Epigenetic Regulation of Circadian Rhythm and Its Possible Role in Diabetes Mellitus



Abstract
:1. Introduction
2. Genetics of Type 2 Diabetes Mellitus
3. Epigenetics of Type 2 Diabetes Mellitus
4. Epigenetic Control of Circadian Rhythms
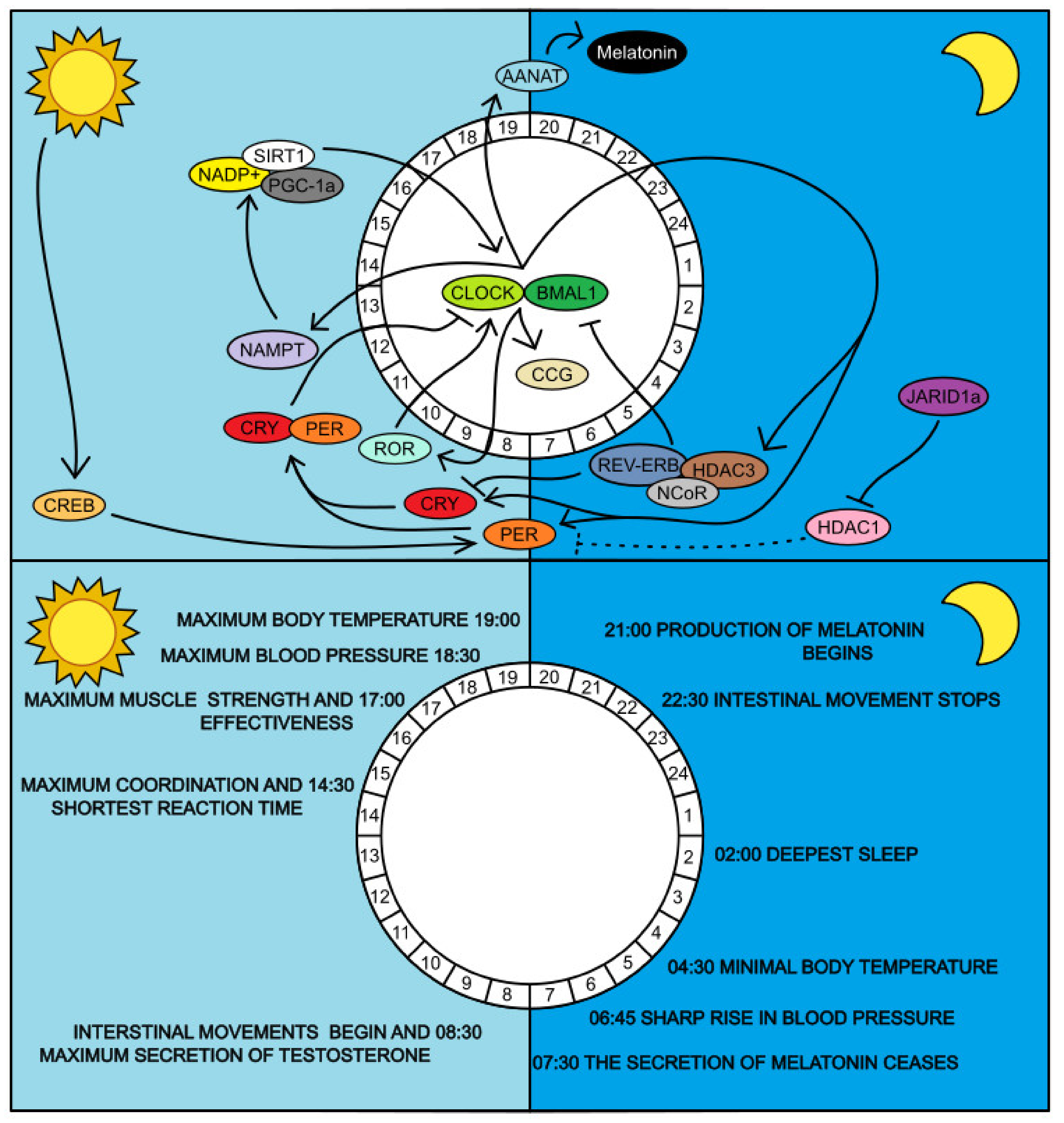
4.1. Regulatory Genes CLOCK and BMAL1
4.2. Regulation of BMAL1 Expression
4.3. The Role of SIRT1 in Regulating Circadian Rhythms
5. Circadian Rhythms and Aging
6. Interindividual Variability of Circadian Preference
6.1. Advanced Sleep-Wake Phase Disorder
6.2. Delayed Sleep-Wake Phase Disorder
7. Pathology Resulting from Decompensation of Circadian Rhythms
7.1. Metabolic Syndrome, T2DM and Circadian Rhythm Disorders
7.2. The Role of REV-ERB Nuclear Receptors in Metabolism Regulation
7.3. Circadian Rhythm of Blood Pressure
8. Conclusions and Possible Treatments
Author Contributions
Funding
Conflicts of Interest
References
- Stapel, L.C.; Zechner, C.; Vastenhouw, N.L. Uniform gene expression in embryos is achieved by temporal averaging of transcription noise. Genes Dev. 2017, 31, 1635–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Relógio, A.; Westermark, P.O.; Wallach, T.; Schellenberg, K.; Kramer, A.; Herzel, H. Tuning the Mammalian Circadian Clock: Robust Synergy of Two Loops. PLoS Comput. Biol. 2011, 7, e1002309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell-Pedersen, D.; Cassone, V.M.; Earnest, D.J.; Golden, S.S.; Hardin, P.E.; Thomas, T.L.; Zoran, M.J. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 2005, 6, 544–556. [Google Scholar] [CrossRef]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Tsang, A.H.; Astiz, M.; Friedrichs, M.; Oster, H. Endocrine regulation of circadian physiology. J. Endocrinol. 2016, 230, R1–R11. [Google Scholar] [CrossRef]
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803. [Google Scholar] [CrossRef]
- Facer-Childs, E.R.; Middleton, B.; Skene, D.J.; Bagshaw, A.P. Resetting the late timing of ‘night owls’ has a positive impact on mental health and performance. Sleep Med. 2019, 60, 236–247. [Google Scholar] [CrossRef]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Kudielka, B.M.; Federenko, I.S.; Hellhammer, D.H.; Wüst, S. Morningness and eveningness: The free cortisol rise after awakening in “early birds” and “night owls”. Biol. Psychol. 2006, 72, 141–146. [Google Scholar] [CrossRef]
- Leliavski, A.; Dumbell, R.; Ott, V.; Oster, H. Adrenal Clocks and the Role of Adrenal Hormones in the Regulation of Circadian Physiology. J. Biol. Rhythms 2015, 30, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Madrid, J.A.; Tan, D.-X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef] [PubMed]
- Rajaratnam, S.M.W.; Arendt, J. Health in a 24-h society. Lancet 2001, 358, 999–1005. [Google Scholar] [CrossRef]
- Ayas, N.T.; White, D.P.; Al-Delaimy, W.K.; Manson, J.E.; Stampfer, M.J.; Speizer, F.E.; Patel, S.; Hu, F.B. A Prospective Study of Self-Reported Sleep Duration and Incident Diabetes in Women. Diabetes Care 2003, 26, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, K.L.; Spiegel, K.; Penev, P.; Van Cauter, E. The metabolic consequences of sleep deprivation. Sleep Med. Rev. 2007, 11, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Kader, F.; Ghai, M. DNA methylation and application in forensic sciences. Forensic Sci. Int. 2015, 249, 255–265. [Google Scholar] [CrossRef]
- Scheer, F.A.J.L.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. 2009, 106, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- Suwazono, Y.; Dochi, M.; Sakata, K.; Okubo, Y.; Oishi, M.; Tanaka, K.; Kobayashi, E.; Kido, T.; Nogawa, K. A Longitudinal Study on the Effect of Shift Work on Weight Gain in Male Japanese Workers. Obesity 2008, 16, 1887–1893. [Google Scholar] [CrossRef]
- Tam, B.T.; Morais, J.A.; Santosa, S. Obesity and ageing: Two sides of the same coin. Obes. Rev. 2020, 21, 1–21, e12991. [Google Scholar] [CrossRef]
- Zylka, M.J.; Shearman, L.P.; Weaver, D.R.; Reppert, S.M. Three period Homologs in Mammals: Differential Light Responses in the Suprachiasmatic Circadian Clock and Oscillating Transcripts Outside of Brain. Neuron 1998, 20, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Travnickova-Bendova, Z.; Cermakian, N.; Reppert, S.M.; Sassone-Corsi, P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. 2002, 99, 7728–7733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Minguez, J.; Gómez-Abellán, P.; Garaulet, M. Circadian rhythms, food timing and obesity. Proc. Nutr. Soc. 2016, 75, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, K.L. An hPer2 Phosphorylation Site Mutation in Familial Advanced Sleep Phase Syndrome. Science 2001, 291, 1040–1043. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Etchegaray, J.-P.; Cagampang, F.R.A.; Loudon, A.S.I.; Reppert, S.M. Posttranslational Mechanisms Regulate the Mammalian Circadian Clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef]
- Taheri, S.; Lin, L.; Austin, D.; Young, T.; Mignot, E. Short Sleep Duration Is Associated with Reduced Leptin, Elevated Ghrelin, and Increased Body Mass Index. PLoS Med. 2004, 1, e62. [Google Scholar] [CrossRef] [PubMed]
- Turek, F.W. Obesity and Metabolic Syndrome in Circadian Clock Mutant Mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimba, S.; Ishii, N.; Ohta, Y.; Ohno, T.; Watabe, Y.; Hayashi, M.; Wada, T.; Aoyagi, T.; Tezuka, M. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 12071–12076. [Google Scholar] [CrossRef] [Green Version]
- Curtis, A.M.; Cheng, Y.; Kapoor, S.; Reilly, D.; Price, T.S.; FitzGerald, G.A. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc. Natl. Acad. Sci. USA 2007, 104, 3450–3455. [Google Scholar] [CrossRef] [Green Version]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-erb and Rev-erb coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Doi, M.; Takahashi, Y.; Komatsu, R.; Yamazaki, F.; Yamada, H.; Haraguchi, S.; Emoto, N.; Okuno, Y.; Tsujimoto, G.; Kanematsu, A.; et al. Salt-sensitive hypertension in circadian clock–deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat. Med. 2010, 16, 67–74. [Google Scholar] [CrossRef]
- Feng, D.; Liu, T.; Sun, Z.; Bugge, A.; Mullican, S.E.; Alenghat, T.; Liu, X.S.; Lazar, M.A. A Circadian Rhythm Orchestrated by Histone Deacetylase 3 Controls Hepatic Lipid Metabolism. Science 2011, 331, 1315–1319. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Padiath, Q.S.; Shapiro, R.E.; Jones, C.R.; Wu, S.C.; Saigoh, N.; Saigoh, K.; Ptáček, L.J.; Fu, Y.-H. Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 2005, 434, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, E.D. Delayed Sleep Phase Syndrome. Arch. Gen. Psychiatry 1981, 38, 737. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado de Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear Trapping of the Forkhead Transcription Factor FoxO1 via Sirt-dependent Deacetylation Promotes Expression of Glucogenetic Genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Kharroubi, A.T. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; McCarthy, M.I. Human Genetics of Obesity and Type 2 Diabetes Mellitus. Circ. Genomic Precis. Med. 2018, 11, e002090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karalliedde, J.; Gnudi, L. Diabetes mellitus, a complex and heterogeneous disease, and the role of insulin resistance as a determinant of diabetic kidney disease. Nephrol. Dial. Transplant. 2014, 6, 206–213. [Google Scholar] [CrossRef]
- Freedman, B.I.; Tuttle, A.B.; Spray, B.J. Familial predisposition to nephropathy in African-Americans with non-insulin-dependent diabetes mellitus. Am. J. Kidney Dis. 1995, 25, 710–713. [Google Scholar] [CrossRef]
- Pettitt, D.J.; Saad, M.F.; Bennett, P.H.; Nelson, R.G.; Knowler, W.C. Familial predisposition to renal disease in two generations of Pima Indians with Type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1990, 33, 438–443. [Google Scholar] [CrossRef] [Green Version]
- Medici, F.; Hawa, M.; Ianari, A.; Pyke, D.A.; Leslie, R.D.G. Concordance rate for Type II diabetes mellitus in monozygotic twins: actuarial analysis. Diabetologia 1999, 42, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Bhattacharya, J.; Butte, A.J. An Environment-Wide Association Study (EWAS) on Type 2 Diabetes Mellitus. PLoS One 2010, 5, e10746. [Google Scholar] [CrossRef] [PubMed]
- Hivert, M.-F.; Jablonski, K.A.; Perreault, L.; Saxena, R.; McAteer, J.B.; Franks, P.W.; Hamman, R.F.; Kahn, S.E.; Haffner, S.; Meigs, J.B.; et al. Updated Genetic Score Based on 34 Confirmed Type 2 Diabetes Loci Is Associated With Diabetes Incidence and Regression to Normoglycemia in the Diabetes Prevention Program. Diabetes 2011, 60, 1340–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onaolapo, A.Y.; Onaolapo, O.J. Circadian dysrhythmia-linked diabetes mellitus: Examining melatonin’s roles in prophylaxis and management. World J. Diabetes 2018, 9, 99–114. [Google Scholar] [CrossRef]
- Quinn, M.; Angelico, M.C.; Warram, J.H.; Krolewski, A.S. Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 1996, 39, 940–945. [Google Scholar] [CrossRef]
- Ng, D.; Krolewski, A. Molecular Genetic Approaches for Studying the Etiology of Diabetic Nephropathy. Curr. Mol. Med. 2005, 5, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Bostrom, M.; Daeihagh, P.; Bowden, D.W. Genetic Factors in Diabetic Nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S. Genetics of diabetic nephropathy. Ther. Adv. Cardiovasc. Dis. 2008, 2, 363–371. [Google Scholar] [CrossRef]
- Divers, J.; Freedman, B.I. Susceptibility genes in common complex kidney disease. Curr. Opin. Nephrol. Hypertens. 2010, 19, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Epigenetic Reprogramming in Mammalian Development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Vaag, A.A.; Grunnet, L.G.; Arora, G.P.; Brøns, C. The thrifty phenotype hypothesis revisited. Diabetologia 2012, 55, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef] [Green Version]
- El-Maarri, O.; Becker, T.; Junen, J.; Manzoor, S.S.; Diaz-Lacava, A.; Schwaab, R.; Wienker, T.; Oldenburg, J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum. Genet. 2007, 122, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, Y.; Asada, H.; Tamura, I.; Lee, L.; Maekawa, R.; Taniguchi, K.; Taketani, T.; Matsuoka, A.; Tamura, H.; Sugino, N. DNA methyltransferase expression in the human endometrium: down-regulation by progesterone and estrogen. Hum. Reprod. 2009, 24, 1126–1132. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [Green Version]
- Kaminsky, Z.A.; Tang, T.; Wang, S.-C.; Ptak, C.; Oh, G.H.T.; Wong, A.H.C.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef]
- Provencio, I.; Rodriguez, I.R.; Jiang, G.; Hayes, W.P.; Moreira, E.F.; Rollag, M.D. A Novel Human Opsin in the Inner Retina. J. Neurosci. 2000, 20, 600–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berson, D.M. Phototransduction by Retinal Ganglion Cells That Set the Circadian Clock. Science 2002, 295, 1070–1073. [Google Scholar] [CrossRef] [Green Version]
- Abrahamson, E.E.; Moore, R.Y. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 2001, 916, 172–191. [Google Scholar] [CrossRef]
- King, D.P.; Zhao, Y.; Sangoram, A.M.; Wilsbacher, L.D.; Tanaka, M.; Antoch, M.P.; Steeves, T.D.L.; Vitaterna, M.H.; Kornhauser, J.M.; Lowrey, P.L.; et al. Positional Cloning of the Mouse Circadian Clock. Gene Cell 1997, 89, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK Protein in the Mammalian Circadian Mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Vitaterna, M.H.; Selby, C.P.; Todo, T.; Niwa, H.; Thompson, C.; Fruechte, E.M.; Hitomi, K.; Thresher, R.J.; Ishikawa, T.; Miyazaki, J.; et al. Differential regulation of mammalian Period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. USA 1999, 96, 12114–12119. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef]
- Kondratov, R.V. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003, 17, 1921–1932. [Google Scholar] [CrossRef] [Green Version]
- Cardone, L. Circadian Clock Control by SUMOylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.E.; Kay, S.A. Clocks not winding down: Unravelling circadian networks. Nat. Rev. Mol. Cell Biol. 2010, 11, 764–776. [Google Scholar] [CrossRef]
- Milagro, F.I.; Gómez-Abellán, P.; Campión, J.; Martínez, J.A.; Ordovás, J.M.; Garaulet, M. CLOCK, PER2 and BMAL1 DNA Methylation: Association with Obesity and Metabolic Syndrome Characteristics and Monounsaturated Fat Intake. Chronobiol. Int. 2012, 29, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK Regulates the Circadian Clock by Cryptochrome Phosphorylation and Degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowrey, P.L. Positional Syntenic Cloning and Functional Characterization of the Mammalian Circadian Mutation tau. Science 2000, 288, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busino, L.; Bassermann, F.; Maiolica, A.; Lee, C.; Nolan, P.M.; Godinho, S.I.H.; Draetta, G.F.; Pagano, M. SCFFbxl3 Controls the Oscillation of the Circadian Clock by Directing the Degradation of Cryptochrome Proteins. Science 2007, 316, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhu, Y.; Goldberg, J.; Vaccarino, V.; Zhao, J. DNA Methylation of Five Core Circadian Genes Jointly Contributes to Glucose Metabolism: A Gene-Set Analysis in Monozygotic Twins. Front. Genet. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Oishi, Y.; Hayashi, S.; Isagawa, T.; Oshima, M.; Iwama, A.; Shimba, S.; Okamura, H.; Manabe, I. Bmal1 regulates inflammatory responses in macrophages by modulating enhancer RNA transcription. Sci. Rep. 2017, 7, 7086. [Google Scholar] [CrossRef] [Green Version]
- Nawrot, T.S.; Saenen, N.D.; Schenk, J.; Janssen, B.G.; Motta, V.; Tarantini, L.; Cox, B.; Lefebvre, W.; Vanpoucke, C.; Maggioni, C.; et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ. Int. 2018, 114, 231–241. [Google Scholar] [CrossRef]
- Preitner, N.; Damiola, F.; Luis-Lopez-Molina; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The Orphan Nuclear Receptor REV-ERBα Controls Circadian Transcription within the Positive Limb of the Mammalian Circadian Oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Yin, L.; Wu, N.; Curtin, J.C.; Qatanani, M.; Szwergold, N.R.; Reid, R.A.; Waitt, G.M.; Parks, D.J.; Pearce, K.H.; Wisely, G.B.; et al. Rev-erb, a Heme Sensor That Coordinates Metabolic and Circadian Pathways. Science 2007, 318, 1786–1789. [Google Scholar] [CrossRef]
- Yin, L.; Lazar, M.A. The Orphan Nuclear Receptor Rev-erbα Recruits the N-CoR/Histone Deacetylase 3 Corepressor to Regulate the Circadian Bmal1 Gene. Mol. Endocrinol. 2005, 19, 1452–1459. [Google Scholar] [CrossRef]
- Yang, W.; Kang, X.; Liu, J.; Li, H.; Ma, Z.; Jin, X.; Qian, Z.; Xie, T.; Qin, N.; Feng, D.; et al. Clock Gene Bmal1 Modulates Human Cartilage Gene Expression by Crosstalk With Sirt1. Endocrinology 2016, 157, 3096–3107. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism that Decays with Aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: the need to relaxThis paper is one of a selection of papers in a Special Issue entitled 31st Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, D.; Gold, B. Origin of mutations in genes associated with human glioblastoma multiform cancer: random polymerase errors versus deamination. Heliyon 2019, 5, e01265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M.L.; Van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [Green Version]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The Mammalian SIR2α Protein Has a Role in Embryogenesis and Gametogenesis. Mol. Cell. Biol. 2003, 23, 38–54. [Google Scholar]
- Zwerger, M.; Medalia, O. From lamins to lamina: A structural perspective. Histochem. Cell Biol. 2013, 140, 3–12. [Google Scholar]
- White, A.J.; Kresovich, J.K.; Xu, Z.; Sandler, D.P.; Taylor, J.A. Shift work, DNA methylation and epigenetic age. Int. J. Epidemiol. 2019, 48, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Amir, S. The aging clock: circadian rhythms and later life. J. Clin. Invest. 2017, 127, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Kretzschmar, D.; Rakshit, K.; Chow, E.; Giebultowicz, J.M. The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging 2009, 1, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Hurd, M.W.; Ralph, M.R. The Significance of Circadian Organization for Longevity in the Golden Hamster. J. Biol. Rhythms 1998, 13, 430–436. [Google Scholar] [CrossRef]
- Cai, A.; Scarbrough, K.; Hinkle, D.A.; Wise, P.M. Fetal grafts containing suprachiasmatic nuclei restore the diurnal rhythm of CRH and POMC mRNA in aging rats. Am. J. Physiol. Integr. Comp. Physiol. 1997, 273, R1764–R1770. [Google Scholar] [CrossRef] [Green Version]
- Roenneberg, T.; Kuehnle, T.; Juda, M.; Kantermann, T.; Allebrandt, K.; Gordijn, M.; Merrow, M. Epidemiology of the human circadian clock. Sleep Med. Rev. 2007, 11, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Lombardi, D.A.; Marucci-Wellman, H.; Roenneberg, T. Chronotypes in the US – Influence of age and sex. PLoS One 2017, 12, e0178782. [Google Scholar] [CrossRef] [Green Version]
- Knutson, K.L.; von Schantz, M. Associations between chronotype, morbidity and mortality in the UK Biobank cohort. Chronobiol. Int. 2018, 35, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Reutrakul, S.; Hood, M.M.; Crowley, S.J.; Morgan, M.K.; Teodori, M.; Knutson, K.L.; Van Cauter, E. Chronotype Is Independently Associated With Glycemic Control in Type 2 Diabetes. Diabetes Care 2013, 36, 2523–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merikanto, I.; Lahti, T.; Puolijoki, H.; Vanhala, M.; Peltonen, M.; Laatikainen, T.; Vartiainen, E.; Salomaa, V.; Kronholm, E.; Partonen, T. Associations of Chronotype and Sleep With Cardiovascular Diseases and Type 2 Diabetes. Chronobiol. Int. 2013, 30, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Makarem, N.; Paul, J.; Giardina, E.-G.V.; Liao, M.; Aggarwal, B. Evening chronotype is associated with poor cardiovascular health and adverse health behaviors in a diverse population of women. Chronobiol. Int. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Makarem, N.; Zuraikat, F.M.; Aggarwal, B.; Jelic, S.; St-Onge, M.-P. Variability in Sleep Patterns: an Emerging Risk Factor for Hypertension. Curr. Hypertens. Rep. 2020, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Roenneberg, T.; Merrow, M. The Circadian Clock and Human Health. Curr. Biol. 2016, 26, R432–R443. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Rex, K.M.; Nievergelt, C.M.; Kelsoe, J.R.; Kripke, D.F. Delayed sleep phase syndrome is related to seasonal affective disorder. J. Affect. Disord. 2011, 133, 573–579. [Google Scholar] [CrossRef] [Green Version]
- Ebisawa, T.; Uchiyama, M.; Kajimura, N.; Mishima, K.; Kamei, Y.; Katoh, M.; Watanabe, T.; Sekimoto, M.; Shibui, K.; Kim, K.; et al. Association of structural polymorphisms in the human period3 gene with delayed sleep phase syndrome. EMBO Rep. 2001, 2, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.N.; Robilliard, D.L.; Skene, D.J.; Smits, M.; Williams, A.; Arendt, J.; von Schantz, M. A Length Polymorphism in the Circadian Clock Gene Per3 is Linked to Delayed Sleep Phase Syndrome and Extreme Diurnal Preference. Sleep 2003, 26, 413–415. [Google Scholar] [CrossRef]
- Bhatti, P.; Zhang, Y.; Song, X.; Makar, K.W.; Sather, C.L.; Kelsey, K.T.; Houseman, E.A.; Wang, P. Nightshift work and genome-wide DNA methylation. Chronobiol. Int. 2015, 32, 103–112. [Google Scholar] [CrossRef]
- Bukowska-Damska, A.; Reszka, E.; Kaluzny, P.; Wieczorek, E.; Przybek, M.; Zienolddiny, S.; Peplonska, B. Sleep quality and methylation status of core circadian rhythm genes among nurses and midwives. Chronobiol. Int. 2017, 34, 1211–1223. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. The metabolic syndrome—a new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Lewis, P.; Oster, H.; Korf, H.W.; Foster, R.G.; Erren, T.C. Food as a circadian time cue — evidence from human studies. Nat. Rev. Endocrinol. 2020, 16, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Minguez, J.; Dashti, H.S.; Madrid-Valero, J.J.; Madrid, J.A.; Saxena, R.; Scheer, F.A.J.L.; Ordoñana, J.R.; Garaulet, M. Heritability of the timing of food intake. Clin. Nutr. 2019, 38, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Yildiz, B.O.; Suchard, M.A.; Wong, M.-L.; McCann, S.M.; Licinio, J. Alterations in the dynamics of circulating ghrelin, adiponectin, and leptin in human obesity. Proc. Natl. Acad. Sci. 2004, 101, 10434–10439. [Google Scholar] [CrossRef] [Green Version]
- Laposky, A.D.; Bradley, M.A.; Williams, D.L.; Bass, J.; Turek, F.W. Sleep-wake regulation is altered in leptin-resistant ( db/db ) genetically obese and diabetic mice. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R2059–R2066. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur. Heart J. 2004, 25, 735–741. [Google Scholar] [CrossRef]
- Reppert, S.M.; Godson, C.; Mahle, C.D.; Weaver, D.R.; Slaugenhaupt, S.A.; Gusella, J.F. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc. Natl. Acad. Sci. 1995, 92, 8734–8738. [Google Scholar] [CrossRef] [Green Version]
- Hunt, A.E.; Al-Ghoul, W.M.; Gillette, M.U.; Dubocovich, M.L. Activation of MT 2 melatonin receptors in rat suprachiasmatic nucleus phase advances the circadian clock. Am. J. Physiol. Physiol. 2001, 280, C110–C118. [Google Scholar] [CrossRef] [Green Version]
- Bouatia-Naji, N.; Bonnefond, A.; Cavalcanti-Proença, C.; Sparsø, T.; Holmkvist, J.; Marchand, M.; Delplanque, J.; Lobbens, S.; Rocheleau, G.; Durand, E.; et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet. 2009, 41, 89–94. [Google Scholar] [CrossRef]
- Peschke, E.; Bach, A.G.; Muhlbauer, E. Parallel signaling pathways of melatonin in the pancreatic β-cell. J. Pineal Res. 2006, 40, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Peschke, D.; Hammer, T.; Csernus, V. Influence of melatonin and serotonin on glucose-stimulated insulin release from perifused rat pancreatic islets in vitro. J. Pineal Res. 1997, 23, 156–163. [Google Scholar] [CrossRef]
- Sun, Z.; Feng, D.; Everett, L.J.; Bugge, A.; Lazar, M.A. Circadian Epigenomic Remodeling and Hepatic Lipogenesis: Lessons from HDAC3. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 49–55. [Google Scholar] [CrossRef]
- Shi, S.; Hida, A.; McGuinness, O.P.; Wasserman, D.H.; Yamazaki, S.; Johnson, C.H. Circadian Clock Gene Bmal1 Is Not Essential; Functional Replacement with its Paralog, Bmal2. Curr. Biol. 2010, 20, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Storch, K.-F.; Lipan, O.; Leykin, I.; Viswanathan, N.; Davis, F.C.; Wong, W.H.; Weitz, C.J. Extensive and divergent circadian gene expression in liver and heart. Nature 2002, 417, 78–83. [Google Scholar] [CrossRef]
- Gangwisch, J.E.; Heymsfield, S.B.; Boden-Albala, B.; Buijs, R.M.; Kreier, F.; Pickering, T.G.; Rundle, A.G.; Zammit, G.K.; Malaspina, D. Short Sleep Duration as a Risk Factor for Hypertension. Hypertension 2006, 47, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Makarem, N.; Shechter, A.; Carnethon, M.R.; Mullington, J.M.; Hall, M.H.; Abdalla, M. Sleep Duration and Blood Pressure: Recent Advances and Future Directions. Curr. Hypertens. Rep. 2019, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Ushijima, T. Diagnostic and Therapeutic Applications of Epigenetics. Jpn. J. Clin. Oncol. 2005, 35, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Gršković, B.; Zrnec, D.; Vicković, S.; Popović, M.; Mršić, G. DNA methylation: the future of crime scene investigation? Mol. Biol. Rep. 2013, 40, 4349–4360. [Google Scholar] [CrossRef]
- Plikus, M.V.; Van Spyk, E.N.; Pham, K.; Geyfman, M.; Kumar, V.; Takahashi, J.S.; Andersen, B. The Circadian Clock in Skin. J. Biol. Rhythms 2015, 30, 163–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Zhao, X.; Bai, J.; Gery, S.; Sun, H.; Lin, D.-C.; Chen, Q.; Chen, Z.; Mack, L.; Yang, H.; et al. Circadian clock cryptochrome proteins regulate autoimmunity. Proc. Natl. Acad. Sci. USA 2017, 114, 12548–12553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomilehto, J.; Lindström, J.; Eriksson, J.G.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; Louheranta, A.; Rastas, M.; et al. Prevention of Type 2 Diabetes Mellitus by Changes in Lifestyle among Subjects with Impaired Glucose Tolerance. N. Engl. J. Med. 2001, 344, 1343–1350. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research Group Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar]
- Singhal, N.; Misra, A. A School-Based Intervention for Diabetes Risk Reduction. N. Engl. J. Med. 2010, 363, 1769–1770. [Google Scholar] [PubMed] [Green Version]
- Florez, J.C.; Jablonski, K.A.; Bayley, N.; Pollin, T.I.; de Bakker, P.I.W.; Shuldiner, A.R.; Knowler, W.C.; Nathan, D.M.; Altshuler, D. TCF7L2 Polymorphisms and Progression to Diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 2006, 355, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Morales-Lara, D.; De-la-Peña, C.; Murillo-Rodríguez, E. Dad’s Snoring May Have Left Molecular Scars in Your DNA: the Emerging Role of Epigenetics in Sleep Disorders. Mol. Neurobiol. 2018, 55, 2713–2724. [Google Scholar] [CrossRef]
- Van den Berg, C.B.; Chaves, I.; Herzog, E.M.; Willemsen, S.P.; van der Horst, G.T.J.; Steegers-Theunissen, R.P.M. Early- and late-onset preeclampsia and the DNA methylation of circadian clock and clock-controlled genes in placental and newborn tissues. Chronobiol. Int. 2017, 34, 921–932. [Google Scholar] [CrossRef]
- Smarr, B.L.; Grant, A.D.; Perez, L.; Zucker, I.; Kriegsfeld, L.J. Maternal and Early-Life Circadian Disruption Have Long-Lasting Negative Consequences on Offspring Development and Adult Behavior in Mice. Sci. Rep. 2017, 7, 3326. [Google Scholar] [CrossRef]


{kind=link}
| Name of CCG | Manifestation of the Disease |
|---|---|
| CLOCK | Hyperphagia, hyperlipidemia, hyperinsulinemic hyperglycemia, sleep disorders [26] |
| BMAL1 | Induction of adipogenesis in adipocytes of adipose tissue, hypotension [27,28] |
| PPARγ | Reduction of circadian blood pressure oscillation [29] |
| CRY | Hypertension, aldosterone overproduction [30] |
| HDAC3 | Disruption in lipid homeostasis of the liver and severe steatosis [31] |
| PER | Circadian period change, Advanced Sleep-Wake Phase Disorder (ASWPD), Delayed Sleep-Wake Phase Disorder (DSWPD) [23,32,33] |
| SIRT1 | Regulation of adipocyte differentiation, gluconeogenesis and insulin secretion, changes in fat burning during sleep [34,35,36] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudec, M.; Dankova, P.; Solc, R.; Bettazova, N.; Cerna, M. Epigenetic Regulation of Circadian Rhythm and Its Possible Role in Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 3005. https://doi.org/10.3390/ijms21083005
Hudec M, Dankova P, Solc R, Bettazova N, Cerna M. Epigenetic Regulation of Circadian Rhythm and Its Possible Role in Diabetes Mellitus. International Journal of Molecular Sciences. 2020; 21(8):3005. https://doi.org/10.3390/ijms21083005
Chicago/Turabian StyleHudec, Michael, Pavlina Dankova, Roman Solc, Nardjas Bettazova, and Marie Cerna. 2020. "Epigenetic Regulation of Circadian Rhythm and Its Possible Role in Diabetes Mellitus" International Journal of Molecular Sciences 21, no. 8: 3005. https://doi.org/10.3390/ijms21083005
APA StyleHudec, M., Dankova, P., Solc, R., Bettazova, N., & Cerna, M. (2020). Epigenetic Regulation of Circadian Rhythm and Its Possible Role in Diabetes Mellitus. International Journal of Molecular Sciences, 21(8), 3005. https://doi.org/10.3390/ijms21083005