; Zanna, P.; Cocco, T.; Guida, M.; Guida, G. New insight into the role of metabolic reprogramming in melanoma cells harboring BRAF mutations. Biochim. Biophys. Acta 2016, 1863, 2710–2718. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Lai, F.; Wilmott, J.S.; Yan, X.G.; Liu, X.Y.; Luan, Q.; Guo, S.T.; Jiang, C.C.; Tseng, H.Y.; Scolyer, R.A.; et al. Noxa upregulation by oncogenic activation of MEK/ERK through CREB promotes autophagy in human melanoma cells. Oncotarget 2014, 30, 11237–11251. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, W.; Min, I.; Wyrwas, B.; Moore, M.; Zarnegar, R.; Fahey, T.J. BRAF V600E-dependent role of autophagy in uveal melanoma. J. Cancer Res. Clin. Oncol. 2017, 143, 447–455. [Google Scholar] [CrossRef]
- Marino, M.L.; Pellegrini, P.; Di Lernia, G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hägg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef]
- Matsuo, T.; Sadzuka, Y. Extracellular acidification by lactic acid suppresses glucose deprivation-induced cell death and autophagy in B16 melanoma cells. Biochem. Biophys. Res. Commun. 2018, 496, 1357–1361. [Google Scholar] [CrossRef]
- Matsuo, T.; Daishaku, S.; Sadzuka, Y. Lactic acid promotes cell survival by blocking autophagy of B16F10 mouse melanoma cells under glucose deprivation and hypoxic conditions. Biol. Pharm. Bull. 2019, 42, 837–839. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Wu, J.; Wang, W.; Zhang, Y. Oxygen partial pressure plays a crucial role in B16 melanoma cell survival by regulating autophagy and mitochondrial functions. Biochem. Biophys. Res. Commun. 2019, 510, 643–648. [Google Scholar] [CrossRef]
- Kraya, A.A.; Piao, S.; Xu, X.; Zhang, G.; Herlyn, M.; Gimotty, P.; Levine, B.; Amaravadi, R.K.; Speicher, D.W. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; da Silva Pereira, G.J.; de Freitas Saito, R.; Gil, C.D.; Zanatta, D.B.; Smaili, S.S.; Chammas, R. Galectin-3 sensitized melanoma cell lines to vemurafenib (PLX4032) induced cell death through prevention of autophagy. Oncotarget 2018, 9, 14567–14579. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Jiao, L.; Wang, Y.; Yu, Y.; Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 2018, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Ndiaye, M.A.; Singh, C.K.; Chhabra, G.; Huang, W.; Ahmad, N. SIRT6 histone deacetylase functions as a potential oncogene in human melanoma. Genes Cancer 2017, 8, 701–712. [Google Scholar] [CrossRef]
- Wang, L.; Guo, W.; Ma, J.; Dai, W.; Liu, L.; Guo, S.; Chen, J.; Wang, H.; Yang, Y.; Yi, X.; et al. Aberrant SIRT6 expression contributes to melanoma growth: Role of the autophagy paradox and IGF-AKT signaling. Autophagy 2018, 14, 518–533. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, F.; Li, M. Upregulation of BCL2 19 kD Protein-Interacting Protein 3 (BNIP3) is predictive of unfavorable prognosis in uveal melanoma. Med. Sci. Monit. 2018, 24, 4711–4717. [Google Scholar] [CrossRef]
- Li, P.; He, J.; Yang, Z.; Ge, S.; Zhang, H.; Zhong, Q.; Fan, X. ZNNT1 long noncoding RNA induces autophagy to inhibit tumorigenesis of uveal melanoma by regulating key autophagy gene expression. Autophagy 2019, 1–14. [Google Scholar] [CrossRef]
- Maes, H.; Martin, S.; Verfaillie, T.; Agostinis, P. Dynamic interplay between autophagic flux and Akt during melanoma progression in vitro. Exp. Dermatol. 2014, 23, 101–106. [Google Scholar] [CrossRef]
- García-Fernández, M.; Karras, P.; Checinska, A.; Cañón, E.; Calvo, G.T.; Gómez-López, G.; Cifdaloz, M.; Colmenar, A.; Espinosa-Hevia, L.; Olmeda, D.; et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 2016, 12, 1776–1790. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Yang, Y.; Guo, S.; Zhang, W.; Liu, Y.; Yi, X.; Ma, J.; Zhao, T.; Liu, L.; et al. Down-regulated miR-23a contributes to the metastasis of cutaneous melanoma by promoting autophagy. Theranostics 2017, 7, 2231–2249. [Google Scholar] [CrossRef]
- Karras, P.; Riveiro-Falkenbach, E.; Cañón, E.; Tejedo, C.; Calvo, T.G.; Martínez-Herranz, R.; Alonso-Curbelo, D.; Cifdaloz, M.; Perez-Guijarro, E.; Gómez-López, G.; et al. p62/SQSTM1 fuels melanoma progression by opposing mRNA decay of a selective set of pro-metastatic factors. Cancer Cell 2019, 35, 46–63.e10. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Giampietri, C.; Muscio, M.; Scatozza, F.; Facchiano, F.; Facchiano, A. WIPI1, BAG1, and PEX3 autophagy-related genes are relevant melanoma markers. Oxid. Med. Cell. Longev. 2018, 2018, 1471682. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Y.; Ellis, R.A.; Lovat, P.E. Prognostic impact of autophagy biomarkers for cutaneous melanoma. Front. Oncol. 2016, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Scatozza, F.; D’Arcangelo, D.; Giampietri, C.; Facchiano, F.; Facchiano, A. Melanogenesis and autophagy in melanoma. Melanoma Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Jin, L.; Su, Y.; Liu, Y.; Li, C.; Wang, Z. Development and validation of autophagy-related-gene biomarker and nomogram for predicting the survival of cutaneous melanoma. IUBMB Life 2020. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Luo, L.; Thompson, T.A.; Torres, S.; Hu, C.A.; Thomas, N.E.; Lilyquist, J.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Variants in autophagy-related genes and clinical characteristics in melanoma: A population-based study. Cancer Med. 2016, 5, 3336–3345. [Google Scholar] [CrossRef]
- Li, J.; Yang, R.; Yang, H.; Chen, S.; Wang, L.; Li, M.; Yang, S.; Feng, Z.; Bi, J. NCAM regulates the proliferation, apoptosis, autophagy, EMT, and migration of human melanoma cells via the Src/Akt/mTOR/cofilin signaling pathway. J. Cell. Biochem. 2020, 121, 1192–1204. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, S.; Ye, Z.; Zhou, X. MCM7 silencing promotes cutaneous melanoma cell autophagy and apoptosis by inactivating the AKT1/mTOR signaling pathway. J. Cell. Biochem. 2020, 121, 1283–1294. [Google Scholar] [CrossRef]
- Brown, K.; Yang, P.; Salvador, D.; Kulikauskas, R.; Ruohola-Baker, H.; Robitaille, A.M.; Chien, A.J.; Moon, R.T.; Sherwood, V. WNT/β-catenin signaling regulates mitochondrial activity to alter the oncogenic potential of melanoma in a PTEN-dependent manner. Oncogene 2017, 36, 3119–3136. [Google Scholar] [CrossRef]
- Ndoye, A.; Budina-Kolomets, A.; Kugel, C.H.; Webster, M.R.; Kaur, A.; Behera, R.; Rebecca, V.W.; Li, L.; Brafford, P.A.; Liu, Q.; et al. ATG5 mediates a positive feedback loop between Wnt signaling and autophagy in melanoma. Cancer Res. 2017, 77, 5873–5885. [Google Scholar] [CrossRef]
- Starobinets, H.; Ye, J.; Broz, M.; Barry, K.; Goldsmith, J.; Marsh, T.; Rostker, F.; Krummel, M.; Debnath, J. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Investig. 2016, 126, 4417–4429. [Google Scholar] [CrossRef] [PubMed]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving Natural Killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, e1452583. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rufo, N.; Korf, H.; Mathieu, C.; Garg, A.D.; Agostinis, P. BNIP3 modulates the interface between B16-F10 melanoma cells and immune cells. Oncotarget 2018, 9, 17631–17644. [Google Scholar] [CrossRef]
- Ndoye, A.; Weeraratna, A.T. Autophagy-an emerging target for melanoma therapy. F1000Resesrch 2016, 5, 1000. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.X.; Yao, M.; Zhang, X.D.; Xu, H.X.; Dong, Q. ER stress-induced autophagy in melanoma. Clin. Exp. Pharmacol. Physiol. 2015, 42, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Pereira, G.J.S.; Saito, R.F.; Buri, M.V.; Gagliardi, M.; Bincoletto, C.; Chammas, R.; Fimia, G.M.; Piacentini, M.; Corazzari, M.; et al. Effective synergy of sorafenib and nutrient shortage in inducing melanoma cell death through energy stress. Cells 2020, 9, 640. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The role of autophagy in the resistance to BRAF inhibition in BRAF-mutated melanoma. Target. Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef]
- Buart, S.; Terry, S.; Noman, M.Z.; Lanoy, E.; Boutros, C.; Fogel, P.; Dessen, P.; Meurice, G.; Gaston-Mathé, Y.; Vielh, P.; et al. Transcriptional response to hypoxic stress in melanoma and prognostic potential of GBE1 and BNIP3. Oncotarget 2017, 8, 108786–108801. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Dudek-Perić, A.M.; Maes, H.; Garg, A.D.; Gabrysiak, M.; Demirsoy, S.; Swinnen, J.V.; Agostinis, P. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in vemurafenib-resistant melanoma cells. Biochem. Pharmacol. 2015, 93, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Wu, S.; O’Connell, D.; Gao, T.; Zhong, H.; Yang, Y. miR-216b enhances the efficacy of vemurafenib by targeting Beclin-1, UVRAG and ATG5 in melanoma. Cell. Signal. 2018, 42, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiang, N.; Lin, M.; Huang, J.W.; Zhang, J.; Cheng, B.; Ji, C. miR- 26a sensitizes melanoma cells To dabrafenib via targeting HMGB1-dependent autophagy pathways. Drug Des. Devel. Ther. 2019, 13, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER Translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019, 9, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAFV600E inhibitor-resistant metastatic melanoma cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wu, C.; Shah, S.S.; Chen, S.M.; Wangpaichitr, M.; Kuo, M.T.; Feun, L.G.; Han, X.; Suarez, M.; Prince, J.; et al. Degradation of AMPK-α1 sensitizes BRAF inhibitor-resistant melanoma cells to arginine deprivation. Mol. Oncol. 2017, 11, 1806–1825. [Google Scholar] [CrossRef]
- Guerriero, L.; Palmieri, G.; De Marco, M.; Cossu, A.; Remondelli, P.; Capunzo, M.; Turco, M.C.; Rosati, A. The anti-apoptotic BAG3 protein is involved in BRAF inhibitor resistance in melanoma cells. Oncotarget 2017, 8, 80393–80404. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ren, W.; Yao, F.; Wang, H.; Zhang, K.; Luo, M.; Shang, Y.; O’Connell, D.; Bei, Z.; Wang, H.; et al. YY1 cooperates with TFEB to regulate autophagy and lysosomal biogenesis in melanoma. Mol. Carcinog. 2019, 58, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Möller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Osrodek, M.; Hartman, M.L. Plasticity of drug-naïve and vemurafenib- or trametinib-resistant melanoma cells in execution of differentiation/pigmentation program. J. Oncol. 2019, 2019, 1697913. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Czyz, M. Dissecting mechanisms of melanoma resistance to BRAF and MEK inhibitors revealed genetic and non-genetic patient- and drug-specific alterations and remarkable phenotypic plasticity. Cells 2020, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 23, 100. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Florean, C.; Song, S.; Dicato, M.; Diederich, M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 2019, 134, 177–189. [Google Scholar] [CrossRef]
- Degterev, A.; Maki, J.L.; Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013, 20, 366. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the pathophysiology of disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Miller, G. Molecular pathways: The ncrosome-a target for cancer therapy. Clin. Cancer Res. 2017, 23, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.C.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; van Delft, M.F.; Liu, Z.; Conos, S.A.; Zhang, J.G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 2017, 47, 51–65.e7. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Ke, H.; Augustine, C.K.; Gandham, V.D.; Jin, J.Y.; Tyler, D.S.; Akiyama, S.K.; Hall, R.P.; Zhang, J.Y. CYLD inhibits melanoma growth and progression through suppression of the JNK/AP-1 and β1-integrin signaling pathways. J. Investig. Dermatol. 2013, 133, 221–229. [Google Scholar] [CrossRef]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 10, e1884. [Google Scholar] [CrossRef]
- Rossi, A.; Pakhomova, O.N.; Pakhomov, A.G.; Weygandt, S.; Bulysheva, A.A.; Murray, L.E.; Mollica, P.A.; Muratori, C. Mechanisms and immunogenicity of nsPEF-induced cell death in B16F10 melanoma tumors. Sci. Rep. 2019, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.N.; Hei, T.K. Regulation of viability, differentiation and death of human melanoma cells carrying neural stem cell biomarkers: A possibility for neural trans-differentiation. Apoptosis 2015, 20, 996–1015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luan, Q.; Jin, L.; Jiang, C.C.; Tay, K.H.; Lai, F.; Liu, X.Y.; Liu, Y.L.; Guo, S.T.; Li, C.Y.; Yan, X.G.; et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy 2015, 11, 975–994. [Google Scholar] [CrossRef]
- Podder, B.; Guttà, C.; Rožanc, J.; Gerlach, E.; Feoktistova, M.; Panayotova-Dimitrova, D.; Alexopoulos, L.G.; Leverkus, M.; Rehm, M. TAK1 suppresses RIPK1-dependent cell death and is associated with disease progression in melanoma. Cell Death Differ. 2019, 26, 2520–2534. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Webster, J.D.; Varfolomeev, E.; Kwon, Y.C.; Cheng, J.H.; Zhang, J.; Dugger, D.L.; Wickliffe, K.E.; Maltzman, A.; Sujatha-Bhaskar, S.; et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020, 27, 161–175. [Google Scholar] [CrossRef]
- Hou, J.; Ju, J.; Zhang, Z.; Zhao, C.; Li, Z.; Zheng, J.; Sheng, T.; Zhang, H.; Hu, L.; Yu, X.; et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 2019, 10, 493. [Google Scholar] [CrossRef]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Cruz, S.A.; Qin, Z.; Stewart, A.F.R.; Chen, H.H. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural. Regen. Res. 2018, 13, 252–256. [Google Scholar] [CrossRef]
- Tahseen, A.I.; Patel, N.B. Successful dabrafenib transition after vemurafenib-induced toxic epidermal necrolysis in a patient with metastatic melanoma. JAAD Case Rep. 2018, 4, 930–933. [Google Scholar] [CrossRef]
- Werthmöller, N.; Frey, B.; Wunderlich, R.; Fietkau, R.; Gaipl, U.S. Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death Dis. 2015, 14, e1761. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhan, X.; Xiong, J.; Peng, S.; Huang, W.; Joshi, R.; Cai, Y.; Liu, Y.; Li, R.; Yuan, K.; et al. Temperature-dependent cell death patterns induced by functionalized gold nanoparticle photothermal therapy in melanoma cells. Sci. Rep. 2018, 8, 8720. [Google Scholar] [CrossRef]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Medrano, R.F.V.; Hunger, A.; Catani, J.P.P.; Strauss, B.E. Uncovering the immunotherapeutic cycle initiated by p19Arf and interferon-β gene transfer to cancer cells: An inducer of immunogenic cell death. Oncoimmunology 2017, 6, e1329072. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Raes, L.; Stremersch, S.; Brans, T.; Fraire, J.C.; Roelandt, R.; Declercq, W.; Vandenabeele, P.; Raemdonck, K.; Braeckmans, K.; et al. Delivery of mixed-lineage kinase domain-like protein by vapor nanobubble photoporation induces necroptotic-like cell death in tumor cells. Int. J. Mol. Sci. 2019, 20, 4254. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell. 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chen, X.; Li, J.; Comish, P.; Kang, R.; Tang, D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in cancer cell biology. Cancers (Basel) 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; Di Filippo, M.; French, L.E.; Beer, H.D. The crosstalk between Nrf2 and inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Toyokuni, S. Iron and thiols as two major players in carcinogenesis: Friends or foes? Front. Pharmacol. 2014, 5, 200. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, D.; Ma, D.; Ji, C.; Wu, W.; Xu, L.; Zhang, L. Ferroptosis suppressed the growth of melanoma that may be related to DNA damage. Dermatol. Ther. 2019, 32, e12921. [Google Scholar] [CrossRef] [PubMed]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.H.; et al. PML-regulated mitochondrial metabolism enhances chemosensitivity in human ovarian cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef]
- Gagliardi, M.; Cotella, D.; Santoro, C.; Corà, D.; Barlev, N.A.; Piacentini, M.; Corazzari, M. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019, 10, 902. [Google Scholar] [CrossRef]
- Osrodek, M.; Hartman, M.L.; Czyz, M. Physiologically relevant oxygen concentration (6% O2) as an important component of the microenvironment impacting melanoma phenotype and melanoma response to targeted therapeutics in vitro. Int. J. Mol. Sci. 2019, 20, 4203. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 27, 2500. [Google Scholar] [CrossRef] [PubMed]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Patrice Maboudou, G.G.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Hanniford, D.; Ulloa-Morales, A.; Karz, A.; Berzoti-Coelho, M.G.; Moubarak, R.S.; Sánchez-Sendra, B.; Kloetgen, A.; Davalos, V.; Imig, J.; Wu, P.; et al. Epigenetic silencing of CDR1as drives IGF2BP3-mediated melanoma invasion and metastasis. Cancer Cell. 2020, 37, 55–70.e15. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.M.; Attardi, L.D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Ou, Y.; Jiang, L.; Gu, W. Ferroptosis: A missing puzzle piece in the p53 blueprint? Mol. Cell. Oncol. 2015, 3, e1046581. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Liao, P.; Luo, S.; Zhang, M.; Hu, G.; Liu, H.; Zhang, Y.; Cao, B.; Baddoo, M.; et al. Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. Elife 2016, 5, e15099. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Shibazaki, M.; Kasai, S.; Yasuhira, S.; Watanabe, A.; Inoue, T.; Kageshita, Y.; Tsunoda, K.; Takahashi, K.; Akasaka, T.; et al. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J. Investig. Dermatol. 2014, 134, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Trush, M.A.; Li, Y.R. Nrf2 deficiency promotes melanoma growth and lung metastasis. React. Oxyg. Species (Apex) 2016, 2, 308–314. [Google Scholar] [CrossRef][Green Version]
- Hämäläinen, M.; Teppo, H.R.; Skarp, S.; Haapasaari, K.M.; Porvari, K.; Vuopala, K.; Kietzmann, T.; Karihtala, P. NRF1 and NRF2 mRNA and protein expression decrease early during melanoma carcinogenesis: An insight into survival and microRNAs. Oxid. Med. Cell. Longev. 2019, 2019, 2647068. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Liu, X.L.; Zhao, R. Induction of pyroptosis and its implications in cancer management. Front. Oncol. 2019, 9, 971. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem. Biol. Interact. 2020, 109052. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 6, 99–103. [Google Scholar] [CrossRef]
- Yu, P.; Wang, H.Y.; Tian, M.; Li, A.X.; Chen, X.S.; Wang, X.L.; Zhang, Y.; Cheng, Y. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol. Sin. 2019, 40, 1237–1244. [Google Scholar] [CrossRef]
- Lage, H.; Helmbach, H.; Grottke, C.; Dietel, M.; Schadendorf, D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001, 6, 54–59. [Google Scholar] [CrossRef]
- Colunga, A.G.; Laing, J.M.; Aurelian, L. The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther. 2010, 17, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, J.Y.; Liu, X.S.; Chen, H.Z.; Ai, Y.L.; Cheng, K.; Sun, R.Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Emran, A.A.; Tseng, H.Y.; Coleman, M.C.; Tiffen, J.; Cook, S.; McGuire, H.M.; Gallagher, S.; Feng, C.; Hersey, P. Do innate killing mechanisms activated by inflammasomes have a role in treating melanoma? Pigment Cell Melanoma Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Luo, Y.; Dunn, J.H.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J. Investig. Dermatol. 2013, 133, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Watabe, K.; Ito, A.; Asada, H.; Endo, Y.; Kobayashi, T.; Nakamoto, K.; Itami, S.; Takao, S.; Shinomura, Y.; Aikou, T.; et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn. J. Cancer Res. 2001, 92, 140–151. [Google Scholar] [CrossRef]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. (Landmark Ed.) 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, J.; David, K.K.; Yang, Z.J.; Li, X.; Dawson, T.M.; Dawson, V.L.; Koehler, R.C. Endonuclease G does not play an obligatory role in poly(ADP-ribose) polymerase-dependent cell death after transient focal cerebral ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R215–R221. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Moriceau, G.; Sun, L.; Lomeli, S.; Piva, M.; Damoiseaux, R.; Holmen, S.L.; Sharpless, N.E.; Hugo, W.; Lo, R.S. Exploiting drug addiction mechanisms to select against MAPKi-resistant melanoma. Cancer Discov. 2018, 8, 74–93. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Liguoro, D.; Mancini, R.; Ciliberto, G. Single cell analysis to dissect molecular heterogeneity and disease evolution in metastatic melanoma. Cell Death Dis. 2019, 10, 827. [Google Scholar] [CrossRef]
- Luebker, S.A.; Koepsell, S.A. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | Autophagy | Necroptosis | Ferroptosis | Pyroptosis | |
|---|---|---|---|---|---|
| morphology | membrane blebbing | formation of intracellular vesicles | cell swelling | shrunken mitochondria with reduced crista | cell swelling; pore formation |
| DNA fragmentation | + | - | + (random) | - | + |
| caspase activation | + | - | - | - | + |
| positive staining with Annexin-V | + | - | + | + | + |
| inflammation | - | - | + | + | + |
| other typical features | PARP cleavage | LC3-I to LC3-II conversion; p62/SQSTM1 degradation | RIPK3/MLKL activation; drop in ATP level | iron accumulation and lipid peroxidation | osmotic lysis of cell |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, M.L. Non-Apoptotic Cell Death Signaling Pathways in Melanoma. Int. J. Mol. Sci. 2020, 21, 2980. https://doi.org/10.3390/ijms21082980
Hartman ML. Non-Apoptotic Cell Death Signaling Pathways in Melanoma. International Journal of Molecular Sciences. 2020; 21(8):2980. https://doi.org/10.3390/ijms21082980
Chicago/Turabian StyleHartman, Mariusz L. 2020. "Non-Apoptotic Cell Death Signaling Pathways in Melanoma" International Journal of Molecular Sciences 21, no. 8: 2980. https://doi.org/10.3390/ijms21082980
APA StyleHartman, M. L. (2020). Non-Apoptotic Cell Death Signaling Pathways in Melanoma. International Journal of Molecular Sciences, 21(8), 2980. https://doi.org/10.3390/ijms21082980