Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
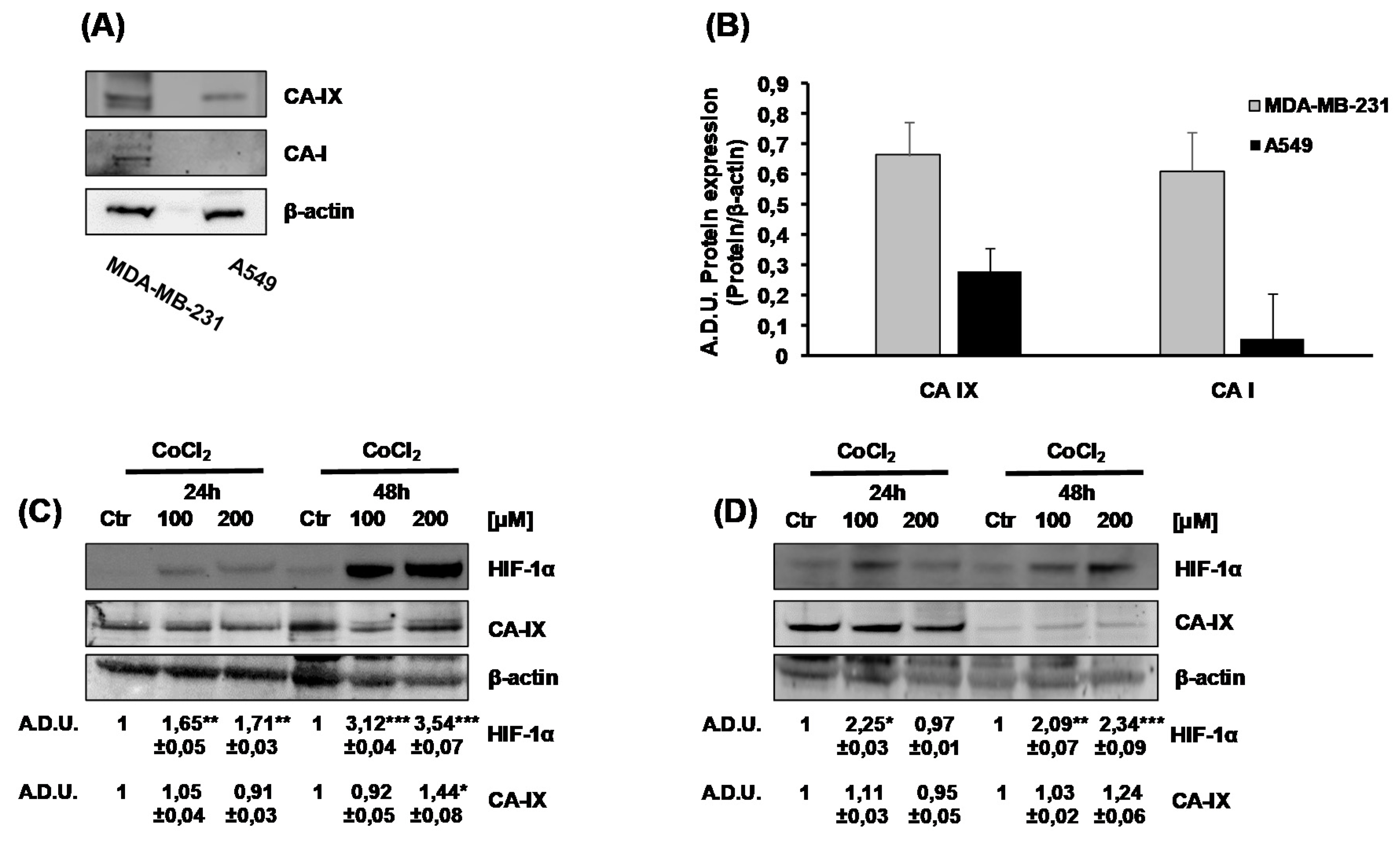
2.1. Characterization of CA-I, CA-II and CA-IX Expression in Tumor Cells
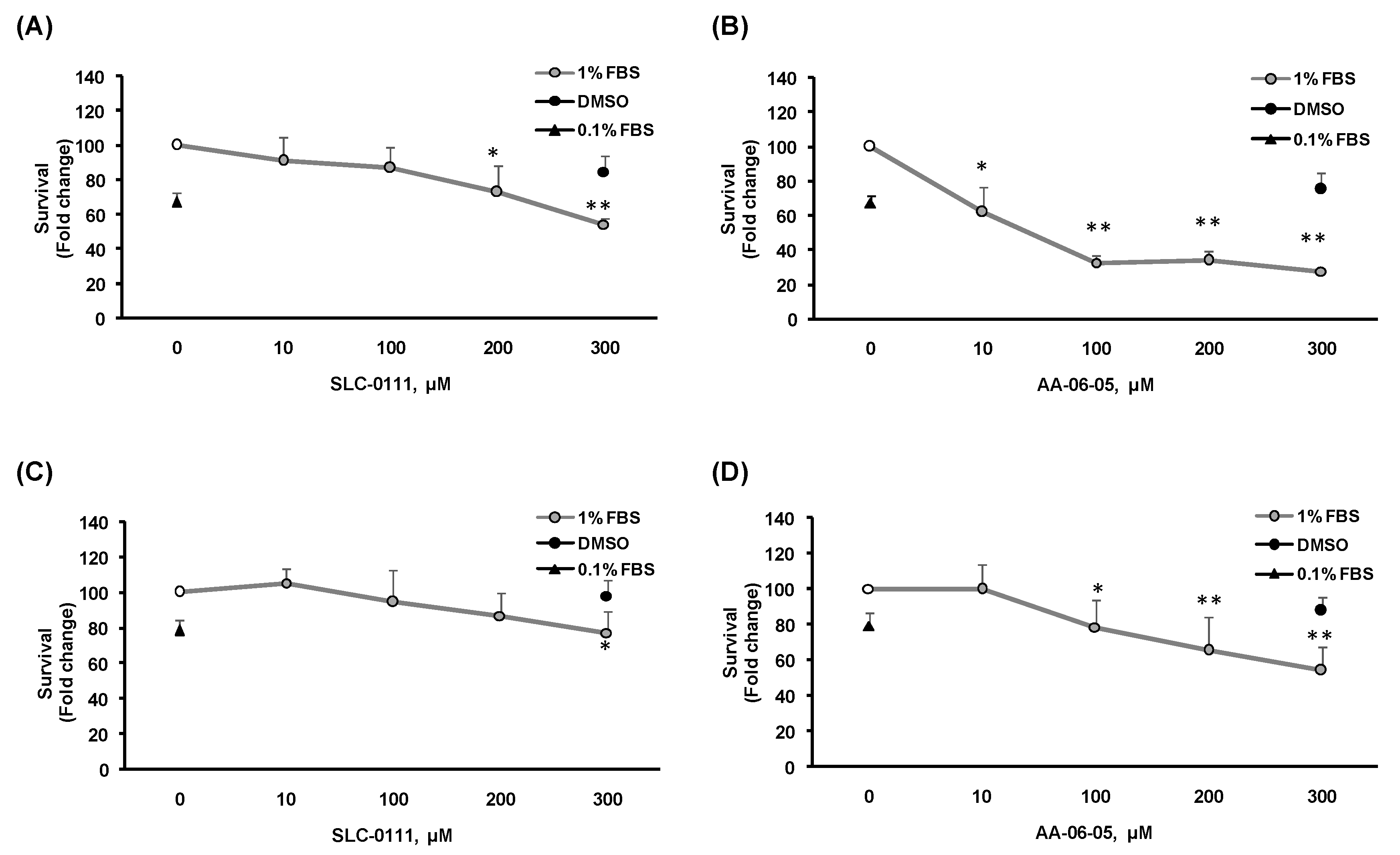
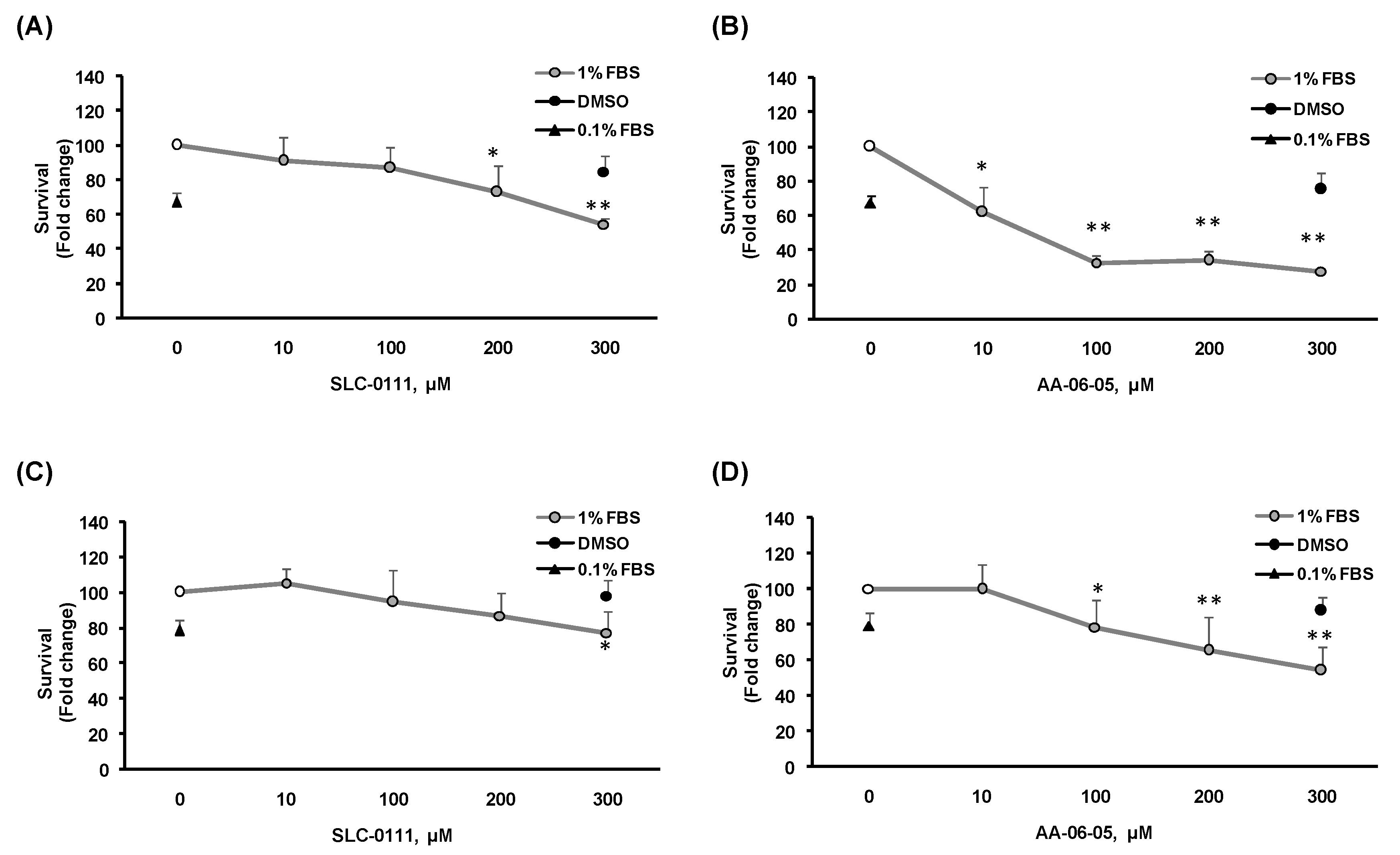
2.2. CA-IX Pharmacological Inhibition Induces Cell Death in Tumor Cells
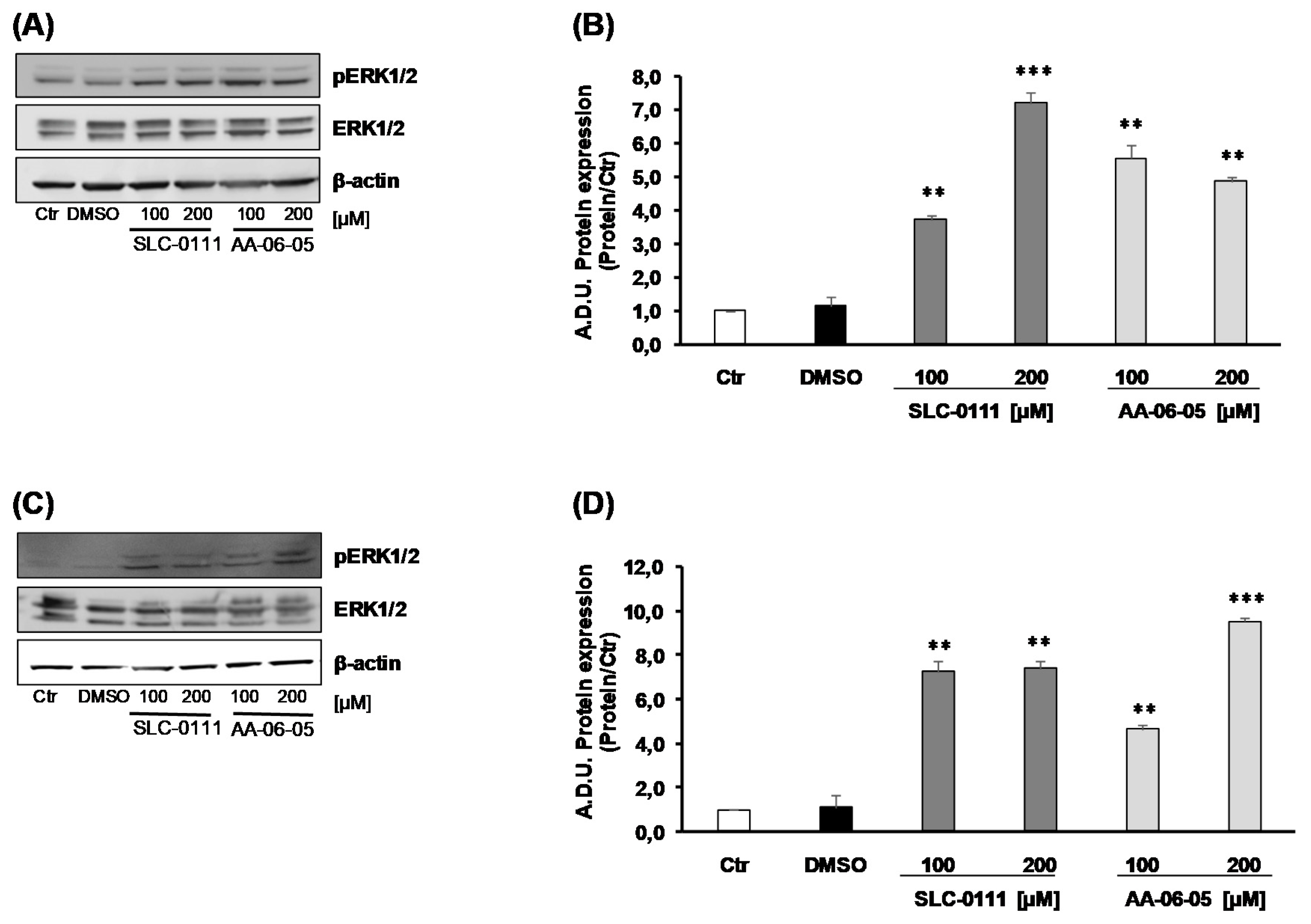
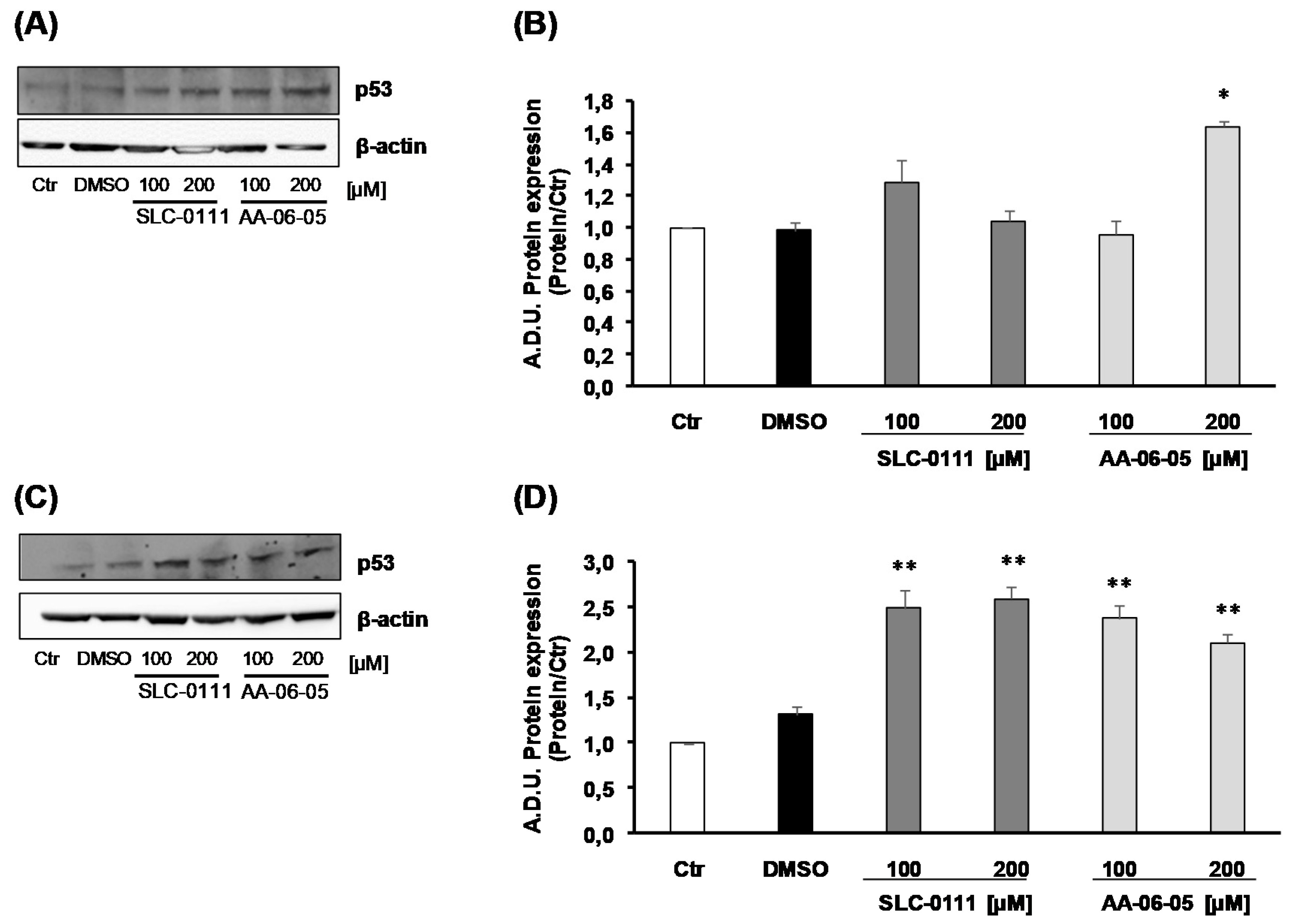
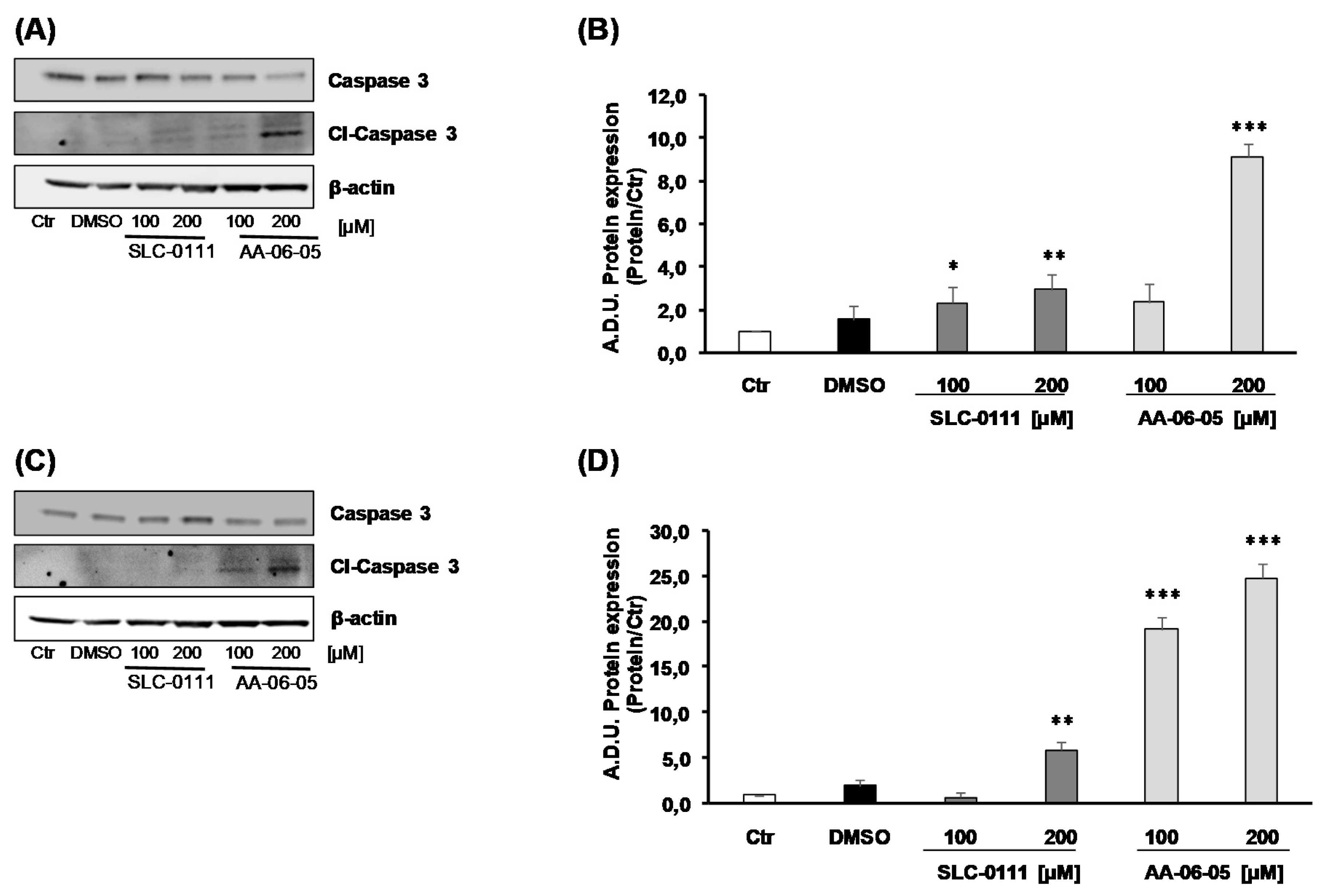
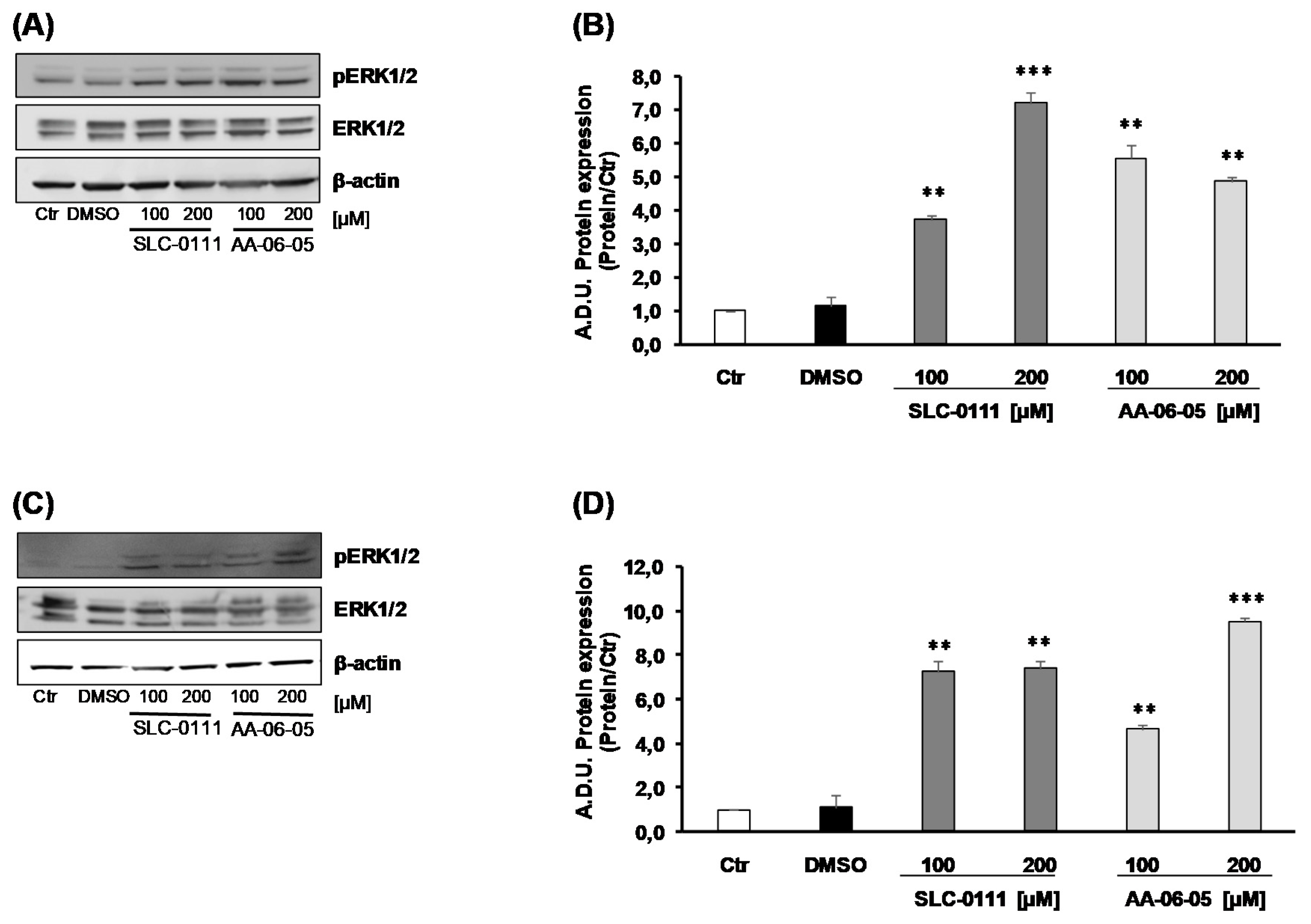
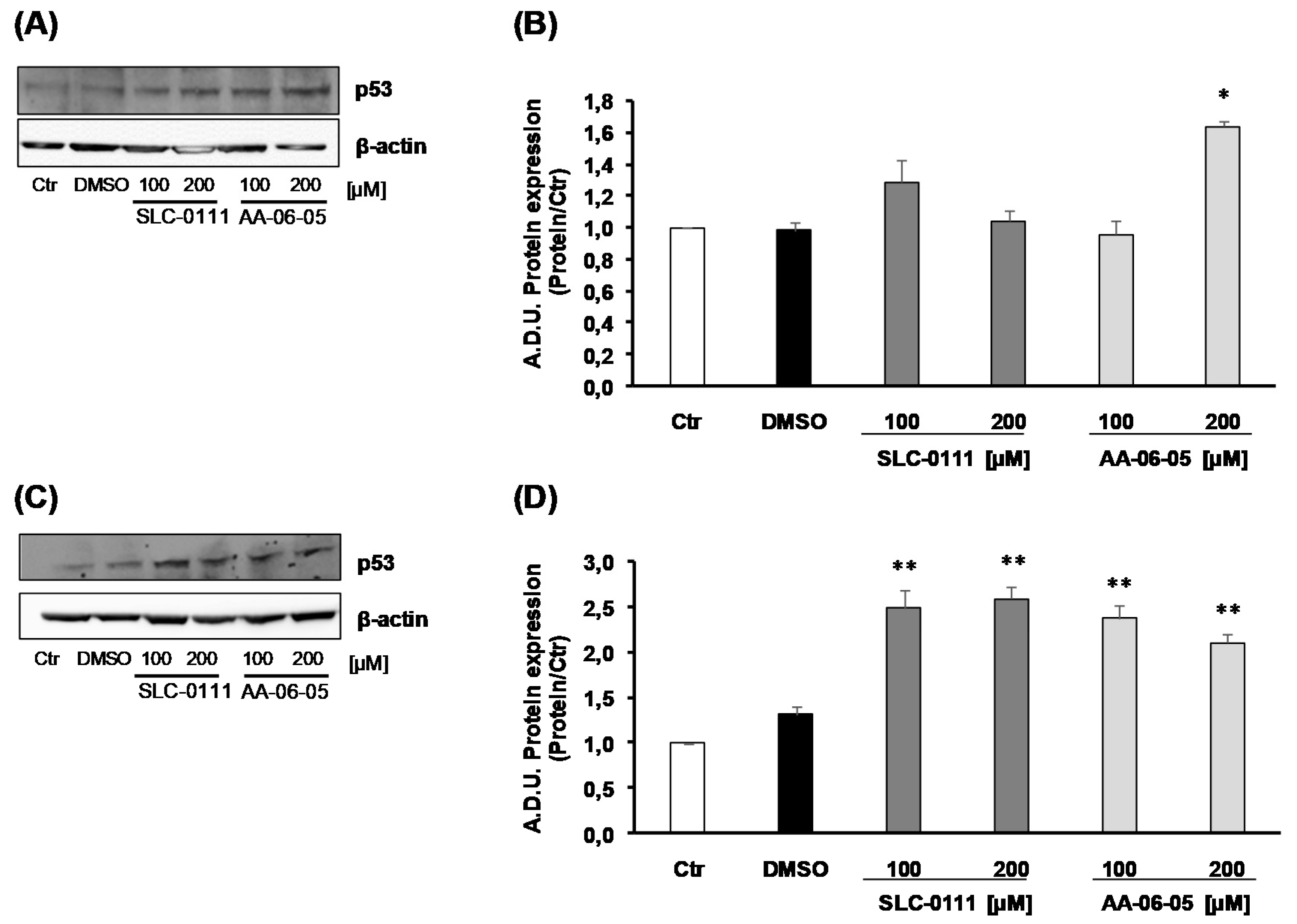
2.3. CA-IX Pharmacological Inhibition Activates Apoptotic Pathway in Tumor Cells
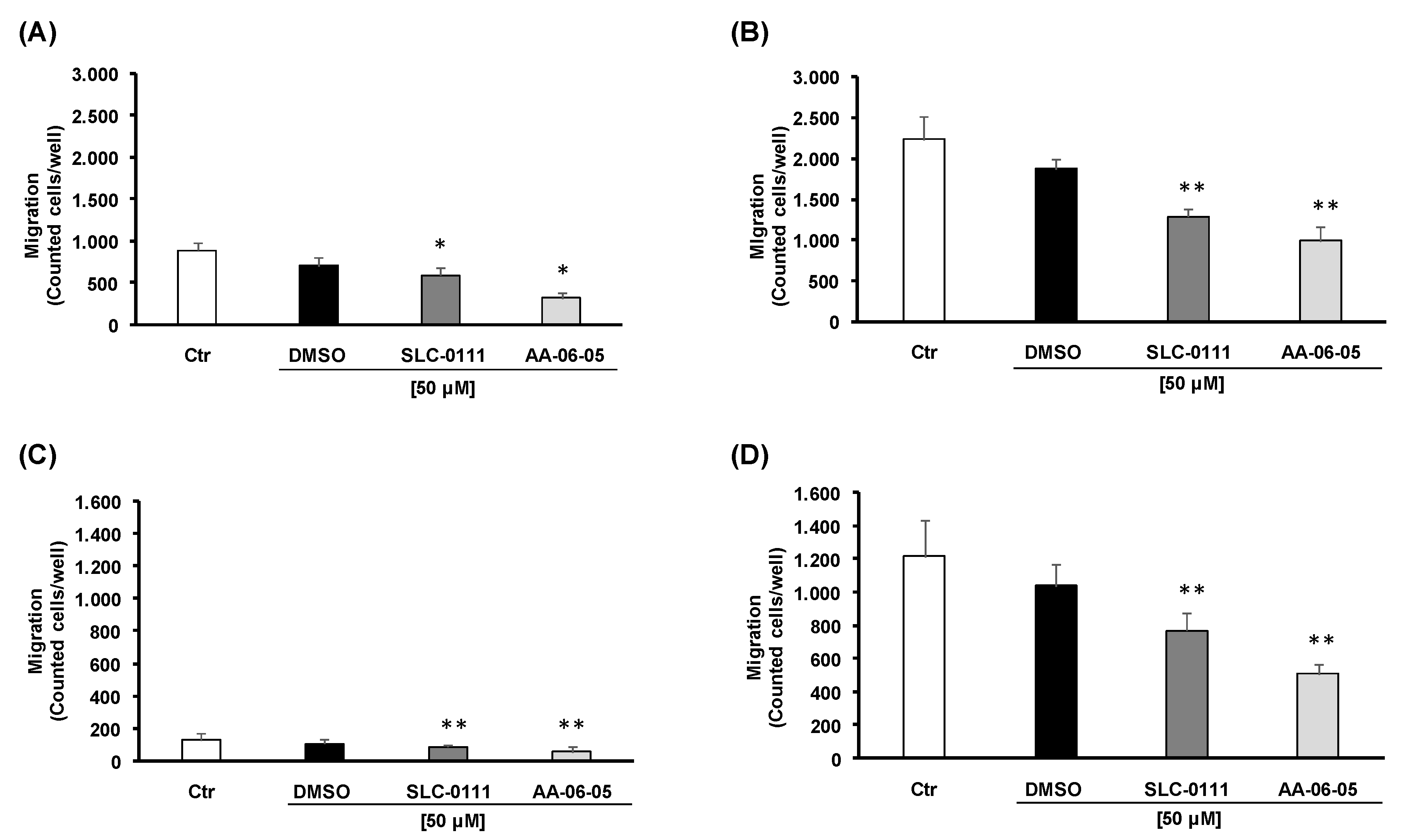
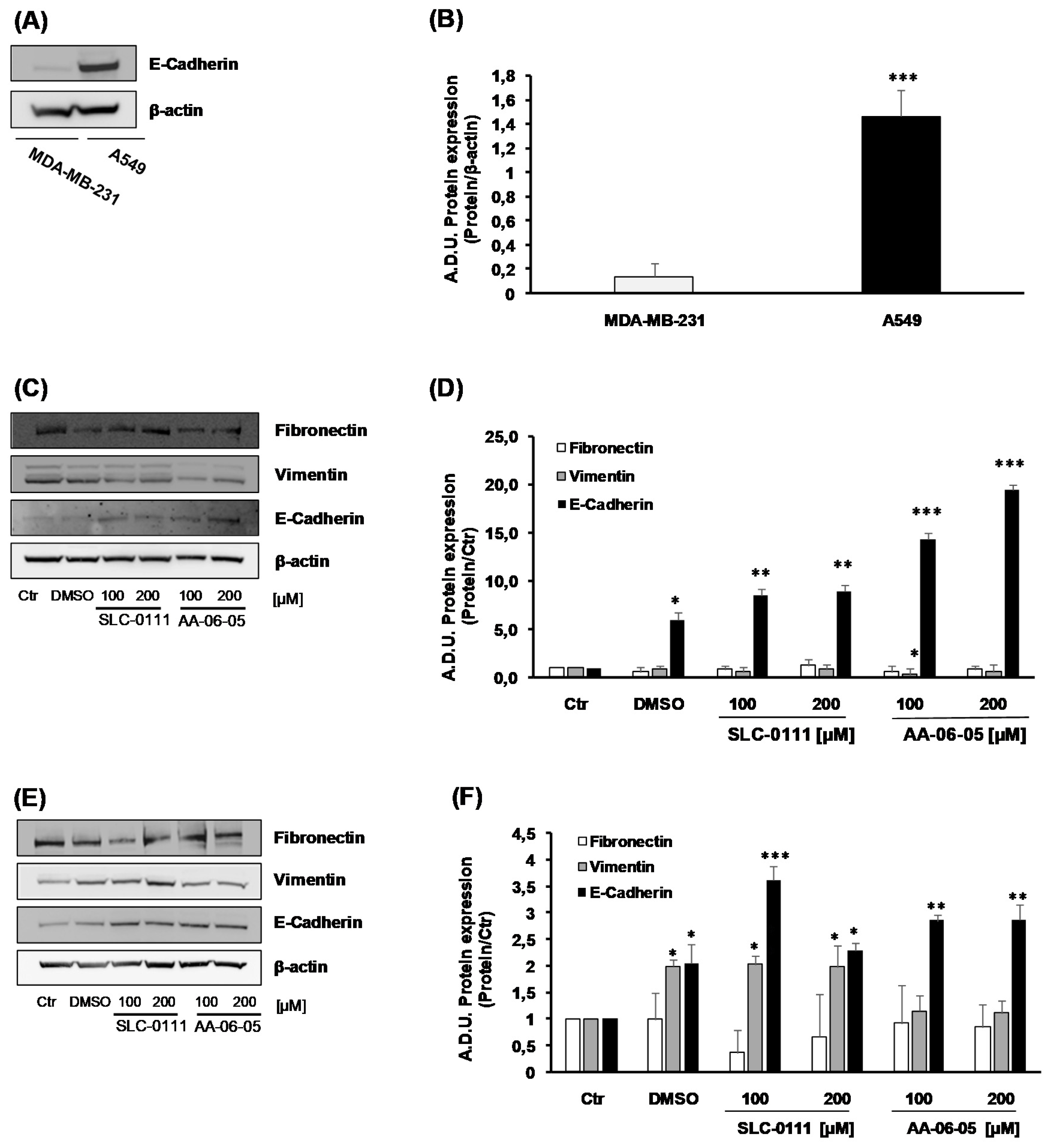
2.4. Pharmacological Inhibition of CA-IX Reduces Tumor Cell Migration and Invasion Ability
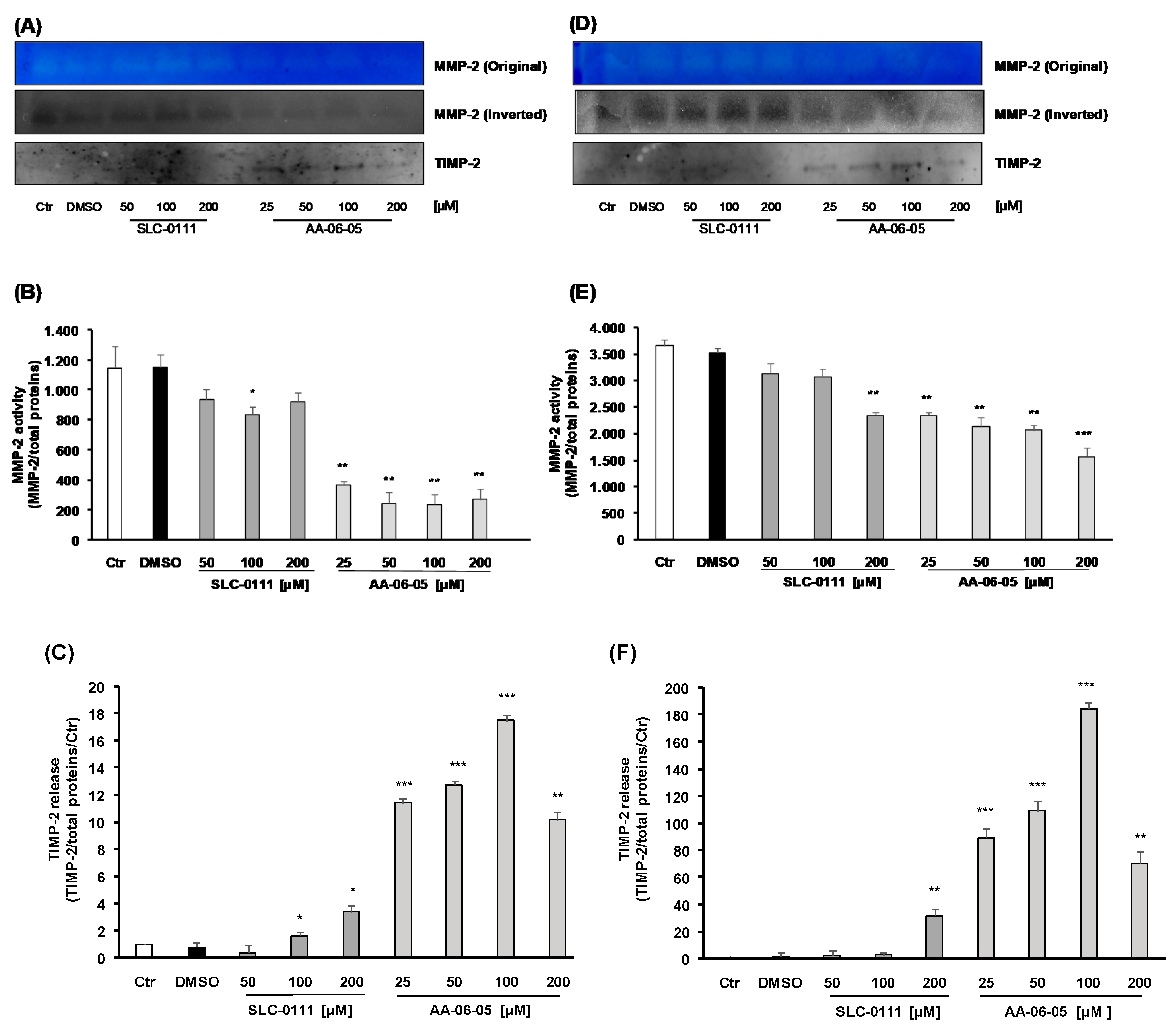
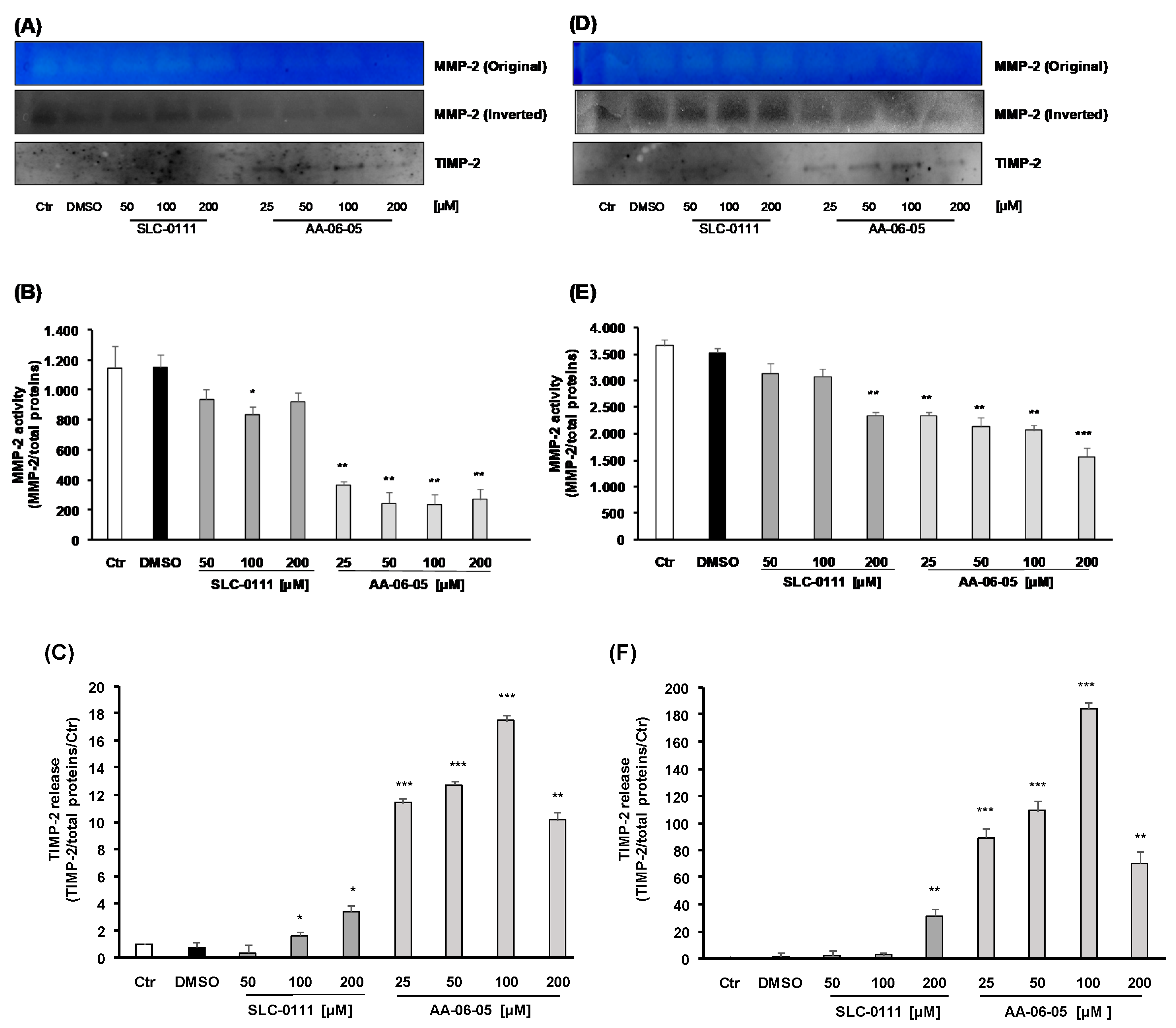
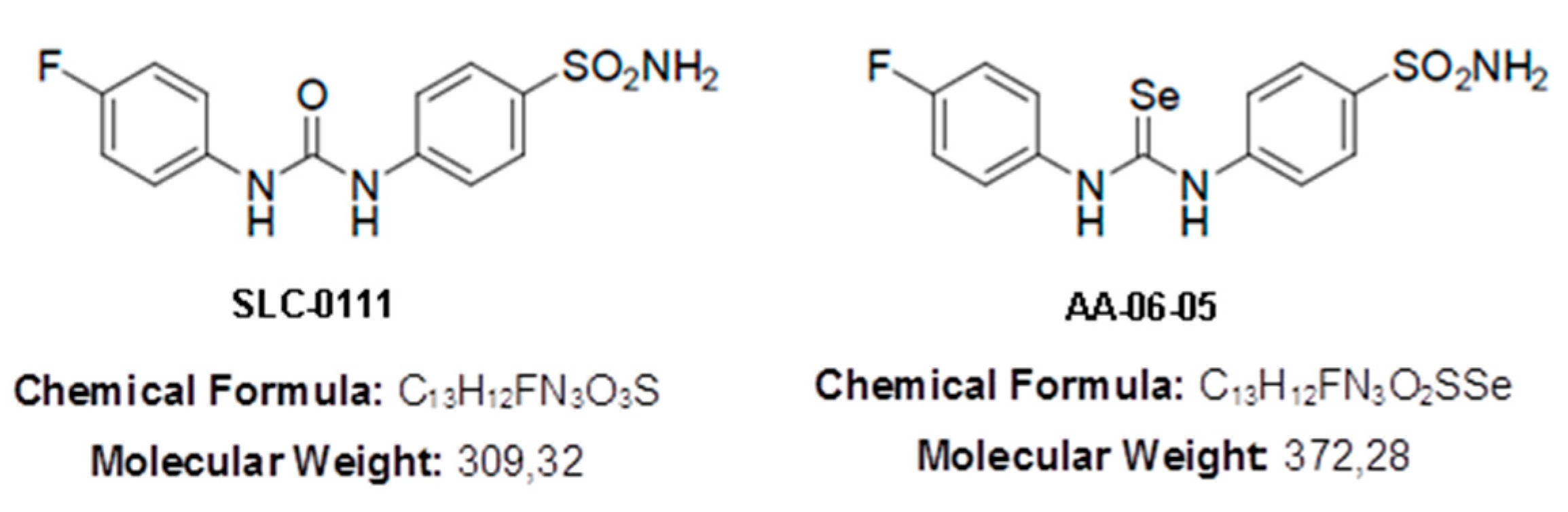
2.5. Inhibition of CA-IX is Related to Inhibition of MMP-2 Activity
3. Discussion
4. Materials and Methods
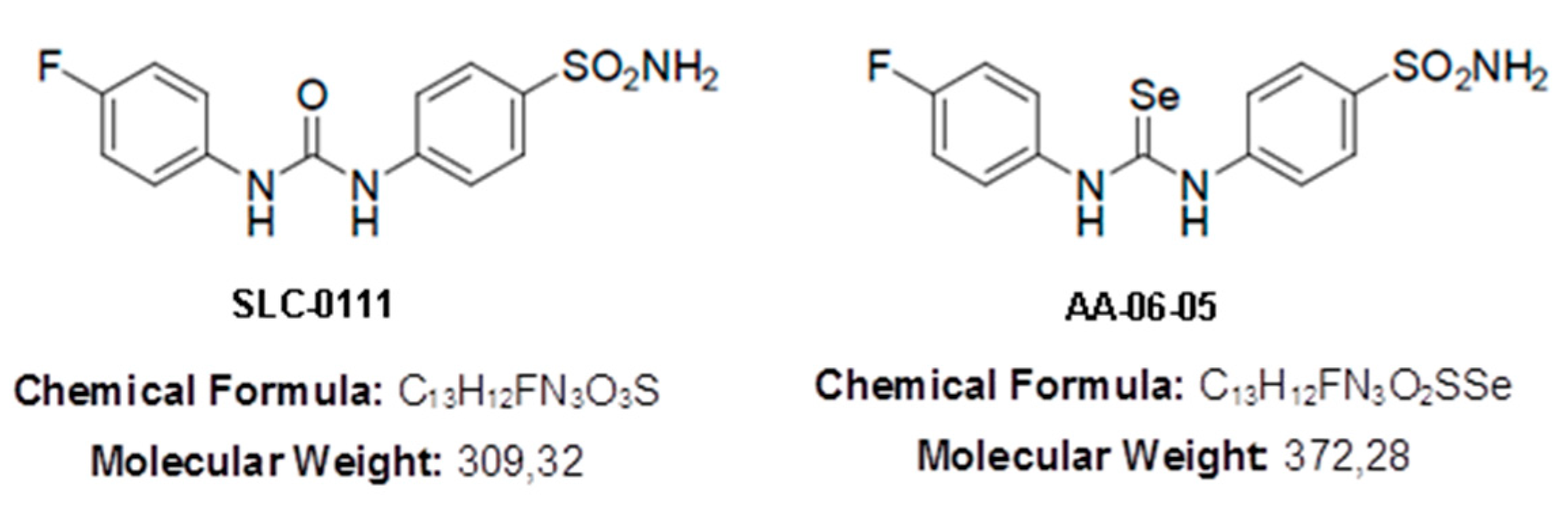
4.1. Drugs and Reagents
4.2. Cell Cultures
4.3. Tumor Cell Viability
4.4. Tumor Cell Invasion
4.5. Protein Expression
4.6. Gelatin Zymography
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CA | Carbonic anhydrase |
| HIF-1α | Hypoxia inducible factor 1- α |
| FBS | Fetal Bovine Serum |
| ECM | Extracellular matrix |
| EMT | Epithelial-Mesenchymal Transition |
| pHi | Intracellular pH |
| pHe | Extracellular pH |
| MMP | Matrix Metalloproteinase |
| TIMP | Tissue Inhibitor of Metalloproteinase |
References
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Astekar, M.; Soi, S.; Manjunatha, B.S.; Shetty, D.C.; Radhakrishnan, R. pH Gradient Reversal: An Emerging Hallmark of Cancers. Recent Pat. Anticancer Drug Discov. 2015, 10, 244–258. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflugers Arch. 2009, 458, 981–992. [Google Scholar] [CrossRef]
- Suzuki, A.; Maeda, T.; Baba, Y.; Shimamura, K.; Kato, Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int. 2014, 14, 129. [Google Scholar] [CrossRef] [Green Version]
- Riemann, A.; Rauschner, M.; Gießelmann, M.; Reime, S.; Haupt, V.; Thews, O. Extracellular Acidosis Modulates the Expression of Epithelial-Mesenchymal Transition (EMT) Markers and Adhesion of Epithelial and Tumor Cells. Neoplasia 2019, 21, 450–458. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Saarnio, J.; Parkkila, S.; Parkkila, A.K.; Waheed, A.; Casey, M.C.; Zhou, X.Y.; Pastoreková, S.; Pastorek, J.; Karttunen, T.; Haukipuro, K.; et al. Immunohistochemistry of carbonic anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J. Histochem. Cytochem. 1998, 46, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar]
- Forker, L.; Gaunt, P.; Sioletic, S.; Shenjere, P.; Potter, R.; Roberts, D.; Irlam, J.; Valentine, H.; Hughes, D.; Hughes, A.; et al. The hypoxia marker CA-IX is prognostic in the UK phase III VorteX-Biobank cohort: An important resource for translational research in soft tissue sarcoma. Br. J. Cancer 2018, 118, 698–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [Green Version]
- Ilie, M.; Mazure, N.M.; Hofman, V.; Ammadi, R.E.; Ortholan, C.; Bonnetaud, C.; Havet, K.; Venissac, N.; Mograbi, B.; Mouroux, J.; et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br. J. Cancer 2010, 102, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kuijk, S.J.; Yaromina, A.; Houben, R.; Niemans, R.; Lambin, P.; Dubois, L.J. Prognostic Significance of Carbonic Anhydrase IX Expression in Cancer Patients: A Meta-Analysis. Front. Oncol 2016, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Tanini, D.; Viglianisi, C.; Panzella, L.; Capperucci, A.; Menichetti, S.; Supuran, C.T. Evaluation of selenide, diselenide and selenoheterocycle derivatives as carbonic anhydrase I, II, IV, VII and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 2518–2523. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; Dedhar, S. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Peat, T.S.; Di Cesare Mannelli, L.; Bartolucci, G.; Capperucci, A.; Ghelardini, C.; Supuran, C.T.; Carta, F. Discovery of New Selenoureido Analogues of 4-(4-Fluorophenylureido)benzenesulfonamide as Carbonic Anhydrase Inhibitors. ACS Med. Chem. Lett. 2017, 8, 963–968. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Beasley, N.; Watson, P.H.; Campo, L.; Chia, S.K.; English, R.; Pastorek, J.; Sly, W.S.; Ratcliffe, P.; Harris, A.L. Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast. Am. J. Pathol. 2001, 158, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Tülüce, Y.; Ahmed, B.A.; Koyuncu, İ.; Durgun, M. The cytotoxic, apoptotic and oxidative effects of carbonic anhydrase IX inhibitor on colorectal cancer cells. J. Bioenerg. Biomembr. 2018, 50, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Monti, M.; Monzani, E.; Casella, L.; Morbidelli, L. The metal-nonoateNi(SalPipNONO) inhibits. Oncotarget 2018, 9, 13353–13365. [Google Scholar] [PubMed] [Green Version]
- Monti, M.; Terzuoli, E.; Ziche, M.; Morbidelli, L. The sulphydryl containing ACE inhibitor Zofenoprilat protects coronary endothelium from Doxorubicin-induced apoptosis. Pharmacol. Res. 2013, 76, 171–181. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. Polarity proteins in migration and invasion. Oncogene 2008, 27, 6970–6980. [Google Scholar] [CrossRef] [Green Version]
- Mbalaviele, G.; Dunstan, C.R.; Sasaki, A.; Williams, P.J.; Mundy, G.R.; Yoneda, T. E-cadherin expression in human breast cancer cells suppresses the development of osteolytic bone metastases in an experimental metastasis model. Cancer Res. 1996, 56, 4063–4070. [Google Scholar]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Meehan, J.; Gray, M.; Kunkler, I.H.; Langdon, S.P.; Argyle, D.J. Carbonic Anhydrase IX (CA-IX), Cancer, and Radiation Responsiveness. Metabolites 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Langdon, S.P.; Mullen, P.; Harris, A.L.; Harrison, D.J.; Supuran, C.T.; Kunkler, I.H. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat. Rev. 2013, 39, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Svastová, E.; Zilka, N.; Zat’ovicová, M.; Gibadulinová, A.; Ciampor, F.; Pastorek, J.; Pastoreková, S. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with beta-catenin. Exp. Cell Res. 2003, 290, 332–345. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Horne, H.N.; Oh, H.; Sherman, M.E.; Palakal, M.; Hewitt, S.M.; Schmidt, M.K.; Milne, R.L.; Hardisson, D.; Benitez, J.; Blomqvist, C.; et al. E-cadherin breast tumor expression, risk factors and survival: Pooled analysis of 5,933 cases from 12 studies in the Breast Cancer Association Consortium. Sci. Rep. 2018, 8, 6574. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar] [CrossRef]
- Li, C.L.; Yang, D.; Cao, X.; Wang, F.; Hong, D.Y.; Wang, J.; Shen, X.C.; Chen, Y. Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain. Oncol. Lett. 2017, 13, 3889–3895. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Rho, S.B.; Jung, D.C.; Han, I.O.; Oh, E.S.; Kim, J.Y. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J. Cell Sci. 2011, 124, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.Z.; Feng, W.Y.; Zhou, Q.; Liu, Y.W.; Qi, S.T. The Impact of MMP-2 and Its Specific Inhibitor TIMP-2 Expression on the WHO Grade and Prognosis of Gliomas in Chinese Population: A Meta-Analysis. Mol. Neurobiol. 2017, 54, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.M.; Fridman, R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem. J. 2003, 374, 739–745. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Correction to: Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 45. [Google Scholar] [CrossRef] [Green Version]
- Morbidelli, L.; Parenti, A.; Giovannelli, L.; Granger, H.J.; Ledda, F.; Ziche, M. B1 receptor involvement in the effect of bradykinin on venular endothelial cell proliferation and potentiation of FGF-2 effects. Br. J. Pharmacol. 1998, 124, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, V.; Monti, M.; Antonini, G.; Mattoli, L.; Burico, M.; Marini, F.; Maidecchi, A.; Morbidelli, L. Efficacy of AdipoDren®® in Reducing Interleukin-1-Induced Lymphatic Endothelial Hyperpermeability. J. Vasc. Res. 2016, 53, 255–268. [Google Scholar] [CrossRef]
- Terzuoli, E.; Nannelli, G.; Giachetti, A.; Morbidelli, L.; Ziche, M.; Donnini, S. Targeting endothelial-to-mesenchymal transition: The protective role of hydroxytyrosol sulfate metabolite. Eur. J. Nutr. 2020, 59, 517–527. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccone, V.; Filippelli, A.; Angeli, A.; Supuran, C.T.; Morbidelli, L. Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. Int. J. Mol. Sci. 2020, 21, 2983. https://doi.org/10.3390/ijms21082983
Ciccone V, Filippelli A, Angeli A, Supuran CT, Morbidelli L. Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. International Journal of Molecular Sciences. 2020; 21(8):2983. https://doi.org/10.3390/ijms21082983
Chicago/Turabian StyleCiccone, Valerio, Arianna Filippelli, Andrea Angeli, Claudiu T. Supuran, and Lucia Morbidelli. 2020. "Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness" International Journal of Molecular Sciences 21, no. 8: 2983. https://doi.org/10.3390/ijms21082983
APA StyleCiccone, V., Filippelli, A., Angeli, A., Supuran, C. T., & Morbidelli, L. (2020). Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. International Journal of Molecular Sciences, 21(8), 2983. https://doi.org/10.3390/ijms21082983