A Comprehensive Proteomic and Phosphoproteomic Analysis of Retinal Pigment Epithelium Reveals Multiple Pathway Alterations in Response to the Inflammatory Stimuli

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
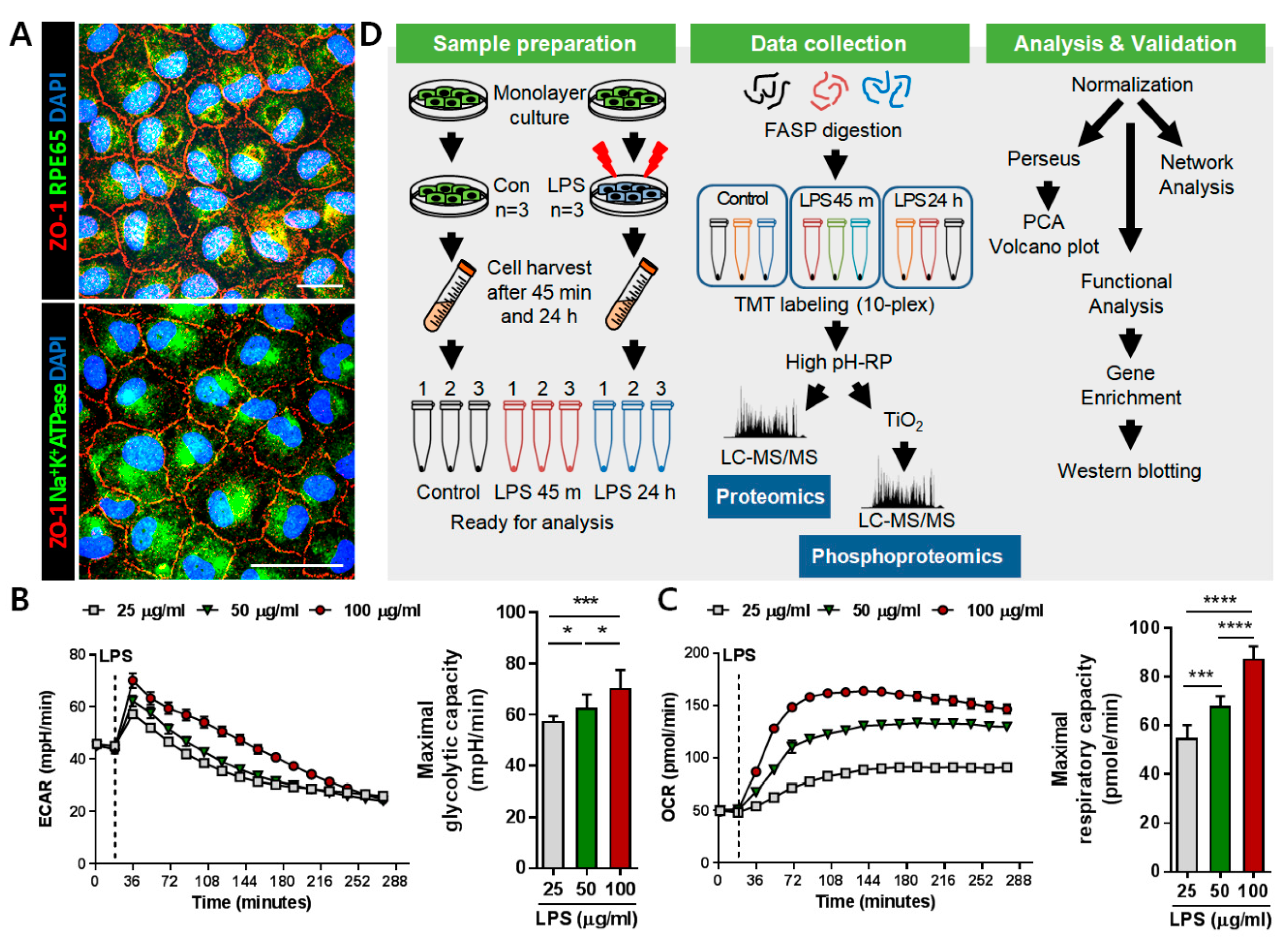
2.1. Induction of Inflammation on ARPE-19 Monolayer for Proteomic Analysis
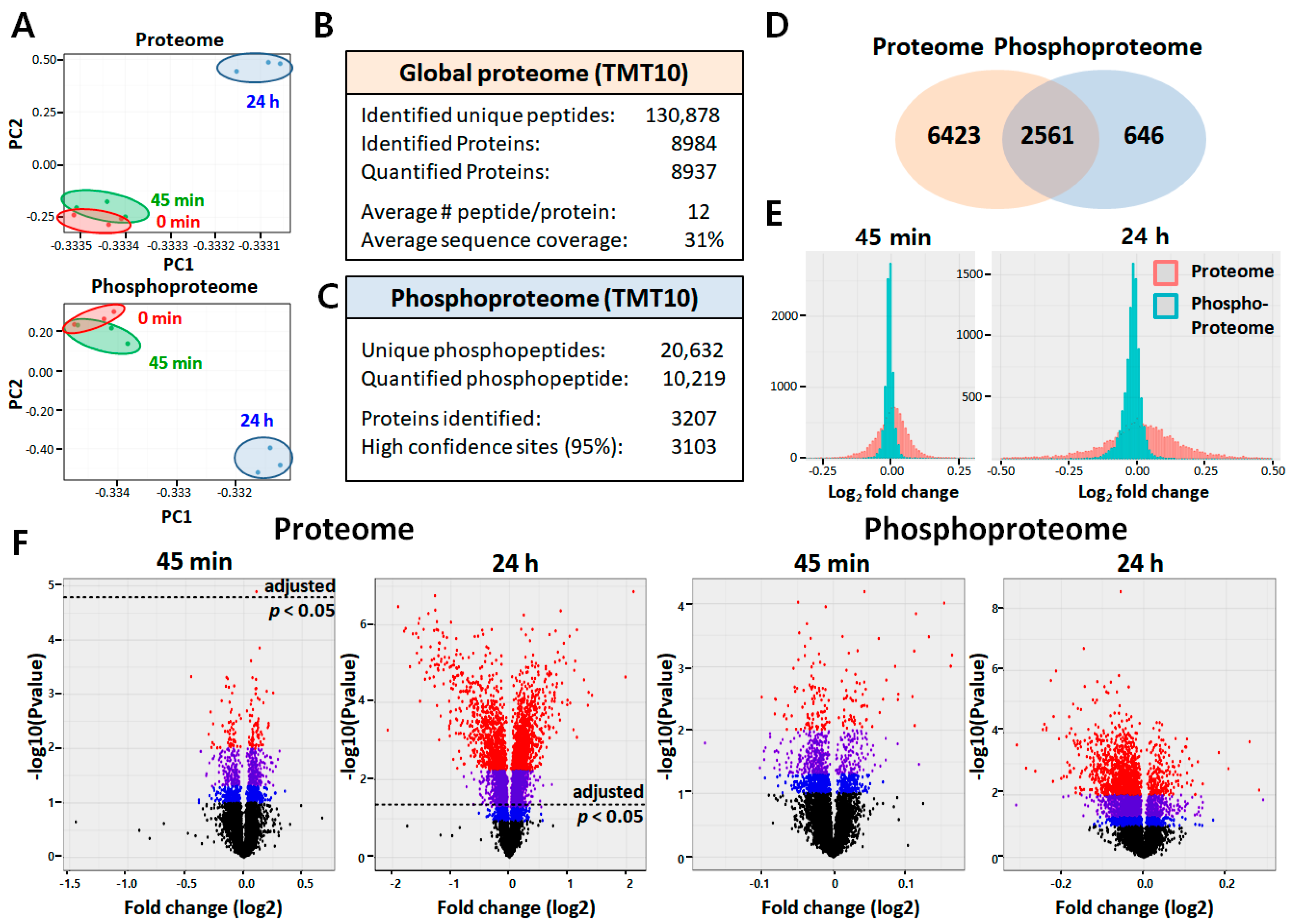
2.2. Quantitative Proteomic and Phosphoproteomic Analyses of ARPE-19 Cells
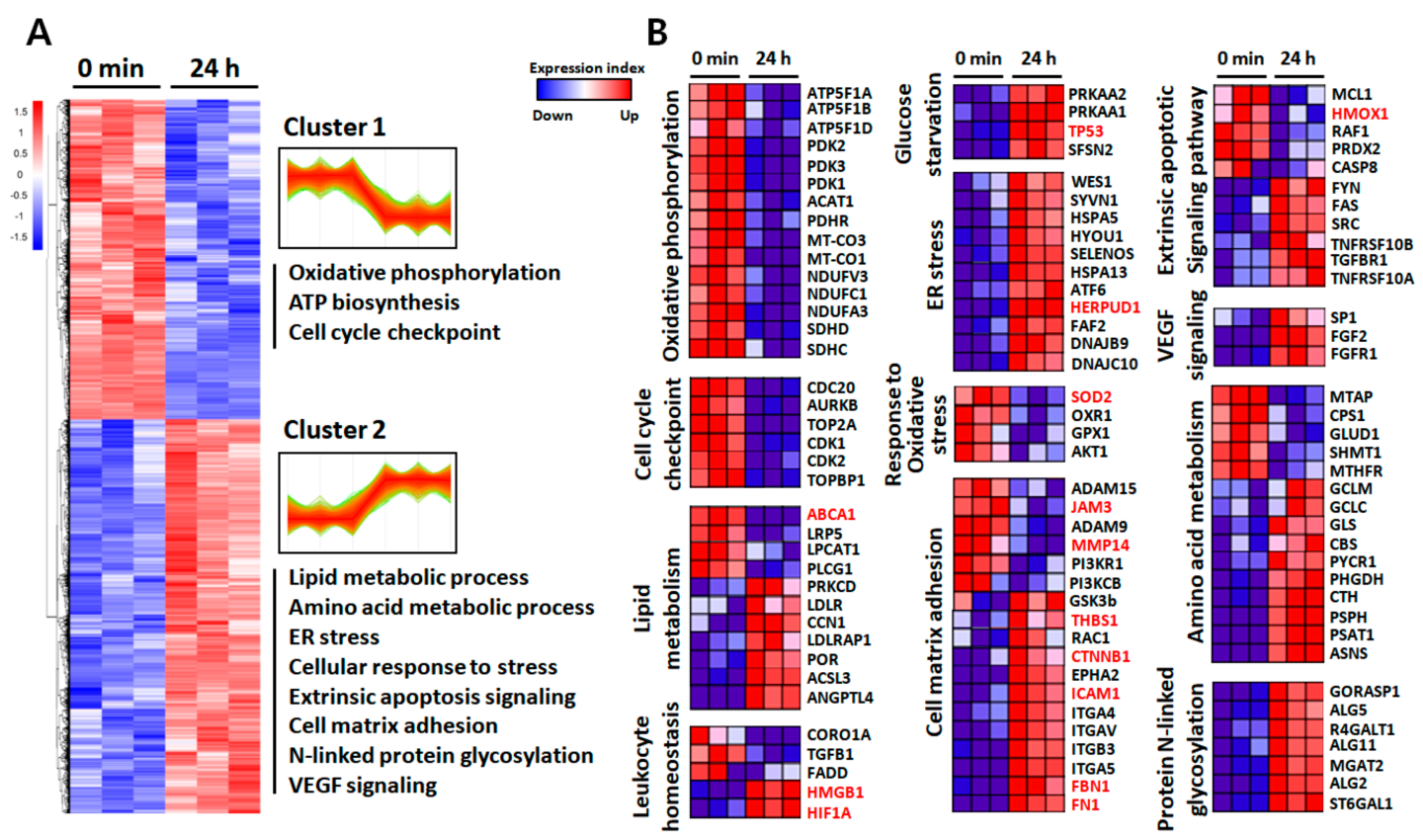
2.3. Functional Annotation of the Identified Proteins
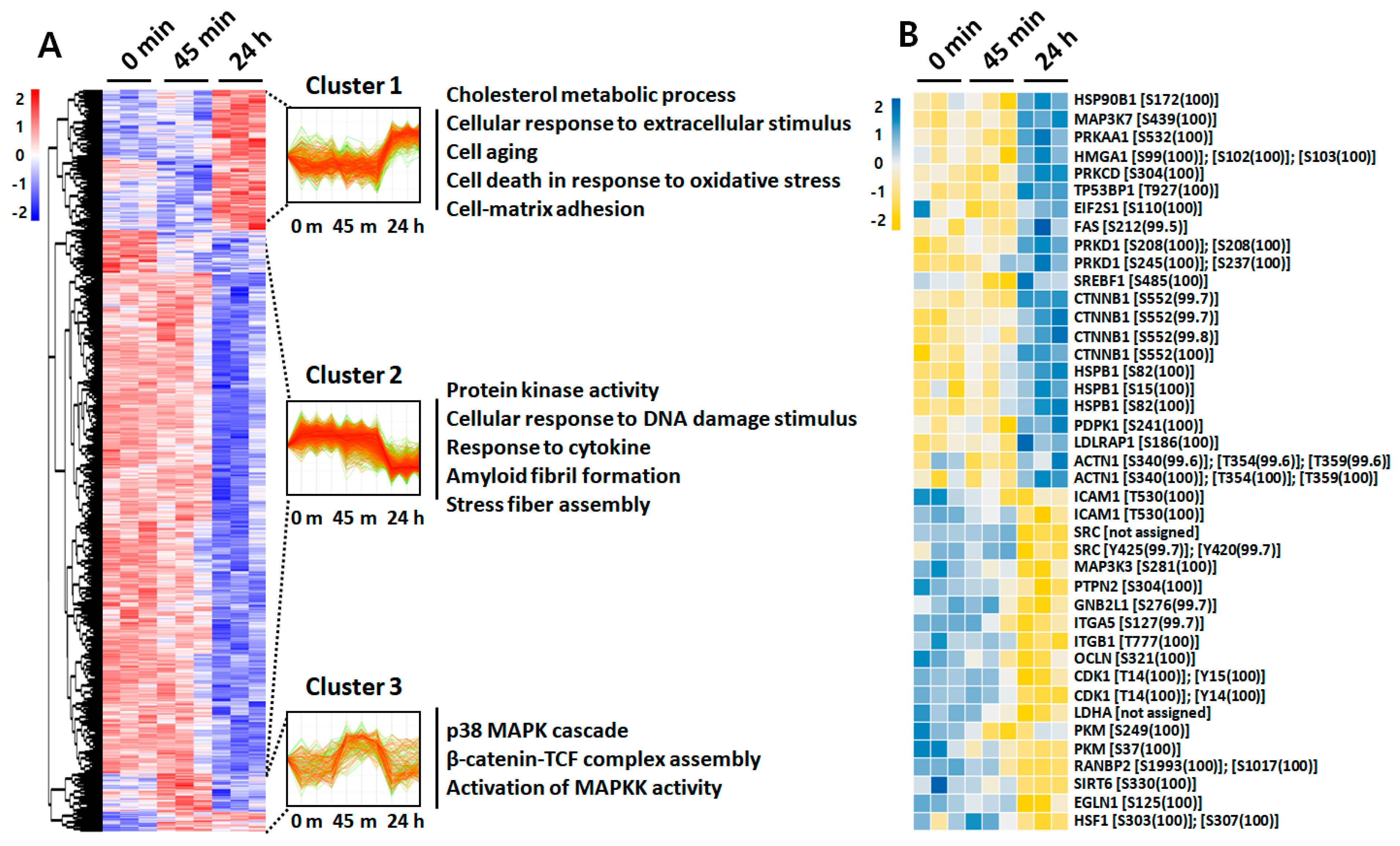
2.4. Phosphoproteome Analysis
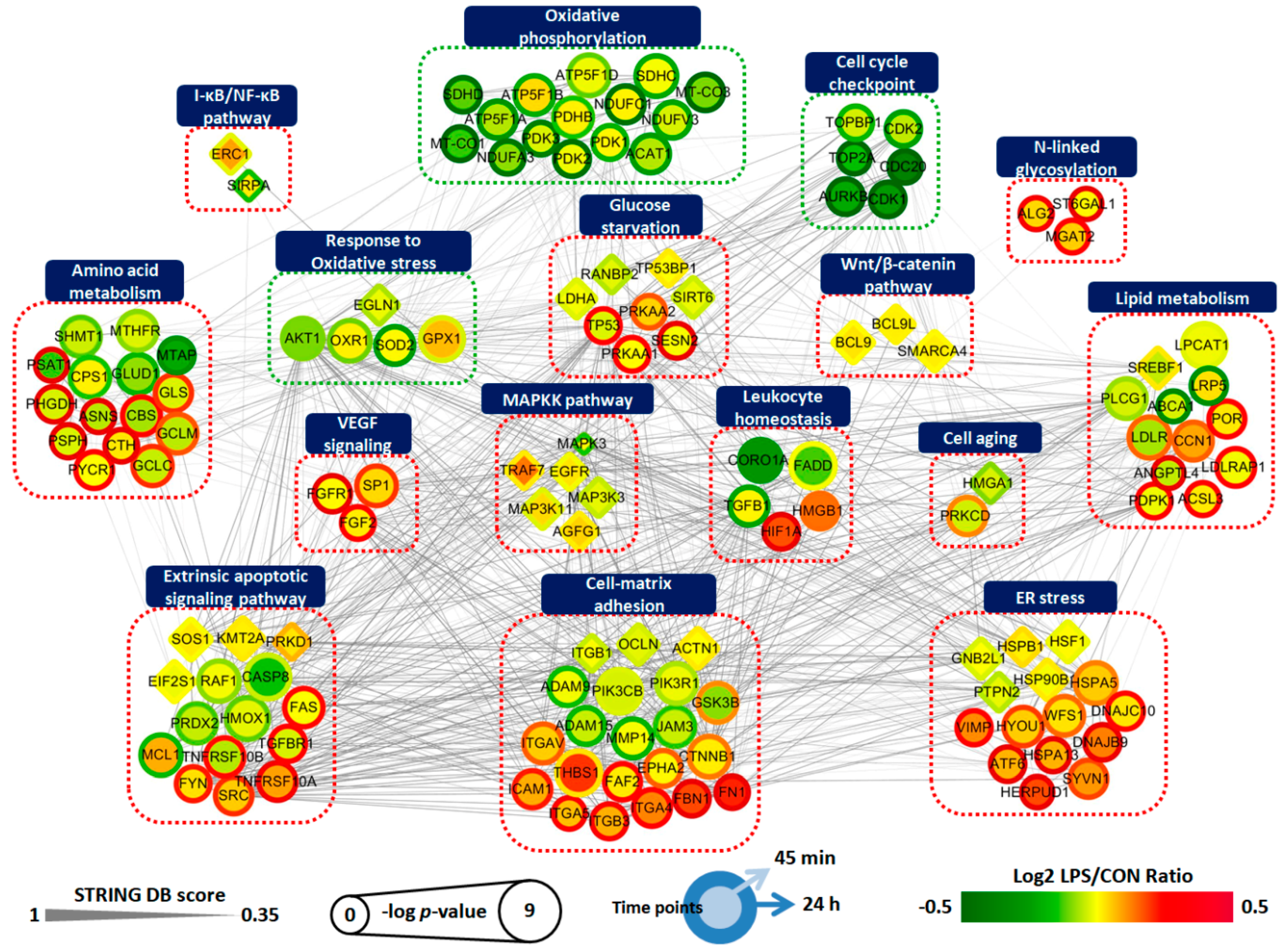
2.5. Protein Interaction Network of LPS-Treated ARPE-19 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Line Culture
4.2. Immunofluorescence Staining
4.3. LPS Treatment and Sample Preparation
4.4. Protein Digestion
4.5. TMT 10-Plex Labeling
4.6. High-pH Peptide Fractionation
4.7. Phosphopeptide Enrichment Using TiO2
4.8. Mass Spectrometric Analysis and Database Search
4.9. Bioinformatics Analysis
4.10. ECAR and OCR Measurements
4.11. Cell Viability Assay
4.12. ELISA
4.13. Western Blotting
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boulton, M.; Dayhaw-Barker, P. The role of the retinal pigment epithelium: Topographical variation and ageing changes. Eye (Lond) 2001, 15 Pt 3, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzolo, L.J. Barrier properties of cultured retinal pigment epithelium. Exp. Eye Res. 2014, 126, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J Neurosci Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, M.; Sugita, S.; Kamoi, K. Immunological homeostasis of the eye. Prog. Retin. Eye Res. 2013, 33, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Sugita, S.; Futagami, Y.; Yamada, Y.; Mochizuki, M. Human retinal pigment epithelium-induced CD4+CD25+ regulatory T cells suppress activation of intraocular effector T cells. Clin. Immunol. 2010, 136, 83–95. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N Engl J Med 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Wang, Y.; Hanus, J.W.; Abu-Asab, M.S.; Shen, D.; Ogilvy, A.; Ou, J.; Chu, X.K.; Shi, G.; Li, W.; Wang, S.; et al. NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2016, 17, 73. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Kubota, S.; Kurihara, T.; Mochimaru, H.; Satofuka, S.; Noda, K.; Ozawa, Y.; Oike, Y.; Ishida, S.; Tsubota, K. Prevention of ocular inflammation in endotoxin-induced uveitis with resveratrol by inhibiting oxidative damage and nuclear factor-kappaB activation. Invest. OphthalMol. Vis. Sci. 2009, 50, 3512–3519. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Chen, J.T.; Liang, C.M.; Tai, M.C.; Lu, D.W.; Chen, Y.H. Silibinin treatment prevents endotoxin-induced uveitis in rats in vivo and in vitro. PLoS ONE 2017, 12, e0174971. [Google Scholar] [CrossRef] [Green Version]
- Vagaja, N.N.; Binz, N.; McLenachan, S.; Rakoczy, E.P.; McMenamin, P.G. Influence of endotoxin-mediated retinal inflammation on phenotype of diabetic retinopathy in Ins2 Akita mice. Br. J. OphthalMol. 2013, 97, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.; Tenconi, P.E.; Giusto, N.M.; Mateos, M.V. Lipopolysaccharide-Induced Autophagy Mediates Retinal Pigment Epithelium Cells Survival. Modulation by the Phospholipase D Pathway. Front. Cell NeuroSci. 2019, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Ozal, S.A.; Turkekul, K.; Gurlu, V.; Guclu, H.; Erdogan, S. Esculetin Protects Human Retinal Pigment Epithelial Cells from Lipopolysaccharide-induced Inflammation and Cell Death. Curr. Eye Res. 2018, 43, 1169–1176. [Google Scholar] [CrossRef]
- Gao, J.; Liu, R.T.; Cao, S.; Cui, J.Z.; Wang, A.; To, E.; Matsubara, J.A. NLRP3 inflammasome: Activation and regulation in age-related macular degeneration. Mediat. Inflamm. 2015, 2015, 690243. [Google Scholar] [CrossRef] [Green Version]
- Paeng, S.H.; Park, W.S.; Jung, W.K.; Lee, D.S.; Kim, G.Y.; Choi, Y.H.; Seo, S.K.; Jang, W.H.; Choi, J.S.; Lee, Y.M.; et al. YCG063 inhibits Pseudomonas aeruginosa LPS-induced inflammation in human retinal pigment epithelial cells through the TLR2-mediated AKT/NF-kappaB pathway and ROS-independent pathways. Int. J. Mol. Med. 2015, 36, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Dai, B.; Li, Y.; Peng, H. C5a and toll-like receptor 4 crosstalk in retinal pigment epithelial cells. Mol. Vis. 2015, 21, 1122–1129. [Google Scholar]
- Zou, X.L.; Wang, G.F.; Li, D.D.; Chen, J.X.; Zhang, C.L.; Yu, Y.Z.; Zhou, W.J.; Zou, Y.P.; Rao, B.Q. Protection of tight junction between RPE cells with tissue factor targeting peptide. Int. J. OphthalMol. 2018, 11, 1594–1599. [Google Scholar]
- Handa, J.T.; Bowes Rickman, C.; Dick, A.D.; Gorin, M.B.; Miller, J.W.; Toth, C.A.; Ueffing, M.; Zarbin, M.; Farrer, L.A. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 2019, 10, 3347. [Google Scholar] [CrossRef] [PubMed]
- Decanini, A.; Karunadharma, P.R.; Nordgaard, C.L.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Human retinal pigment epithelium proteome changes in early diabetes. Diabetologia 2008, 51, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Hung, C.T.; Keller, J.J.; Lin, H.C.; Wu, Y.J. Proteomic analysis of retinal pigment epithelium cells after exposure to UVA radiation. BMC OphthalMol. 2019, 19, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, S.; Hadrian, K.; Schlatt, S.; Wistuba, J.; Thanos, S.; Bohm, M.R.R. Topographic protein profiling of the age-related proteome in the retinal pigment epithelium of Callithrix jacchus with respect to macular degeneration. J. Proteom. 2019, 191, 1–15. [Google Scholar] [CrossRef]
- Meyer, J.G.; Garcia, T.Y.; Schilling, B.; Gibson, B.W.; Lamba, D.A. Proteome and Secretome Dynamics of Human Retinal Pigment Epithelium in Response to Reactive Oxygen Species. Sci. Rep. 2019, 9, 15440. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Ramos, H.; Shannon, P.; Aebersold, R. The protein information and property explorer: An easy-to-use, rich-client web application for the management and functional analysis of proteomic data. Bioinformatics 2008, 24, 2110–2111. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Leung, K.W.; Barnstable, C.J.; Tombran-Tink, J. Bacterial endotoxin activates retinal pigment epithelial cells and induces their degeneration through IL-6 and IL-8 autocrine signaling. Mol. Immunol. 2009, 46, 1374–1386. [Google Scholar] [CrossRef]
- Tao, L.; Qiu, Y.; Fu, X.; Lin, R.; Lei, C.; Wang, J.; Lei, B. Angiotensin-converting enzyme 2 activator diminazene aceturate prevents lipopolysaccharide-induced inflammation by inhibiting MAPK and NF-kappaB pathways in human retinal pigment epithelium. J. Neuroinflamm. 2016, 13, 35. [Google Scholar] [CrossRef] [Green Version]
- Dridi, S.; Hirano, Y.; Tarallo, V.; Kim, Y.; Fowler, B.J.; Ambati, B.K.; Bogdanovich, S.; Chiodo, V.A.; Hauswirth, W.W.; Kugel, J.F.; et al. ERK1/2 activation is a therapeutic target in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 13781–13786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Hu, Y.; Chen, Y.; Zhou, K.K.; Zhang, B.; Gao, G.; Ma, J.X. The pathogenic role of the canonical Wnt pathway in age-related macular degeneration. Investig. OphthalMol. Vis. Sci. 2010, 51, 4371–4379. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Wang, Y.; Cheng, R.; Li, Y.; Chen, M.; Qiu, F.; Qian, H.; Shen, D.; Penalva, R.; Xu, H.; et al. Wnt signaling in age-related macular degeneration: Human macular tissue and mouse model. J. Transl Med. 2015, 13, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yue, W.; Zhu, M.J.; Sreejayan, N.; Du, M. AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of beta-catenin at Ser 552. BioChem. Biophys. Res. Commun. 2010, 395, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J. Mol. NeuroSci. 2017, 62, 368–379. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [Green Version]
- Vallee, A.; Lecarpentier, Y.; Vallee, R.; Guillevin, R.; Vallee, J.N. Circadian Rhythms in Exudative Age-Related Macular Degeneration: The Key Role of the Canonical WNT/beta-Catenin Pathway. Int. J. Mol. Sci. 2020, 21, 820. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Lin, M.; Lee, K.; Mott, R.A.; Ma, J.X. Pathogenic role of the Wnt signaling pathway activation in laser-induced choroidal neovascularization. Investig. OphthalMol. Vis. Sci. 2013, 54, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, J.; Palczewski, K. Human aging and disease: Lessons from age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 2866–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. OphthalMol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijanto, J.; Philp, N.J. Cultured primary human fetal retinal pigment epithelium (hfRPE) as a model for evaluating RPE metabolism. Exp. Eye Res. 2014, 126, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Kong, L.; Wang, J.; Ash, J.D. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2018, 115, 10475–10480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the Sod2 (+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef] [PubMed]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef]
- Bretillon, L.; Acar, N.; Seeliger, M.W.; Santos, M.; Maire, M.A.; Juaneda, P.; Martine, L.; Gregoire, S.; Joffre, C.; Bron, A.M.; et al. ApoB100,LDLR-/- mice exhibit reduced electroretinographic response and cholesteryl esters deposits in the retina. Investig. OphthalMol. Vis. Sci. 2008, 49, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, E.M.; Emri, E.; Merle, B.M.J.; Colijn, J.M.; Kersten, E.; Cougnard-Gregoire, A.; Dammeier, S.; Meester-Smoor, M.; Pool, F.M.; De Jong, E.K.; et al. A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res. 2018, 67, 56–86. [Google Scholar] [CrossRef] [PubMed]
- Storti, F.; Klee, K.; Todorova, V.; Steiner, R.; Othman, A.; Van der Velde-Visser, S.; Samardzija, M.; Meneau, I.; Barben, M.; Karademir, D.; et al. Impaired ABCA1/ABCG1-mediated lipid efflux in the mouse retinal pigment epithelium (RPE) leads to retinal degeneration. Elife 2019, 8, e45100. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Liao, D.F.; Tang, C.K. ATP-binding membrane cassette transporter A1 (ABCA1): A possible link between inflammation and reverse cholesterol transport. Mol. Med. 2010, 16, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Li, W.; Wang, N.; Zhu, Y.; Wang, X. ROS and NF-kappaB but not LXR mediate IL-1beta signaling for the downregulation of ATP-binding cassette transporter A1. Am. J. Physiol. Cell Physiol. 2007, 292, C1493–C1501. [Google Scholar] [CrossRef]
- Gopinath, B.; Flood, V.M.; Rochtchina, E.; Wang, J.J.; Mitchell, P. Homocysteine, folate, vitamin B-12, and 10-y incidence of age-related macular degeneration. Am. J. Clin. Nutr 2013, 98, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.S.; Mander, S.; Hussein, K.A.; Elsherbiny, N.M.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia disrupts retinal pigment epithelial structure and function with features of age-related macular degeneration. Oncotarget 2016, 7, 8532–8545. [Google Scholar] [CrossRef]
- Jin, E.; Bai, Y.; Huang, L.; Zhao, M.; Zhang, C.; Zhao, M.; Li, X. Evidence of a novel gene HERPUD1 in polypoidal choroidal vasculopathy. Int. J. Clin. Exp. Pathol 2015, 8, 13928–13944. [Google Scholar]
- Tawfik, A.; Mohamed, R.; Elsherbiny, N.M.; DeAngelis, M.M.; Bartoli, M.; Al-Shabrawey, M. Homocysteine: A Potential Biomarker for Diabetic Retinopathy. J. Clin. Med. 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Pinna, A.; Zaccheddu, F.; Boscia, F.; Carru, C.; Solinas, G. Homocysteine and risk of age-related macular degeneration: A systematic review and meta-analysis. Acta OphthalMol. 2018, 96, e269–e276. [Google Scholar] [CrossRef] [Green Version]
- Ahmado, A.; Carr, A.J.; Vugler, A.A.; Semo, M.; Gias, C.; Lawrence, J.M.; Chen, L.L.; Chen, F.K.; Turowski, P.; Da Cruz, L.; et al. Induction of differentiation by pyruvate and DMEM in the human retinal pigment epithelium cell line ARPE-19. Investig. OphthalMol. Vis. Sci. 2011, 52, 7148–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.; Min, H.; Rahaman, K.A.; Muresan, A.R.; Kim, H.; Han, D.; Kwon, O.S. Quantitative Proteome Analysis of Brain Subregions and Spinal Cord from Experimental Autoimmune Encephalomyelitis Mice by TMT-Based Mass Spectrometry. Proteomics 2019, 19, e1800355. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Han, D.; Park, J.; Choi, H.; Park, J.C.; Cha, M.Y.; Woo, J.; Byun, M.S.; Lee, D.Y.; Kim, Y.; et al. Deep proteome profiling of the hippocampus in the 5XFAD mouse model reveals biological process alterations and a novel biomarker of Alzheimer’s disease. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef]
- Jin, M.S.; Lee, H.; Woo, J.; Choi, S.; Do, M.S.; Kim, K.; Song, M.J.; Kim, Y.; Park, I.A.; Han, D.; et al. Integrated Multi-Omic Analyses Support Distinguishing Secretory Carcinoma of the Breast from Basal-Like Triple-Negative Breast Cancer. Proteomics Clin. Appl. 2018, 12, e1700125. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef]
- Parkin, M.; Nectoux, J.; Stiller, C.; Draper, G. Incidence of cancer in children throughout the world. Pediatrie 1989, 44, 725–736. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Calton, M.A.; Beaulieu, M.O.; Benchorin, G.; Vollrath, D. Method for measuring extracellular flux from intact polarized epithelial monolayers. Mol. Vis. 2018, 24, 425–433. [Google Scholar] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Han, D.; Lee, H.; Kim, D.J.; Cho, J.-Y.; Park, J.-H.; Seok, S.H. A Comprehensive Proteomic and Phosphoproteomic Analysis of Retinal Pigment Epithelium Reveals Multiple Pathway Alterations in Response to the Inflammatory Stimuli. Int. J. Mol. Sci. 2020, 21, 3037. https://doi.org/10.3390/ijms21093037
Song J, Han D, Lee H, Kim DJ, Cho J-Y, Park J-H, Seok SH. A Comprehensive Proteomic and Phosphoproteomic Analysis of Retinal Pigment Epithelium Reveals Multiple Pathway Alterations in Response to the Inflammatory Stimuli. International Journal of Molecular Sciences. 2020; 21(9):3037. https://doi.org/10.3390/ijms21093037
Chicago/Turabian StyleSong, Juha, Dohyun Han, Heonyi Lee, Da Jung Kim, Joo-Youn Cho, Jae-Hak Park, and Seung Hyeok Seok. 2020. "A Comprehensive Proteomic and Phosphoproteomic Analysis of Retinal Pigment Epithelium Reveals Multiple Pathway Alterations in Response to the Inflammatory Stimuli" International Journal of Molecular Sciences 21, no. 9: 3037. https://doi.org/10.3390/ijms21093037