Abstract
DNA glycosylases are enzymes that initiate the base excision repair pathway, a major biochemical process that protects the genomes of all living organisms from intrinsically and environmentally inflicted damage. Recently, base excision repair inhibition proved to be a viable strategy for the therapy of tumors that have lost alternative repair pathways, such as BRCA-deficient cancers sensitive to poly(ADP-ribose)polymerase inhibition. However, drugs targeting DNA glycosylases are still in development and so far have not advanced to clinical trials. In this review, we cover the attempts to validate DNA glycosylases as suitable targets for inhibition in the pharmacological treatment of cancer, neurodegenerative diseases, chronic inflammation, bacterial and viral infections. We discuss the glycosylase inhibitors described so far and survey the advances in the assays for DNA glycosylase reactions that may be used to screen pharmacological libraries for new active compounds.
1. Introduction: Base Excision Repair
DNA in living cells is always exposed to many damaging factors of environmental and endogenous origin. These insults produce various modifications of nucleobases and lead to the formation of apurinic/apyrimidinic (AP) sites and DNA strand breaks. Accumulating damage significantly affects genomic stability and may ultimately end in mutations or cell death [1,2].
Mechanisms that correct genomic damage, commonly known as DNA repair systems, exist in all forms of life [1]. The most abundant DNA lesions—deaminated, oxidized, and alkylated bases, AP sites, single-strand breaks—are repaired by the base excision repair (BER) system [1,3]. Initially, the damaged base is recognized by a DNA glycosylase (of which, 11 are known in humans, and 8 in Escherichia coli), which cleaves the N-glycosidic bond between the nucleobase and the C1′ deoxyribose atom, forming an AP site. Afterwards, an AP endonuclease hydrolyzes the phosphodiester bond 5′ to the AP site. The repair proceeds with the incorporation of the correct dNMP by a DNA polymerase and the ligation of the remaining single-strand break (Figure 1).
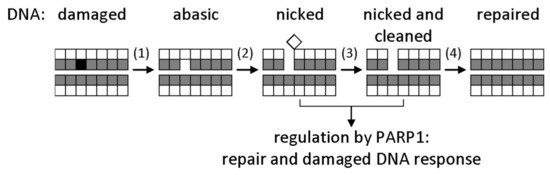
Figure 1.
General base excision repair (BER) scheme. Bases are represented by shaded squares, and sugars by white squares. A damaged base (black) is excised by a DNA glycosylase (1); the resulting apurinic/apyrimidinic (AP) site is cut by an AP endonuclease (2); the deoxyribose fragment is removed by a deoxyribophosphate lyase (3); a correct dNMP is incorporated by a DNA polymerase, and the nick is sealed by a DNA ligase (4). Nicked DNA also activates signaling by poly(ADP-ribose)polymerase 1 (PARP1), which initiates poly(ADP-ribosyl)ation of many chromatin proteins to facilitate the access of DNA repair factors to the site of damage.
DNA glycosylases belong to several major structural superfamilies and have different, sometimes overlapping, substrate specificities (Table 1). They all, however, have a common reaction mechanism: the C1′ of the damaged nucleotide is attacked by a nucleophilic moiety (either an activated water molecule in monofunctional DNA glycosylases or an enzyme’s amino group in bifunctional ones), the damaged base departs, and either an AP site is generated (monofunctional glycosylases), or a Schiff base covalent intermediate forms (bifunctional glycosylases) [4,5,6]. The Schiff base undergoes β- or β,δ-elimination and is then hydrolyzed, leaving a nick with either an α,β-unsaturated aldehyde or a phosphate as the 3′-terminal group. To access C1′, most glycosylases flip the target nucleotide from the DNA stack into the enzyme’s active site, which is equipped with a deep lesion recognition pocket, representing a convenient druggable target [7].

Table 1.
Examples of DNA glycosylases found in humans, Escherichia coli, and other species.
In human cells, BER is tightly regulated at several levels. One of the best-studied players orchestrating the BER process is poly(ADP-ribose)polymerase 1 (PARP1), together with its homologs PARP2 and PARP3, which act as nick sensors and regulate the access of repair factors to the damage sites through modification of acceptor proteins and DNA ends by poly(ADP-ribose) [8,9]. PARPs attracted attention as potential targets for cancer treatment after PARP inhibitors were discovered to be highly toxic for cells with inactivated homologous recombination repair pathway [10,11]. In human tumors, recombination repair deficiency is often associated with inactivating mutations in the BRCA1 and BRCA2 genes, the main driver mutations in hereditary breast and ovarian cancers. BRCA1 and BRCA2 proteins regulate the DNA break response through a pathway that does not overlap with BER [12]. Blocking both these pathways is lethal for the cell, while normal cells with active recombination repair survive PARP1 inhibition. The lethal effect of PARP inhibitors is largely mediated by PARP trapping at nicks [13,14], which mainly originate from ribonucleotides misincorporated during DNA replication [15]. Several PARP inhibitors are presently approved for clinical use, and several hundred clinical trials are ongoing.
2. Inhibitors of DNA Glycosylases: General Considerations
The example of PARP inhibitors highlights the concept of synthetic lethality, which underlies most of the attempts to develop BER inhibitors into practically useful drugs. Two conditions must be satisfied for such compounds to be effective. First, the target cells must experience genotoxic stress, induced either directly by DNA-damaging factors or indirectly through some kind of metabolic stress (nucleotide pool imbalance, oxidative stress, etc.). Second, if DNA damage caused by this type of stress can be repaired bypassing BER, the bypass must be blocked by mutations or another drug used in combination with a BER inhibitor. The second requirement is often satisfied in cancers, where mutations in DNA repair genes are usually among driver mutations. Genotoxic treatment may also be tuned to produce lesions repaired predominantly by BER (such as uracil accumulated through antimetabolite treatment, uracil analogs used as drugs or prodrugs, or oxidized purines appearing through MTH1 inhibition or induced by photodynamic therapy), in which case BER impairment alone could be sufficient to effect considerable cytotoxicity.
Two considerations are crucial when assessing the cytotoxic potential of DNA glycosylase inhibitors. First, unlike the enzymes underlying common BER steps, such as break signaling by PARPs or AP site cleavage by AP endonucleases, DNA glycosylases are specific for damaged bases, and their inhibition will affect only a subset of BER reactions. This in fact may be advantageous for fine-tuning or selection of concurrently used DNA damaging agents, many of which produce specific primary lesions rather than AP sites of strand breaks [16]. Second, DNA glycosylases are often ambivalent with respect to cell-killing effects of DNA damage (as discussed below in sections about specific types of lesions and their repair): they may either counteract the damage by repairing the induced lesions or potentiate the damage by converting damaged bases to AP sites or strand breaks, which are generally more cytotoxic. Thus, the inhibition of DNA glycosylases is not always warranted for inducing synthetic lethality in cancer cells or bacteria. It is always desirable to validate a particular DNA glycosylase as a drug target by knockout or knockdown approaches in an appropriate cell line or pathogen.
The inhibitors discussed in the remaining parts of this paper are mostly small-molecule compounds. Almost all DNA glycosylases are inhibited to a certain degree by non-specific single- or double-stranded DNA, competing for binding with substrate DNA [17,18,19,20,21], and a number of modified nucleotides tightly bound but not cleaved when incorporated into oligonucleotides have been described [22,23,24,25,26,27,28]. Moreover, binding and inhibition of DNA glycosylases by polyanions such as heparin [29,30,31,32] likely stems from the ability of these enzymes to bind nucleic acids. Minor-groove ligands of various chemical nature also interfere with DNA glycosylase binding [33,34]. Despite the obvious importance of such interactions for the biological functions of DNA glycosylases, delivery and targeting problems thus far prevent the therapeutic use of oligonucleotides and other macromolecular polyanions as mass action-driven inhibitors of intracellular enzymes. However, one strategy known for a while and recently applied to DNA glycosylases is the use of prodrugs that are metabolized to nucleoside triphosphate analogs and incorporated into DNA [35,36]. For example, 1′-cyano-2′-deoxyuridine triphosphate is a good substrate for DNA polymerases and, when incorporated into DNA, inhibits E. coli uracil-DNA glycosylase (Ung) and human UNG, displaying nanomolar Ki values [37]. Interestingly, some lesions, such as 2-deoxyribonolactone and 5-hydroxy-5-methylhydantoin [27,38,39], demonstrate an intrinsic ability to trap bifunctional DNA glycosylases covalently on DNA, reminiscent of the PARP-trapping potential of cytotoxic PARP inhibitors. Thus, the development of nucleotides that can be incorporated into DNA and trap DNA glycosylases may be an interesting direction of the glycosylase inhibitor design.
3. Uracil in DNA: Synergism of Glycosylase Inhibitors and Antimetabolites
Antimetabolites, the class of drugs interfering with nucleotide metabolism pathways and thereby with DNA or RNA synthesis, are one of the staples of therapeutic interventions against cancer and bacterial and protozoan infections and are especially useful in combination therapy [40,41]. Many clinically used antimetabolites, such as antifolates, interfere with thymine biosynthesis and cause the accumulation of uracil (or its analogs) in genomic DNA [42,43]. The repair of drug-induced genomic uracil is a double-edge sword: while it protects cells from the effect of this non-canonical nucleobase at low levels of substitution, extensive uracil buildup and excision are toxic and may be the primary reason of cell death after exposure to antifolates [44,45]. Therefore, the inhibition of uracil repair may have different consequences depending on the level of DNA modification and, possibly, on the nature of the modification (if different from uracil).
Human cells possess four DNA glycosylases capable of excising uracil from DNA. However, for three of them (TDG, SMUG1, and MBD4), uracil either is not the main substrate or is removed only from specific contexts (for example, methylated CpG dinucleotides). The main enzyme responsible for uracil repair, UNG, is among the most important factors limiting the efficiency of antifolates and fludarabine, whose action is based on the accumulation of uracil in genomic DNA [46,47,48]. UNG knockdown in human prostate cancer cell lines increases their sensitivity to H2O2 and doxorubicin [49]. Non-small cell carcinoma and lung adenocarcinoma cells develop spontaneous resistance to pemetrexed, a folic acid analog inhibiting dihydrofolate reductase, thymidylate synthase, and glycinamide ribonucleotide formyltransferase, due to a significant increase in the level of UNG, and the suppression of UNG expression returns the sensitivity to normal [50,51]. Some uracil analogs that are accumulated in DNA (such as 5-fluorouracil) are more toxic for cells when SMUG1, rather than UNG, is downregulated [52,53]. However, due to the structural similarity between UNG, TDG, and SMUG1, low-molecular-weight inhibitors will most likely be active against all three enzymes; therefore, the nature of the glycosylase that removes uracil during treatment with antimetabolites is not of primary importance.
UNG inhibitors in combination with genotoxic stress effectively suppress the growth of Plasmodium falciparum [54], Trypanosoma brucei [55,56], and Trypanosoma cruzi [57,58], which makes BER a promising target for drug intervention in protozoan infections. Importantly, some inhibitors preferentially suppress the activity of UNG from infections agents but have little effect on the human enzyme (Table 2).
Table 2.
Properties of selected DNA glycosylase inhibitors discussed in this review.
Low-molecular-weight UNG inhibitors are still at a preclinical stage. The literature describes three main classes of such inhibitors. All of them are competitive, mechanism-based and mimic certain features of the transition state of the UNG-catalyzed reaction [6,79]. Free uracil and its analogs inhibit UNG enzymes from various sources, presenting submillimolar to millimolar IC50 [59,80,81,82,83,84,85], so successful inhibitors required extensive modification of the base to ensure tight binding. Historically, the first class of compounds active against human Plasmodium and herpes simplex type 1 UNGs was 6-(p-alkylanilino)uracils, of which 6-(p-n-octylanilino)uracil showed the strongest affinity, with IC50 ~8 μM for the viral enzyme [54,59,86,87,88] (Table 2). Bipartite inhibitors, structurally similar to the 6-substituted derivatives, consist of a uracil base or its analog linked to a phenolic or benzoic fragment [60,61,62,89] (Table 2). In the structures of bipartite inhibitors bound to UNG, the uracil part occupies the uracil-binding pocket of the enzyme, while the aromatic fragment lies in the DNA-binding groove [61,62] (Table 3). Finally, triskelion inhibitors contain three functional groups at the ends of a branched linker: either one analog of uracil and two aromatic fragments, or two analogs of uracil and one aromatic fragment [63] (Table 2). Interestingly, gentamicin, a clinically used aminoglycoside antibiotic, was reported to inhibit E. coli Ung [64,65] (Table 2). Although the reported IC50 value was quite high (0.4–1.5 mM), this effect may reflect interactions between Ung and the sugar part of DNA [21,90] and suggests another possible direction for inhibitor development.
Table 3.
Known structures of DNA glycosylases bound to their inhibitors.
Thymine–DNA glycosylase (TDG) has recently been validated as a possible drug target in melanoma: its knockdown causes cell cycle arrest, senescence, and cell death in melanoma cell lines but not in normal cells and prevents tumor growth in a xenograft model [91]. Screening of several mid-scale compound libraries yielded about 40 inhibitors with a variety of structures and IC50 >10 μM [91].
Uracil-DNA glycosylases from poxviruses (D4 according to vaccinia virus naming convention) provide a unique drug target. While they possess quite efficient uracil-removing activity, the main role of these enzymes is not in DNA repair, but in viral replication. D4 binds the A20 protein to form a processivity factor for poxviral DNA polymerases [92,93]. Deletion of the D4 gene causes a sharp drop in the ability of vaccinia virus to replicate in cells [94,95,96]. Polycyclic aromatic compounds that disrupt the D4/A20 binding interface are considered a promising class of antiviral drugs active against poxviruses [66,97,98] (Table 2).
Finally, a natural Ung inhibitor, the Ugi protein, is produced by PBS1 and PBS2 bacteriophages [99]. Although Ugi is not considered a therapeutically promising candidate, it has recently found an unexpected use in cell technologies involving CRISPR/Cas9 genome editing. A new generation of Cas9-based tools employs base editors, in which a Cas9 targeting module is fused with a cytosine deaminase to generate C→T mutations [100,101]. Repair by UNG counteracts uracil-mediated targeted mutagenesis, so co-expression of Ugi is commonly used to increase the efficiency of this gene editing procedure [100,101,102].
4. Oxidative Damage Repair: Key to Antibiotic Resistance?
It has been shown that BER is necessary for the survival of certain pathogenic and opportunistic bacteria (Mycobacterium, Neisseria, Pseudomonas, Salmonella) under conditions of genotoxic stress caused either by drugs or by the body’s immune response [103,104,105]. Recently, it was discovered that oxidative stress significantly contributes to the death of bacteria exposed to antibiotics of several classes. Topoisomerase inhibitors, β-lactam antibiotics, membrane-permeabilizing agents, and aminoglycosides induce the generation of hydroxyl radicals in several divergent bacterial species through an iron-dependent Fenton reaction, increasing the lethality of these drugs [106,107,108,109], whereas reducing agents such as H2S or NO protect bacteria from a wide range of antibiotics [110,111]. Possible reasons for the enhanced cell death include translation errors due to oxidative RNA damage [112], oxidation of the nucleotide pool followed by massive chromosome breakage at the sites of damaged nucleotide incorporation [113,114,115], and direct DNA damage by reactive antibiotic molecules, their metabolites, or reactive oxygen species [116,117]. Based on these observations, the systems of antioxidant defense and oxidative damage repair in bacteria are now regarded as promising targets for sensitization towards bactericidal antibiotics, which, if successful, can be a breakthrough in the current antibiotic resistance crisis.
In bacteria, several DNA glycosylases are responsible for oxidative damage repair. In E. coli, the best-studied enzymatic system, termed the “GO system” (for Guanine Oxidation), involves three enzymes: Fpg (MutM), MutY, and MutT, which have complementary functions in countering the mutagenic effect of 8-oxoguanine (oxoG) [118,119,120]. OxoG is an abundant DNA lesion that easily forms stable oxoG(syn):A(anti) Hoogsteen-type pairs, leading to characteristic G:C→T:A transversions [121,122]. Fpg is a DNA glycosylase that excises oxoG from pairs with C; such pairs appear when G is directly oxidized in DNA or when oxodGMP is incorporated opposite C during replication [123,124]. If oxoG remains in DNA and directs dAMP misincorporation, the excision of oxoG by Fpg would lead to a G→T transversion. To safeguard the cell from this mutagenic route, Fpg does not cleave oxoG:A mispairs, which are recognized by MutY, and A is excised instead of oxoG [125]. If repair DNA polymerases insert the correct dCMP opposite oxoG, the second round of repair is carried out by Fpg; otherwise, dAMP is inserted, and MutY-initiated repair is reinitiated. The third enzyme of the system, MutT, hydrolyses oxodGTP and oxoGTP to monophosphates to prevent oxoG incorporation from the oxidized nucleotide pool [126,127]. E. coli also possesses a homolog of Fpg, endonuclease VIII (Nei), which is not considered part of the GO system and preferentially excises oxidized pyrimidines with little opposite-base preference, although it has some activity against oxoG in vitro and prevents G:C→T:A mutations in the absence of Fpg [128,129,130,131]. Finally, endonuclease III (Nth) also removes a wide variety of oxidized pyrimidine bases [132,133].
Although the GO system has been extensively characterized in E. coli, little is known about its functions and the properties of its components in pathogenic bacterial species. Fpg proteins from Salmonella enterica [134], Neisseria meningitidis [135], and Corynebacterium pseudotuberculosis [136] have been cloned and subjected to limited biochemical characterization, which showed essentially Fpg-like properties. Several Fpg homologs from Mycobacterium tuberculosis and Mycobacterium smegmatis were characterized and found to have divergent substrate specificities resembling either E. coli Fpg or Nei [137,138,139,140]. For MutY, limited enzyme characterization has been done for proteins from N. meningitidis [141], Helicobacter pylori [142], and C. pseudotuberculosis [143,144]. The presence of a fully functional GO system with its characteristic antimutator pattern has been confirmed in vivo in their native bacterial cells for Pseudomonas aeruginosa [145,146], N. meningitides [104,135,147], M. smegmatis [137,148,149], and Staphylococcus aureus [150]. Fpg was found to be functional in vivo in S. enterica [134], and MutY in H. pylori [142].
The available information about the relevance of the GO system for the pathogenicity of bacteria shows its dual role. On one hand, it seems that this line of defense indeed assists successful primary infection. MutY deficiency has been shown to compromise mouse stomach colonization by H. pylori [142]. Successful macrophage infection by Brucella abortus requires intact fpg and mutY genes [151], and M. tuberculosis Fpg and Nei are required for lung colonization in a rhesus macaque model [152]. Hypervirulent Neisseria isolates maintain functional fpg and mutY despite having a general mutator phenotype [147]. On the other hand, hypermutability associated with GO system inactivation sometimes provides the variance for selection of highly virulent or drug-resistant strains [153,154,155,156]. This underscores the importance of a thorough characterization of the GO system for a given pathogen to assess it as a possible drug combination target.
Human homologs of Fpg and Nei (NEIL1, NEIL2, and NEIL3) are significantly different from the bacterial proteins in their sequence and structure, making realistic the development of small ligands selectively targeting the bacterial enzymes. MutY and Nth appear to be less selective targets.
Few specific inhibitors of the bacterial GO system have been reported. Fpg is weakly inhibited by free damaged base analogs such as 2,6-diamino-4-oxo-5-(N-methylformamido)pyrimidine, 5-nitroso-2,4,6-triaminopyridine, and 5-nitroso-4,6-diamino-2-oxopyridine [67,157]. The base analog with the strongest ability to suppress Fpg activity (IC50 ~10–100 μM) is 2-thioxanthine (Table 2); however, it is not a true inhibitor but rather a reagent that oxidizes cysteines in the zinc finger in Fpg, Nei, and NEIL2 [67,68]. MutY from C. pseudotuberculosis was reported to be sensitive to suramin, an antiprotozoan and anthelmintic drug that acts intracellularly [32] (Table 2); however, the relevant drug uptake pathways are likely absent in bacteria. Interestingly, Fpg is strongly inhibited by Cibacron Blue F3GA, a dye bearing structural resemblance to suramin [69] (Table 2). Several papers described the inactivation of Fpg, Nth, OGG1, NEIL1, and NEIL2 by NO-producing agents and suggested damage to redox-sensitive groups in the enzyme molecules [158,159,160,161,162]. However, the mechanisms of this reaction remain unclear, since the known redox-sensitive groups in these glycosylases are different or absent altogether. A screen of a natural product library revealed several inhibitors of M. tuberculosis Nei2, the best of which, norlobaric acid, has Ki = 74 nM [70] (Table 2).
5. Oxidative Damage Repair: Cancer Sensitization Strategy
OGG1 is a human DNA glycosylase that initiates the repair of oxidized purine bases, mainly oxoG and formamidopyrimidines, thus being a functional analog of Fpg. Nevertheless, in its sequence and structure, OGG1 is completely different from Fpg (Table 1). Overexpression of OGG1 in fibroblasts, pulmonary epithelial cells, and bone marrow protects them from the toxic effects of thiotepa, carmustine, and mafosfamide, which mainly yield N7-alkylated purines further hydrolyzed in the cell to formamidopyrimidine derivatives [163,164,165]. However, it is unclear whether normal OGG1 levels can reduce the toxicity of these drugs in tumor cells. A similar effect of OGG1 has been described for cisplatin and oxaliplatin [166], although the nature of the damage removed in this case is not entirely clear. Of the antitumor agents that produce oxidative DNA damage, OGG1 downregulation or inhibition sensitizes cells to bleomycin [167] and ionizing radiation [168].
As with uracil incorporation and repair, the activity of OGG1 may not only safeguard cells from genomic damage but also potentiate the action of DNA-damaging agents, converting damaged bases to more cytotoxic strand breaks. For instance, OGG1 downregulation protects several cancer cell lines from β-lapachone, an NAD(P)H dehydrogenase (quinone 1)-dependent redox cycling drug that produces copious amounts of intracellular H2O2 [169]. Hence, a strategy alternative to OGG1 inhibition may consist in saturating the BER capacity with oxidative lesions. In human cells, OGG1 together with mismatched adenine-DNA glycosylase MUTYH and nucleoside triphosphatase MTH1 (NUDT1) forms an analog of the GO system, which prevents the mutagenic effect of oxoG [170]. Recently, the knockdown or inhibition of MTH1, which hydrolyzes oxoG triphosphates and prevents their incorporation into growing DNA and RNA chains, was shown to be toxic to tumor cells, due to the accumulation of oxidized bases and DNA breaks [71,171,172]. Apparently, as in the case of PARP inhibitors, selective toxicity is due to the suppression of the last remaining pathway for the oxidative damage repair in cancer cells. Several low-molecular-weight MTH1 inhibitors were identified, including a clinically approved tyrosine kinase inhibitor, crizotinib [71,171,173] (Table 2). Interestingly, crizotinib possesses a chiral center that gives rise to (R)- and (S)- enantiomers, of which clinically used (R)-crizotinib inhibits c-MET and ALK protein kinases, whereas the (S)-enantiomer preferentially binds to and inhibits MTH1 [71,174,175]. While it is still debated to which extent the cytotoxic activity of crizotinib and other MTH1 inhibitors is dependent on MTH1 and oxidative overload [172,176,177,178], most reports agree that oxidative damage is an important cell-killing factor, although its causes might not be limited to a direct suppression of MTH1 activity (recently reviewed in [179]). As of today, (S)-crizotinib is not used for patient treatment. However, another anti-cancer drug candidate, karonudib, was developed from previously found MTH1 inhibitors [172,180,181]. Presently, two Phase 1 clinical trials of karonudib are registered with the US National Library of Medicine Clinical Studies Database.
Inhibitors of OGG1 have been reported in the literature but have not yet reached clinical trials. Mechanism-based approaches had only limited success: both oxoG base and its analogs proved to be weak inhibitors [182,183], while substituted 2,6-diaminopurines performed slightly better [76]. Experimental and computational screening of small-molecule pharmacological libraries produced several hits that were expanded into inhibitors with submicromolar affinity, structurally unrelated to the OGG1 substrate [72,73,74] (Table 2). Combinatorial design based on the identified OGG1 and MTH1 inhibitors was used to obtain a compound with submicromolar affinity for both these enzymes [75] (Table 2).
Other DNA glycosylases that repair oxidative damage, including NEIL1, NEIL2, NEIL3, MUTYH, and NTHL1, have been targeted less successfully. Purine-analog library and general library screening produced several inhibitors for NEIL1, but their affinities were in the micromolar range, and the target selectivity was quite low when compared with the inhibition of other glycosylases [76,184] (Table 2). Fumarylacetoacetate was reported to inhibit NEIL1 and NEIL2 and to a lesser degree, OGG1 and NTHL1 (Table 2), but the structural reasons under this effect have not been established [77]. Moreover, these enzymes have low experimental support as targets for sensitization to antitumor therapy, although NEIL1 confers some resistance to ionizing radiation and antifolates [185,186].
6. Oxidative Damage Repair: Unexpected Connections
While DNA damage and its repair are well understood in the cancer paradigm, two unexpected connections of oxidative damage with other human pathologies emerged recently. OxoG and its repair by OGG1 are suspected to play a regulatory role in the inflammatory response. Several lines of evidence support this conclusion. Ogg1-null or -depleted mice show a significantly alleviated inflammatory response to many factors, including bacterial endotoxins, Helicobacter infection, foreign protein response, ragweed pollen grain extract-induced allergy, etc. [187,188,189,190]. Interestingly, however, the inflammation associated with UVB or pulmonary hyperoxia is enhanced rather than reduced in Ogg1−/− mice [191,192,193], suggesting that OGG1-dependent inflammation requires foreign antigens. After the excision of oxoG, the OGG1·oxoG complex can bind Ras family GTPases and facilitate the GDP-to-GTP exchange [194,195], which triggers the signaling pathway leading to the activation of NF-κB, the key pro-inflammatory transcription factor [196]. Moreover, OGG1 can bind oxidized G-rich promoters of pro-inflammatory genes in an enzymatically non-productive mode and facilitate their expression by attracting NF-κB [197,198,199]. A small-molecule OGG1 inhibitor, TH5487, was developed that competes with oxoG for binding and downregulates the inflammatory response in a mouse model [73] (Table 2). Although TH5487 has a 4-bromobenzimidazolone moiety, which is structurally similar to oxoG, the crystal structure of its complex with OGG1 [73] (Table 3) unexpectedly revealed that the oxoG-binding pocket is occupied by another moiety of TH5487, p-iodophenylacetamide, whereas 4-bromobenzimidazolone resides in a so-called exo-site, which normally binds undamaged G and serves as a transient binding site for oxoG on its way to the active site [200,201]. Thus, TH5487 functionally resembles bipartite inhibitors of UNG, simultaneously engaging two selective binding sites in the enzyme molecule. Such design may be employed to construct new potent inhibitors of OGG1 and other DNA glycosylases.
In addition, inhibition of OGG1 holds promise to prevent or delay the onset of Huntington’s disease in risk groups. This hereditary condition, which belongs to the class of “trinucleotide repeat” genetic diseases, is caused by expansion of the (CAG)n repeat run in the HTT gene beyond the critical length of ~35 repeats [202]. Before becoming symptomatic, carriers of a pathogenic allele experience an explosive growth of the (CAG)n run up to several hundred repeats in the striatum at the early stage of the disease [203]. This expansion is triggered by the normal repair of oxoG initiated by OGG1 [204,205] and is likely caused by an imbalance of BER enzymes in this part of the brain, which leads to the accumulation of unprocessed repair intermediates [206]. In the Huntington’s disease mouse model, Ogg1 knockout suppresses the repeat number growth in the striatum and delays the onset of motor dysfunction [207]. Thus, in carriers of the pathogenic HTT allele, for whom the penetrance is inevitably 100%, inhibition of OGG1 may be a reasonable therapeutic strategy.
7. Alkylation Damage Repair: Dual Consequences
Alkylating antitumor agents produce many damaged bases, including O6-alkylguanine repaired by O6-methylguanine–DNA methyltransferase (MGMT), and ring-alkylated purines repaired predominantly by BER [16,208]. Unlike other DNA glycosylases, which impart resistance to DNA-damaging agents to cells, N-methylpurine-DNA glycosylase (MPG, alias alkyladenine-DNA glycosylase (AAG), and alkylpurine-DNA N-glycosylase, APNG) may increase the cytotoxicity of alkylating antitumor agents, removing alkylated bases from DNA to form AP sites, which are more dangerous for the cell [208,209,210,211,212,213,214]. A similar sensitization mechanism is also characteristic of UNG, TDG, and MBD4 DNA glycosylases when they repair C5-halogenated uracil derivatives [215,216,217,218]. On the other hand, inhibition of MPG in carcinoma cells sensitizes them to alkylating agents [219], and Mpg−/− murine cells are hypersensitive to 1,3-bis(2-chloroethyl)-1-nitrosourea and mitomycin C (but not to alkylating nitrogen mustards) [220]. An integrative model of temozolomide-induced DNA damage and DNA repair by MGMT and MPG in glioblastoma predicts that inhibition of both enzymes is the most successful sensitization strategy [221]. For temozolomide-resistant forms of glioblastoma, the combination of inhibition of BER enzymes and PARP-dependent signaling is effective [212,222]. In addition to the detoxification of anticancer drug adducts, MPG and OGG1 have been reported to hydrolyze a human cytomegalovirus replication inhibitor, 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole, opening the possibility of antiviral action of drug combinations including DNA glycosylase inhibitors [223]. Bacterial alkA mutants are hypersensitive to methyl methanesulfonate [224,225]; however, alkylating agents are not among clinically used antibacterial drugs, so this vulnerability is hard to exploit.
Alkylbase-removing DNA glycosylases are the least explored group in terms of specific inhibitors. N3-substituted adenine derivatives are competitive inhibitors of bacterial Tag and mammalian MPG [226,227,228,229,230]. Based on this observation, a series of structural analogs has been computationally designed to inhibit TagA from Leptospira interrogans, the infectious agent of leptospirosis, although no experimental evidence was provided for their activity against the enzyme or the pathogen [231]. A natural flavonol, morin, inhibits MPG [78] (Table 2).
8. Assays for DNA Glycosylase Activity
Most basic research on DNA glycosylases was and is still done using radioactively labeled oligonucleotides and analyzing substrates and product by gel electrophoresis. While this assay offers the highest sensitivity, it is labor-intensive, not easily scalable, and inconvenient when screening pharmacological libraries. In the past years, a number of fluorescence-based assays to follow glycosylase activities appeared, some of them coupled with signal amplification to increase the sensitivity.
The first attempts to utilize fluorescent labels for detecting DNA glycosylase activities in a homogeneous mode were based on changes in the signal from a fluorophore incorporated next to a lesion upon the eversion or the excision of the damaged base [232,233] (Figure 2A). Although this approach has been used for inhibitor screening [75], it is not very sensitive, and fluorophores adjacent to a lesion may even inhibit the measured activity. Molecular beacon substrates developed later consist of an oligonucleotide hairpin or a duplex bearing a fluorophore and a quencher at its termini (Figure 2B). Such substrates allow measuring DNA glycosylase activities both in vitro and in living cells [76,234,235,236,237] and have been used in glycosylase inhibitor library screening [73,76]. In this case, the glycosylase must be bifunctional to nick the substrate, or else an AP endonuclease has to be present in the assay. Several types of arrays or beads with immobilized damaged oligonucleotides have been reported, in which only the damaged strand is labeled, and the cleavage produces short DNA fragments that can be washed off [238,239,240,241,242]. While such assays are not homogeneous, they are well suited for multiplexing and parallel screening. Fluorophores can also be incorporated into double-stranded DNA in situ through base excision, AP site cleavage, and gap filling by DNA polymerase β with a labeled dNTP [243].
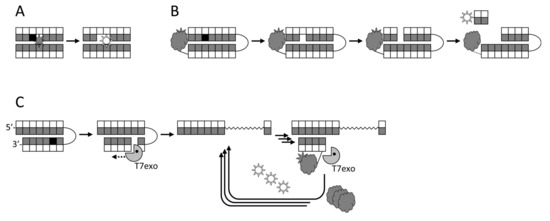
Figure 2.
Schemes of several fluorescence-based approaches to DNA glycosylase detection. (A), fluorescent reporters adjacent to the lesion; (B), molecular beacons with a fluorophore–quencher pair; (C), T7 exonuclease-assisted signal amplification due to cyclic degradation of a probe bearing a fluorophore–quencher pair.
An interesting approach was suggested that employs a DNAzyme inactivated by a strategically placed U residue, and the excision by uracil-DNA glycosylase reactivates the DNAzyme, which cleaves a fluorescent substrate [244]. However, it cannot be applied to other glycosylases that require double-stranded substrates. In a more advanced version, base excision generates a specifically folded quadruplex, which forms a fluorescent complex with quadruplex-selective ligands [245,246,247].
The most sensitive fluorescent assays rely on the formation of a nick after the cleavage by a DNA glycosylase (either bifunctional or coupled with an AP endonuclease), followed by signal amplification. The amplification may be exonuclease-assisted linear isothermal, in which a beacon is annealed and degraded by a double-stranded specific exonuclease in repeated cycles [248,249,250] (Figure 2C). Alternatively, the signal may be enhanced by rolling-circle amplification, using any suitable assay to detect the newly synthesized DNA [251,252,253,254]. Finally, nick formation can serve as a starting point for exponential isothermal amplification [255,256] or cascade hybridization [257].
9. Conclusions
DNA glycosylases, as enzymes that initiate base excision repair, represent an attractive pharmacological target. Their structures reveal mechanism-based features, such as deep pockets for substrate base binding, indicating potential druggability, and several successful attempts of library screening produced tantalizing leads that can be explored further to develop drugs for cancer and infectious diseases. Moreover, recent findings implicate DNA glycosylases not only in genome protection but also in regulatory pathways and suggest that they can be targeted in some inflammatory and neurodegenerative processes. A number of rapid and sensitive assays for screening DNA glycosylase activities were developed in the past few years, which should facilitate the search for their inhibitors.
The most important factor that complicates the targeting of DNA glycosylases in the now well-established framework of synthetic lethality, e.g., in cancer therapy, is their dual function in cell killing. On one hand, glycosylases initiate the repair of genotoxic adducts and, in theory, should potentiate their action. On the other hand, there are many cases in which the main lethal lesions are not adducts per se but intermediates of their repair, such as AP sites or DNA breaks. Such intermediates usually accumulate if the activity of downstream BER enzymes are insufficient to process the inflicted amount of genomic lesions in full. In these situations, DNA glycosylase inhibition would protect rather than sensitize cells to genome damage. Optimally, DNA glycosylases should be targeted in some form of precision therapy, based on the general model of toxicity of various adducts and the specific knowledge of adduct spectra and downstream BER capacity in the affected cells.
Outside of the cancer field, DNA glycosylase inhibition is most likely to find its soonest clinical application in antiviral therapy, since two important groups of human pathogens, poxviruses and herpesviruses, possess their own uracil-DNA glycosylases, a validated target required for replication in host cells, and several promising drug leads are available. Inhibition of OGG1 to prevent somatic trinucleotide repeat expansion in Huntington’s disease also has high priority due to the extreme morbidity and mortality of the condition and the lack of other drugs, although lead compounds capable of brain delivery have not been reported so far. The inflammation-modulating action of OGG1 inhibitors, albeit attracting considerable attention, would still require much research and mechanistic insights to produce drugs comparable with more traditional anti-inflammatory agents. Even if more basic research is required to validate DNA glycosylases as targets for antibacterial combination therapy, yet the payoff in this area may be the largest one. The prospects of bringing DNA glycosylases into the circle of drug targets ultimately depend on our understanding of their action in DNA repair and connection with other cellular pathways.
Author Contributions
Conceptualization, D.O.Z. writing—original draft preparation, G.V.M., A.V.E., E.A.D., and D.O.Z.; writing—review and editing, D.O.Z.; visualization, D.O.Z.; supervision, D.O.Z.; project administration, D.O.Z.; funding acquisition, A.V.E. and D.O.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant 19-74-00068. Partial salary support from the Russian Ministry of Science and Higher Education (State funded budget project АААА-А17-117020210023-1) is acknowledged. Open Access fees were paid by an intramural program of SB RAS Institute of Chemical Biology and Fundamental Medicine.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006; p. 1118. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007; p. 704. [Google Scholar]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1261. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.K.; Dodson, M.L.; Lloyd, R.S. Initiation of base excision repair: Glycosylase mechanisms and structures. Annu. Rev. Biochem. 1999, 68, 255–285. [Google Scholar] [CrossRef] [PubMed]
- Stivers, J.T.; Jiang, Y.L. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003, 103, 2729–2760. [Google Scholar] [CrossRef]
- Huffman, J.L.; Sundheim, O.; Tainer, J.A. DNA base damage recognition and removal: New twists and grooves. Mutat. Res. 2005, 577, 55–76. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPs, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Phillips, D.H. DNA adducts from chemotherapeutic agents. Mutat. Res. 1996, 355, 13–40. [Google Scholar] [CrossRef]
- Thomas, L.; Yang, C.H.; Goldthwait, D.A. Two DNA glycosylases in Escherichia coli which release primarily 3-methyladenine. Biochemistry 1982, 21, 1162–1169. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Vasilenko, N.L.; Sinitsina, O.I.; Yamkovoy, V.I.; Fedorova, O.S.; Douglas, K.T.; Nevinsky, G.A. Thermodynamic, kinetic, and structural basis for recognition and repair of 8-oxoguanine in DNA by Fpg protein from Escherichia Coli. Biochemistry 2002, 41, 7540–7548. [Google Scholar] [CrossRef]
- Fedorova, O.S.; Nevinsky, G.A.; Koval, V.V.; Ishchenko, A.A.; Vasilenko, N.L.; Douglas, K.T. Stopped-flow kinetic studies of the interaction between Escherichia coli Fpg protein and DNA substrates. Biochemistry 2002, 41, 1520–1528. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Ishchenko, A.A.; Douglas, K.T.; Nevinsky, G.A. Recognition of damaged DNA by Escherichia coli Fpg protein: Insights from structural and kinetic data. Mutat. Res. 2003, 531, 141–156. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Mechetin, G.V.; Nevinsky, G.A. Uracil-DNA glycosylase: Structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat. Res. 2010, 685, 11–20. [Google Scholar] [CrossRef]
- Schärer, O.D.; Ortholand, J.-Y.; Ganesan, A.; Ezaz-Nikpay, K.; Verdine, G.L. Specific binding of the DNA repair enzyme AlkA to a pyrrolidine-based inhibitor. J. Am. Chem. Soc. 1995, 117, 6623–6624. [Google Scholar] [CrossRef]
- Schärer, O.D.; Verdine, G.L. A designed inhibitor of base-excision DNA repair. J. Am. Chem. Soc. 1995, 117, 10781–10782. [Google Scholar] [CrossRef]
- Deng, L.; Schärer, O.D.; Verdine, G.L. Unusually strong binding of a designed transition-state analog to a base-excision DNA repair protein. J. Am. Chem. Soc. 1997, 119, 7865–7866. [Google Scholar] [CrossRef]
- Schärer, O.D.; Kawate, T.; Gallinari, P.; Jiricny, J.; Verdine, G.L. Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl. Acad. Sci. USA 1997, 94, 4878–4883. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D.; Nash, H.M.; Jiricny, J.; Laval, J.; Verdine, G.L. Specific binding of a designed pyrrolidine abasic site analog to multiple DNA glycosylases. J. Biol. Chem. 1998, 273, 8592–8597. [Google Scholar] [CrossRef] [PubMed]
- Le Bihan, Y.-V.; Izquierdo, M.A.; Coste, F.; Aller, P.; Culard, F.; Gehrke, T.H.; Essalhi, K.; Carell, T.; Castaing, B. 5-Hydroxy-5-methylhydantoin DNA lesion, a molecular trap for DNA glycosylases. Nucleic Acids Res. 2011, 39, 6277–6290. [Google Scholar] [CrossRef]
- Dai, Q.; Lu, X.; Zhang, L.; He, C. Synthesis of DNA oligos containing 2’-deoxy-2’-fluoro-D-arabinofuranosyl-5-carboxylcytosine as hTDG inhibitor. Tetrahedron 2012, 68, 5145–5151. [Google Scholar] [CrossRef][Green Version]
- Yamagata, Y.; Kato, M.; Odawara, K.; Tokuno, Y.; Nakashima, Y.; Matsushima, N.; Yasumura, K.; Tomita, K.-i.; Ihara, K.; Fujii, Y.; et al. Three-dimensional structure of a DNA repair enzyme, 3-methyladenine DNA glycosylase II, from Escherichia coli. Cell 1996, 86, 311–319. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef]
- Gilboa, R.; Zharkov, D.O.; Golan, G.; Fernandes, A.S.; Gerchman, S.E.; Matz, E.; Kycia, J.H.; Grollman, A.P.; Shoham, G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J. Biol. Chem. 2002, 277, 19811–19816. [Google Scholar] [CrossRef]
- Eberle, R.J.; Coronado, M.A.; Peinado, R.S.; de Moraes, F.R.; Olivier, D.; Dreyer, T.; de Oliveira Lopes, D.; da Luz, B.S.R.; Azevedo, V.; Arni, R.K. The polyanions heparin and suramin impede binding of free adenine to a DNA glycosylase from C. Pseudotuberculosis. Int. J. Biol. Macromol. 2019, 125, 459–468. [Google Scholar] [CrossRef]
- Li, X.; Lu, A.-L. Intact MutY and its catalytic domain differentially contact with A/8-oxoG-containing DNA. Nucleic Acids Res. 2000, 28, 4593–4603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mechetin, G.V.; Dyatlova, E.A.; Sinyakov, A.N.; Ryabinin, V.A.; Vorobjev, P.E.; Zharkov, D.O. Correlated target search by uracil–DNA glycosylase in the presence of bulky adducts and DNA-binding ligands. Russ. J. Bioorg. Chem. 2017, 43, 23–28. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Feng, J.Y. Addressing the selectivity and toxicity of antiviral nucleosides. Antivir. Chem. Chemother. 2018, 26, 2040206618758524. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Stivers, J.T.; Greenberg, M.M. Competitive inhibition of uracil DNA glycosylase by a modified nucleotide whose triphosphate is a substrate for DNA polymerase. J. Am. Chem. Soc. 2009, 131, 1344–1345. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Greenberg, M.M.; Kow, Y.W.; Hwang, J.-T.; Cunningham, R.P. The 2-deoxyribonolactone lesion produced in DNA by neocarzinostatin and other damaging agents forms cross-links with the base-excision repair enzyme endonuclease III. J. Am. Chem. Soc. 2001, 123, 3161–3162. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, K.M.; Hashimoto, M.; Kow, Y.W.; Greenberg, M.M. Cross-linking of 2-deoxyribonolactone and its β-elimination product by base excision repair enzymes. Biochemistry 2003, 42, 2449–2455. [Google Scholar] [CrossRef]
- Peters, G.J.; van der Wilt, C.L.; van Moorsel, C.J.A.; Kroep, J.R.; Bergman, A.M.; Ackland, S.P. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol. Ther. 2000, 87, 227–253. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2280–2293. [Google Scholar] [CrossRef]
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Uracil in DNA—General mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516. [Google Scholar] [CrossRef]
- Sousa, M.M.L.; Krokan, H.E.; Slupphaug, G. DNA-uracil and human pathology. Mol. Asp. Med. 2007, 28, 276–306. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, H.A.; Dickey, L.; Goulian, M. DNA fragmentation and cytotoxicity from increased cellular deoxyuridylate. Biochemistry 1986, 25, 3225–3230. [Google Scholar] [CrossRef] [PubMed]
- Van Triest, B.; Pinedo, H.M.; Giaccone, G.; Peters, G.J. Downstream molecular determinants of response to 5-fluorouracil and antifolate thymidylate synthase inhibitors. Ann. Oncol. 2000, 11, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Bulgar, A.D.; Snell, M.; Donze, J.R.; Kirkland, E.B.; Li, L.; Yang, S.; Xu, Y.; Gerson, S.L.; Liu, L. Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia 2010, 24, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Bulgar, A.D.; Weeks, L.D.; Miao, Y.; Yang, S.; Xu, Y.; Guo, C.; Markowitz, S.; Oleinick, N.; Gerson, S.L.; Liu, L. Removal of uracil by uracil DNA glycosylase limits pemetrexed cytotoxicity: Overriding the limit with methoxyamine to inhibit base excision repair. Cell Death Dis. 2012, 3, e252. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Qing, Y.; Pink, J.J.; Gerson, S.L. Loss of uracil DNA glycosylase selectively resensitizes p53-mutant and -deficient cells to 5-FdU. Mol. Cancer Res. 2018, 16, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.M.K.; Knost, J.A.; Estes, N.; Rao, J.S. Small interfering RNA-directed knockdown of uracil DNA glycosylase induces apoptosis and sensitizes human prostate cancer cells to genotoxic stress. Mol. Cancer Res. 2009, 7, 1285–1293. [Google Scholar] [CrossRef]
- Weeks, L.D.; Fu, P.; Gerson, S.L. Uracil-DNA glycosylase expression determines human lung cancer cell sensitivity to pemetrexed. Mol. Cancer Ther. 2013, 12, 2248–2260. [Google Scholar] [CrossRef]
- Weeks, L.D.; Zentner, G.E.; Scacheri, P.C.; Gerson, S.L. Uracil DNA glycosylase (UNG) loss enhances DNA double strand break formation in human cancer cells exposed to pemetrexed. Cell Death Dis. 2014, 5, e1045. [Google Scholar] [CrossRef]
- An, Q.; Robins, P.; Lindahl, T.; Barnes, D.E. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007, 67, 940–945. [Google Scholar] [CrossRef]
- Nagaria, P.; Svilar, D.; Brown, A.R.; Wang, X.-H.; Sobol, R.W.; Wyatt, M.D. SMUG1 but not UNG DNA glycosylase contributes to the cellular response to recovery from 5-fluorouracil induced replication stress. Mutat. Res. 2013, 743–744, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Suksangpleng, T.; Leartsakulpanich, U.; Moonsom, S.; Siribal, S.; Boonyuen, U.; Wright, G.E.; Chavalitshewinkoon-Petmitr, P. Molecular characterization of Plasmodium falciparum uracil-DNA glycosylase and its potential as a new anti-malarial drug target. Malar. J. 2014, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Acosta, V.M.; Aguilar-Pereyra, F.; Vidal, A.E.; Navarro, M.; Ruiz-Pérez, L.M.; González-Pacanowska, D. Trypanosomes lacking uracil-DNA glycosylase are hypersensitive to antifolates and present a mutator phenotype. Int. J. Biochem. Cell Biol. 2012, 44, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Charret, K.S.; Requena, C.E.; Castillo-Acosta, V.M.; Ruiz-Pérez, L.M.; González-Pacanowska, D.; Vidal, A.E. Trypanosoma brucei AP endonuclease 1 has a major role in the repair of abasic sites and protection against DNA-damaging agents. DNA Repair 2012, 11, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, G.; Barría, C.; Fernández, C.; Sepúlveda, S.; Valenzuela, L.; Kemmerling, U.; Galanti, N. DNA repair BER pathway inhibition increases cell death caused by oxidative DNA damage in Trypanosoma cruzi. J. Cell. Biochem. 2011, 112, 2189–2199. [Google Scholar] [CrossRef]
- Sepúlveda, S.; Valenzuela, L.; Ponce, I.; Sierra, S.; Bahamondes, P.; Ramirez, S.; Rojas, V.; Kemmerling, U.; Galanti, N.; Cabrera, G. Expression, functionality, and localization of apurinic/apyrimidinic endonucleases in replicative and non-replicative forms of Trypanosoma cruzi. J. Cell. Biochem. 2014, 115, 397–409. [Google Scholar] [CrossRef]
- Focher, F.; Verri, A.; Spadari, S.; Manservigi, R.; Gambino, J.; Wright, G.E. Herpes simplex virus type 1 uracil-DNA glycosylase: Isolation and selective inhibition by novel uracil derivatives. Biochem. J. 1993, 292, 883–889. [Google Scholar] [CrossRef][Green Version]
- Jiang, Y.L.; Krosky, D.J.; Seiple, L.; Stivers, J.T. Uracil-directed ligand tethering: An efficient strategy for uracil DNA glycosylase (UNG) inhibitor development. J. Am. Chem. Soc. 2005, 127, 17412–17420. [Google Scholar] [CrossRef]
- Chung, S.; Parker, J.B.; Bianchet, M.; Amzel, L.M.; Stivers, J.T. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat. Chem. Biol. 2009, 5, 407–413. [Google Scholar] [CrossRef]
- Krosky, D.J.; Bianchet, M.A.; Seiple, L.; Chung, S.; Amzel, L.M.; Stivers, J.T. Mimicking damaged DNA with a small molecule inhibitor of human UNG2. Nucleic Acids Res. 2006, 34, 5872–5879. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Chung, S.; Krosky, D.J.; Stivers, J.T. Synthesis and high-throughput evaluation of triskelion uracil libraries for inhibition of human dUTPase and UNG2. Bioorganic Med. Chem. 2006, 14, 5666–5672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Jiang, J.; Yu, R. A highly sensitive electrochemical platform for the assay of uracil-DNA glycosylase activity combined with enzymatic amplification. Anal. Sci. 2013, 29, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Song, P.; Sato, Y.; Nishizawa, S.; Teramae, N.; Tong, A.; Xiang, Y. A label-free and sensitive fluorescent method for the detection of uracil-DNA glycosylase activity. Chem. Commun. 2015, 51, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Nuth, M.; Huang, L.; Saw, Y.L.; Schormann, N.; Chattopadhyay, D.; Ricciardi, R.P. Identification of inhibitors that block vaccinia virus infection by targeting the DNA synthesis processivity factor D4. J. Med. Chem. 2011, 54, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Speina, E.; Ciesla, J.M.; Graziewicz, M.-A.; Laval, J.; Kazimierczuk, Z.; Tudek, B. Inhibition of DNA repair glycosylases by base analogs and tryptophan pyrolysate, Trp-P-1. Acta Biochim. Pol. 2005, 52, 167–178. [Google Scholar] [CrossRef]
- Biela, A.; Coste, F.; Culard, F.; Guerin, M.; Goffinont, S.; Gasteiger, K.; Cieśla, J.; Winczura, A.; Kazimierczuk, Z.; Gasparutto, D.; et al. Zinc finger oxidation of Fpg/Nei DNA glycosylases by 2-thioxanthine: Biochemical and X-ray structural characterization. Nucleic Acids Res. 2014, 42, 10748–10761. [Google Scholar] [CrossRef]
- Chetsanga, C.J.; Frenette, G.P. Excision of aflatoxin B1-imidazole ring opened guanine adducts from DNA by formamidopyrimidine-DNA glycosylase. Carcinogenesis 1983, 4, 997–1000. [Google Scholar] [CrossRef]
- Lata, K.; Afsar, M.; Ramachandran, R. Biochemical characterization and novel inhibitor identification of Mycobacterium tuberculosis Endonuclease VIII 2 (Rv3297). Biochem. Biophys. Rep. 2017, 12, 20–28. [Google Scholar] [CrossRef]
- Huber, K.V.M.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.-S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef]
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Small molecule inhibitors of 8-oxoguanine DNA glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343. [Google Scholar] [CrossRef]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Tahara, Y.-K.; Auld, D.; Ji, D.; Beharry, A.A.; Kietrys, A.M.; Wilson, D.L.; Jimenez, M.; King, D.; Nguyen, Z.; Kool, E.T. Potent and selective inhibitors of 8-oxoguanine DNA glycosylase. J. Am. Chem. Soc. 2018, 140, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Tahara, Y.-K.; Kietrys, A.M.; Hebenbrock, M.; Lee, Y.; Wilson, D.L.; Kool, E.T. Dual inhibitors of 8-oxoguanine surveillance by OGG1 and NUDT1. ACS Chem. Biol. 2019, 14, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.C.; Calkins, M.J.; Jadhav, A.; Dorjsuren, D.; Maloney, D.; Simeonov, A.; Jaruga, P.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Inhibition of DNA glycosylases via small molecule purine analogs. PLoS ONE 2013, 8, e81667. [Google Scholar] [CrossRef]
- Bliksrud, Y.T.; Ellingsen, A.; Bjørås, M. Fumarylacetoacetate inhibits the initial step of the base excision repair pathway: Implication for the pathogenesis of tyrosinemia type I. J. Inherit. Metab. Dis. 2013, 36, 773–778. [Google Scholar] [CrossRef]
- Dixon, M.; Woodrick, J.; Gupta, S.; Karmahapatra, S.K.; Devito, S.; Vasudevan, S.; Dakshanamurthy, S.; Adhikari, S.; Yenugonda, V.M.; Roy, R. Naturally occurring polyphenol, morin hydrate, inhibits enzymatic activity of N-methylpurine DNA glycosylase, a DNA repair enzyme with various roles in human disease. Bioorganic Med. Chem. 2015, 23, 1102–1111. [Google Scholar] [CrossRef]
- Stivers, J.T.; Drohat, A.C. Uracil DNA glycosylase: Insights from a master catalyst. Arch. Biochem. Biophys. 2001, 396, 1–9. [Google Scholar] [CrossRef]
- Lindahl, T.; Ljungquist, S.; Siegert, W.; Nyberg, B.; Sperens, B. DNA N-glycosidases: Properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 1977, 252, 3286–3294. [Google Scholar]
- Krokan, H.; Wittwer, C.U. Uracil DNA-glycosylase from HeLa cells: General properties, substrate specificity and effect of uracil analogs. Nucleic Acids Res. 1981, 9, 2599–2613. [Google Scholar] [CrossRef]
- Leblanc, J.-P.; Martin, B.; Cadet, J.; Laval, J. Uracil-DNA glycosylase: Purification and properties of uracil-DNA glycosylase from Micrococcus luteus. J. Biol. Chem. 1982, 257, 3477–3483. [Google Scholar]
- Williams, M.V.; Pollack, J.D. A mollicute (mycoplasma) DNA repair enzyme: Purification and characterization of uracil-DNA glycosylase. J. Bacteriol. 1990, 172, 2979–2985. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Tordova, M.; Jagadeesh, J.; Drohat, A.C.; Stivers, J.T.; Gilliland, G.L. Crystal structure of Escherichia coli uracil DNA glycosylase and its complexes with uracil and glycerol: Structure and glycosylase mechanism revisited. Proteins 1999, 35, 13–24. [Google Scholar] [CrossRef]
- Duraffour, S.; Ishchenko, A.A.; Saparbaev, M.; Crance, J.-M.; Garin, D. Substrate specificity of homogeneous monkeypox virus uracil-DNA glycosylase. Biochemistry 2007, 46, 11874–11881. [Google Scholar] [CrossRef] [PubMed]
- Pregnolato, M.; Ubiali, D.; Verri, A.; Focher, F.; Spadari, S.; Sun, H.; Zhi, C.; Wright, G.E. Synthesis and molecular modeling of novel HSV1 uracil-DNA glycosylase inhibitors. Nucleosides Nucleotides 1999, 18, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhi, C.; Wright, G.E.; Ubiali, D.; Pregnolato, M.; Verri, A.; Focher, F.; Spadari, S. Molecular modeling and synthesis of inhibitors of herpes simplex virus type 1 uracil-DNA glycosylase. J. Med. Chem. 1999, 42, 2344–2350. [Google Scholar] [CrossRef]
- Hendricks, U.; Crous, W.; Naidoo, K.J. Computational rationale for the selective inhibition of the herpes simplex virus type 1 uracil-DNA glycosylase enzyme. J. Chem. Inf. Model. 2014, 54, 3362–3372. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Cao, C.; Stivers, J.T.; Song, F.; Ichikawa, Y. The merits of bipartite transition-state mimics for inhibition of uracil DNA glycosylase. Bioorgance Chem. 2004, 32, 244–262. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Stivers, J.T. Reconstructing the substrate for uracil DNA glycosylase: Tracking the transmission of binding energy in catalysis. Biochemistry 2001, 40, 7710–7719. [Google Scholar] [CrossRef]
- Mancuso, P.; Tricarico, R.; Bhattacharjee, V.; Cosentino, L.; Kadariya, Y.; Jelinek, J.; Nicolas, E.; Einarson, M.; Beeharry, N.; Devarajan, K.; et al. Thymine DNA glycosylase as a novel target for melanoma. Oncogene 2019, 38, 3710–3728. [Google Scholar] [CrossRef]
- Stanitsa, E.S.; Arps, L.; Traktman, P. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J. Biol. Chem. 2006, 281, 3439–3451. [Google Scholar] [CrossRef]
- Druck Shudofsky, A.M.; Silverman, J.E.Y.; Chattopadhyay, D.; Ricciardi, R.P. Vaccinia virus D4 mutants defective in processive DNA synthesis retain binding to A20 and DNA. J. Virol. 2010, 84, 12325–12335. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.T.; Upton, C.; Higman, M.A.; Niles, E.G.; McFadden, G. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J. Virol. 1993, 67, 2503–2512. [Google Scholar] [CrossRef]
- Millns, A.K.; Carpenter, M.S.; DeLange, A.M. The vaccinia virus-encoded uracil DNA glycosylase has an essential role in viral DNA replication. Virology 1994, 198, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Ellison, K.S.; Peng, W.; McFadden, G. Mutations in active-site residues of the uracil-DNA glycosylase encoded by vaccinia virus are incompatible with virus viability. J. Virol. 1996, 70, 7965–7973. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Sommers, C.I.; Prichard, M.N.; Keith, K.A.; Noah, J.W.; Nuth, M.; Ricciardi, R.P.; Chattopadhyay, D. Identification of protein-protein interaction inhibitors targeting vaccinia virus processivity factor for development of antiviral agents. Antimicrob. Agents Chemother. 2011, 55, 5054–5062. [Google Scholar] [CrossRef] [PubMed]
- Contesto-Richefeu, C.; Tarbouriech, N.; Brazzolotto, X.; Betzi, S.; Morelli, X.; Burmeister, W.P.; Iseni, F. Crystal structure of the vaccinia virus DNA polymerase holoenzyme subunit D4 in complex with the A20 N-terminal domain. PLoS Pathog. 2014, 10, e1003978. [Google Scholar] [CrossRef]
- Putnam, C.D.; Tainer, J.A. Protein mimicry of DNA and pathway regulation. DNA Repair 2005, 4, 1410–1420. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Banno, S.; Nishida, K.; Arazoe, T.; Mitsunobu, H.; Kondo, A. Deaminase-mediated multiplex genome editing in Escherichia Coli. Nat. Microbiol. 2018, 3, 423–429. [Google Scholar] [CrossRef]
- Venkatesh, J.; Kumar, P.; Krishna, P.S.M.; Manjunath, R.; Varshney, U. Importance of uracil DNA glycosylase in Pseudomonas aeruginosa and Mycobacterium smegmatis, G+C-rich bacteria, in mutation prevention, tolerance to acidified nitrite, and endurance in mouse macrophages. J. Biol. Chem. 2003, 278, 24350–24358. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, T.; Tuven, H.K.; Bjørås, M.; Rødland, E.A.; Tønjum, T. Genetic interactions of DNA repair pathways in the pathogen Neisseria meningitidis. J. Bacteriol. 2007, 189, 5728–5737. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.R.; Soliven, K.C.; Castor, M.E.; Barnes, P.D.; Libby, S.J.; Fang, F.C. The base excision repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog. 2009, 5, e1000451. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.J.; Kohanski, M.A.; Hayete, B.; Collins, J.J. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 2007, 3, 91. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.J.; Belenky, P.A.; Yang, J.H.; MacDonald, I.C.; Martell, J.D.; Takahashi, N.; Chan, C.T.Y.; Lobritz, M.A.; Braff, D.; Schwarz, E.G.; et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. USA 2014, 111, E2100–E2109. [Google Scholar] [CrossRef] [PubMed]
- Gusarov, I.; Shatalin, K.; Starodubtseva, M.; Nudler, E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 2009, 325, 1380–1384. [Google Scholar] [CrossRef]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 2008, 135, 679–690. [Google Scholar] [CrossRef]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kottur, J.; Nair, D.T. Reactive oxygen species play an important role in the bactericidal activity of quinolone antibiotics. Angew. Chem. Int. Ed. 2016, 55, 2397–2400. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Gruber, C.C.; Yang, J.H.; Liu, X.; Braff, D.; Yashaswini, C.N.; Bhubhanil, S.; Furuta, Y.; Andreescu, S.; Collins, J.J.; et al. Lethality of MalE-LacZ hybrid protein shares mechanistic attributes with oxidative component of antibiotic lethality. Proc. Natl. Acad. Sci. USA 2017, 114, 9164–9169. [Google Scholar] [CrossRef]
- Kang, T.M.; Yuan, J.; Nguyen, A.; Becket, E.; Yang, H.; Miller, J.H. The aminoglycoside antibiotic kanamycin damages DNA bases in Escherichia coli: Caffeine potentiates the DNA-damaging effects of kanamycin while suppressing cell killing by ciprofloxacin in Escherichia coli and Bacillus anthracis. Antimicrob. Agents Chemother. 2012, 56, 3216–3223. [Google Scholar] [CrossRef]
- Ohnishi, S.; Murata, M.; Ida, N.; Oikawa, S.; Kawanishi, S. Oxidative DNA damage induced by metabolites of chloramphenicol, an antibiotic drug. Free Radic. Res. 2015, 49, 1165–1172. [Google Scholar] [CrossRef]
- Michaels, M.L.; Tchou, J.; Grollman, A.P.; Miller, J.H. A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry 1992, 31, 10964–10968. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Tajiri, T.; Maki, H.; Sekiguchi, M. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 1995, 336, 257–267. [Google Scholar] [CrossRef]
- Moriya, M.; Ou, C.; Bodepudi, V.; Johnson, F.; Takeshita, M.; Grollman, A.P. Site-specific mutagenesis using a gapped duplex vector: A study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat. Res. 1991, 254, 281–288. [Google Scholar] [CrossRef]
- Wood, M.L.; Esteve, A.; Morningstar, M.L.; Kuziemko, G.M.; Essigmann, J.M. Genetic effects of oxidative DNA damage: Comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res. 1992, 20, 6023–6032. [Google Scholar] [CrossRef]
- Tchou, J.; Kasai, H.; Shibutani, S.; Chung, M.-H.; Laval, J.; Grollman, A.P.; Nishimura, S. 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, A.; Jaruga, P.; Bohr, V.A.; Grollman, A.P.; Dizdaroglu, M. Kinetics of excision of purine lesions from DNA by Escherichia coli Fpg protein. Nucleic Acids Res. 1997, 25, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Michaels, M.L.; Cruz, C.; Grollman, A.P.; Miller, J.H. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7022–7025. [Google Scholar] [CrossRef] [PubMed]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar] [CrossRef]
- Taddei, F.; Hayakawa, H.; Bouton, M.; Cirinesi, A.; Matic, I.; Sekiguchi, M.; Radman, M. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 1997, 278, 128–130. [Google Scholar] [CrossRef]
- Jiang, D.; Hatahet, Z.; Blaisdell, J.O.; Melamede, R.J.; Wallace, S.S. Escherichia coli endonuclease VIII: Cloning, sequencing and overexpression of the nei structural gene and characterization of nei and nei nth mutants. J. Bacteriol. 1997, 179, 3773–3782. [Google Scholar] [CrossRef]
- Jiang, D.; Hatahet, Z.; Melamede, R.J.; Kow, Y.W.; Wallace, S.S. Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem. 1997, 272, 32230–32239. [Google Scholar] [CrossRef]
- Blaisdell, J.O.; Hatahet, Z.; Wallace, S.S. A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G→T transversions. J. Bacteriol. 1999, 181, 6396–6402. [Google Scholar] [CrossRef]
- Kropachev, K.Y.; Zharkov, D.O.; Grollman, A.P. Catalytic mechanism of Escherichia coli endonuclease VIII: Roles of the intercalation loop and the zinc finger. Biochemistry 2006, 45, 12039–12049. [Google Scholar] [CrossRef]
- Breimer, L.H.; Lindahl, T. DNA glycosylase activities for thymine residues damaged by ring saturation, fragmentation, or ring contraction are functions of endonuclease III in Escherichia coli. J. Biol. Chem. 1984, 259, 5543–5548. [Google Scholar]
- Dizdaroglu, M.; Laval, J.; Boiteux, S. Substrate specificity of the Escherichia coli endonuclease III: Excision of thymine- and cytosine-derived lesions in DNA produced by radiation-generated free radicals. Biochemistry 1993, 32, 12105–12111. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Matsui, K.; Yamada, M.; Kasai, H.; Sofuni, T.; Nohmi, T. Construction of mutants of Salmonella typhimurium deficient in 8-hydroxyguanine DNA glycosylase and their sensitivities to oxidative mutagens and nitro compounds. Mutat. Res. 1997, 393, 233–246. [Google Scholar] [CrossRef]
- Nagorska, K.; Silhan, J.; Li, Y.; Pelicic, V.; Freemont, P.S.; Baldwin, G.S.; Tang, C.M. A network of enzymes involved in repair of oxidative DNA damage in Neisseria meningitidis. Mol. Microbiol. 2012, 83, 1064–1079. [Google Scholar] [CrossRef]
- Souza Arantes, L.; Gonçalves Vila Nova, L.; Resende, B.C.; Bitar, M.; Vale Coelho, I.E.; Miyoshi, A.; Azevedo, V.A.; dos Santos, L.L.; Machado, C.R.; de Oliveira Lopes, D. The Corynebacterium pseudotuberculosis genome contains two formamidopyrimidine-DNA glycosylase enzymes, only one of which recognizes and excises 8-oxoguanine lesion. Gene 2016, 575, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Kumar, P.; Varshney, U. A distinct role of formamidopyrimidine DNA glycosylase (MutM) in down-regulation of accumulation of G, C mutations and protection against oxidative stress in mycobacteria. DNA Repair 2007, 6, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Rot, M.A.; Filipenko, M.L.; Nevinsky, G.A.; Zharkov, D.O. Novel DNA glycosylases from Mycobacterium tuberculosis. Biochemistry (Mosc) 2008, 73, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Balasingham, S.V.; Davidsen, T.; Debebe, E.; Rødland, E.A.; van Soolingen, D.; Kremer, K.; Alseth, I.; Tønjum, T. Characterization of the major formamidopyrimidine-DNA glycosylase homolog in Mycobacterium tuberculosis and its linkage to variable tandem repeats. FEMS Immunol. Med. Microbiol. 2009, 56, 151–161. [Google Scholar] [CrossRef]
- Guo, Y.; Bandaru, V.; Jaruga, P.; Zhao, X.; Burrows, C.J.; Iwai, S.; Dizdaroglu, M.; Bond, J.P.; Wallace, S.S. The oxidative DNA glycosylases of Mycobacterium tuberculosis exhibit different substrate preferences from their Escherichia coli counterparts. DNA Repair 2010, 9, 177–190. [Google Scholar] [CrossRef]
- Davidsen, T.; Bjørås, M.; Seeberg, E.C.; Tønjum, T. Antimutator role of DNA glycosylase MutY in pathogenic Neisseria species. J. Bacteriol. 2005, 187, 2801–2809. [Google Scholar] [CrossRef]
- Eutsey, R.; Wang, G.; Maier, R.J. Role of a MutY DNA glycosylase in combating oxidative DNA damage in Helicobacter pylori. DNA Repair 2007, 6, 19–26. [Google Scholar] [CrossRef]
- Eberle, R.J.; Coronado, M.A.; Caruso, I.P.; Lopes, D.O.; Miyoshi, A.; Azevedo, V.; Arni, R.K. Chemical and thermal influence of the [4Fe–4S]2+ cluster of A/G-specific adenine glycosylase from Corynebacterium pseudotuberculosis. Biochim. Biophys. Acta 2015, 1850, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Cançado de Faria, R.; Gonçalves Vila-Nova, L.; Bitar, M.; Carvalho Resende, B.; Sousa Arantes, L.; Basso Rebelato, A.; Carvalho Azevedo, V.A.; Franco, G.R.; Machado, C.R.; dos Santos, L.L.; et al. Adenine glycosylase MutY of Corynebacterium pseudotuberculosis presents the antimutator phenotype and evidences of glycosylase/AP lyase activity in vitro. Infect. Genet. Evol. 2016, 44, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.; Sánchez, J.M.; Blázquez, J. Characterization of the GO system of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2002, 217, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; Sudhakaran, J.; Sutton, M.D. The GO system prevents ROS-induced mutagenesis and killing in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2009, 294, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, T.; Amundsen, E.K.; Rødland, E.A.; Tønjum, T. DNA repair profiles of disease-associated isolates of Neisseria meningitidis. FEMS Immunol. Med. Microbiol. 2007, 49, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kurthkoti, K.; Srinath, T.; Kumar, P.; Malshetty, V.S.; Sang, P.B.; Jain, R.; Manjunath, R.; Varshney, U. A distinct physiological role of MutY in mutation prevention in mycobacteria. Microbiology 2010, 156, 88–93. [Google Scholar] [CrossRef]
- Hassim, F.; Papadopoulos, A.O.; Kana, B.D.; Gordhan, B.G. A combinatorial role for MutY and Fpg DNA glycosylases in mutation avoidance in Mycobacterium smegmatis. Mutat. Res. 2015, 779, 24–32. [Google Scholar] [CrossRef]
- Canfield, G.S.; Schwingel, J.M.; Foley, M.H.; Vore, K.L.; Boonanantanasarn, K.; Gill, A.L.; Sutton, M.D.; Gill, S.R. Evolution in fast forward: A potential role for mutators in accelerating Staphylococcus aureus pathoadaptation. J. Bacteriol. 2013, 195, 615–628. [Google Scholar] [CrossRef]
- Eskra, L.; Canavessi, A.; Carey, M.; Splitter, G. Brucella abortus genes identified following constitutive growth and macrophage infection. Infect. Immun. 2001, 69, 7736–7742. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mehra, S.; Didier, P.J.; Roy, C.J.; Doyle, L.A.; Alvarez, X.; Ratterree, M.; Be, N.A.; Lamichhane, G.; Jain, S.K.; et al. Genetic requirements for the survival of tubercle bacilli in primates. J. Infect. Dis. 2010, 201, 1743–1752. [Google Scholar] [CrossRef]
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Mandsberg, L.F.; Ciofu, O.; Kirkby, N.; Christiansen, L.E.; Poulsen, H.E.; Høiby, N. Antibiotic resistance in Pseudomonas aeruginosa strains with increased mutation frequency due to inactivation of the DNA oxidative repair system. Antimicrob. Agents Chemother. 2009, 53, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Couce, A.; Alonso-Rodriguez, N.; Costas, C.; Oliver, A.; Blázquez, J. Intrapopulation variability in mutator prevalence among urinary tract infection isolates of Escherichia coli. Clin. Microbiol. Infect. 2016, 22, 566. [Google Scholar] [CrossRef] [PubMed]
- Perrin, A.; Larsonneur, E.; Nicholson, A.C.; Edwards, D.J.; Gundlach, K.M.; Whitney, A.M.; Gulvik, C.A.; Bell, M.E.; Rendueles, O.; Cury, J.; et al. Evolutionary dynamics and genomic features of the Elizabethkingia anophelis 2015 to 2016 Wisconsin outbreak strain. Nat. Commun. 2017, 8, 15483. [Google Scholar] [CrossRef] [PubMed]
- Chetsanga, C.J.; Lozon, M.; Makaroff, C.; Savage, L. Purification and characterization of Escherichia coli formamidopyrimidine-DNA glycosylase that excises damaged 7-methylguanine from deoxyribonucleic acid. Biochemistry 1981, 20, 5201–5207. [Google Scholar] [CrossRef]
- Wink, D.A.; Laval, J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis 1994, 15, 2125–2129. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Nishioka, N.; Nakabeppu, Y.; Gores, G.J. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001, 61, 6388–6393. [Google Scholar]
- Rogers, P.A.; Eide, L.; Klungland, A.; Ding, H. Reversible inactivation of E. coli endonuclease III via modification of its [4Fe-4S] cluster by nitric oxide. DNA Repair 2003, 2, 809–817. [Google Scholar] [CrossRef]
- Moritz, E.; Pauly, K.; Bravard, A.; Hall, J.; Radicella, J.P.; Epe, B. hOGG1-Cys326 variant cells are hypersensitive to DNA repair inhibition by nitric oxide. Carcinogenesis 2014, 35, 1426–1433. [Google Scholar] [CrossRef]
- Mikhailov, A.A.; Khantakova, D.V.; Nichiporenko, V.A.; Glebov, E.M.; Grivin, V.P.; Plyusnin, V.F.; Yanshole, V.V.; Petrova, D.V.; Kostin, G.A.; Grin, I.R. Photoinduced inhibition of DNA repair enzymes and the possible mechanism of photochemical transformations of the ruthenium nitrosyl complex [RuNO(β-Pic)2(NO2)2OH]. Metallomics 2019, 11, 1999–2009. [Google Scholar] [CrossRef]
- Kobune, M.; Xu, Y.; Baum, C.; Kelley, M.R.; Williams, D.A. Retrovirus-mediated expression of the base excision repair proteins, formamidopyrimidine DNA glycosylase or human oxoguanine DNA glycosylase, protects hematopoietic cells from N,N’,N’’-triethylenethiophosphoramide (thioTEPA)-induced toxicity in vitro and in vivo. Cancer Res. 2001, 61, 5116–5125. [Google Scholar] [PubMed]
- Xu, Y.; Hansen, W.K.; Rosenquist, T.A.; Williams, D.A.; Limp-Foster, M.; Kelley, M.R. Protection of mammalian cells against chemotherapeutic agents thiotepa, 1,3-N,N’-bis(2-chloroethyl)-N-nitrosourea, and mafosfamide using the DNA base excision repair genes Fpg and α-hOgg1: Implications for protective gene therapy applications. J. Pharmacol. Exp. Ther. 2001, 296, 825–831. [Google Scholar] [PubMed]
- He, Y.-H.; Xu, Y.; Kobune, M.; Wu, M.; Kelley, M.R.; Martin, W.J., II. Escherichia coli FPG and human OGG1 reduce DNA damage and cytotoxicity by BCNU in human lung cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L50–L55. [Google Scholar] [CrossRef] [PubMed]
- Preston, T.J.; Henderson, J.T.; McCallum, G.P.; Wells, P.G. Base excision repair of reactive oxygen species-initiated 7,8-dihydro-8-oxo-2’-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol. Cancer Ther. 2009, 8, 2015–2026. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, Z.; Che, W. Suppression of a DNA base excision repair gene, hOGG1, increases bleomycin sensitivity of human lung cancer cell line. Toxicol. Appl. Pharmacol. 2008, 228, 395–402. [Google Scholar] [CrossRef]
- Ramdzan, Z.M.; Ginjala, V.; Pinder, J.B.; Chung, D.; Donovan, C.M.; Kaur, S.; Leduy, L.; Dellaire, G.; Ganesan, S.; Nepveu, A. The DNA repair function of CUX1 contributes to radioresistance. Oncotarget 2017, 8, 19021–19038. [Google Scholar] [CrossRef]
- Chakrabarti, G.; Silvers, M.A.; Ilcheva, M.; Liu, Y.; Moore, Z.R.; Luo, X.; Gao, J.; Anderson, G.; Liu, L.; Sarode, V.; et al. Tumor-selective use of DNA base excision repair inhibition in pancreatic cancer using the NQO1 bioactivatable drug, β-lapachone. Sci. Rep. 2015, 5, 17066. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Tsuzuki, T. Oxidative nucleotide damage: Consequences and prevention. Oncogene 2002, 21, 8895–8904. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Warpman Berglund, U.; Sanjiv, K.; Gad, H.; Kalderén, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef]
- Qing, X.; Shao, Z.; Lv, X.; Pu, F.; Gao, F.; Liu, L.; Shi, D. Anticancer effect of (S)-crizotinib on osteosarcoma cells by targeting MTH1 and activating reactive oxygen species. Anticancer. Drugs 2018, 29, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Pan, D.; Shi, D.; Bai, Q.; Liu, H.; Yao, X. Influence of chirality of crizotinib on its MTH1 protein inhibitory activity: Insight from molecular dynamics simulations and binding free energy calculations. PLoS ONE 2015, 10, e0145219. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Chen, P.; Li, D.; Li, Y.; Hou, T. Directly binding rather than induced-fit dominated binding affinity difference in (S)- and (R)-crizotinib bound MTH1. J. Chem. Theory Comput. 2016, 12, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Guo, G.; Zou, P.; Cui, R.; Chen, W.; Chen, X.; Yin, C.; He, W.; Vinothkumar, R.; Yang, F.; et al. (S)-crizotinib induces apoptosis in human non-small cell lung cancer cells by activating ROS independent of MTH1. J. Exp. Clin. Cancer Res. 2017, 36, 120. [Google Scholar] [CrossRef]
- Ji, J.; Chen, W.; Lian, W.; Chen, R.; Yang, J.; Zhang, Q.; Weng, Q.; Hu, Z.K.J.; Chen, X.; Zou, P.; et al. (S)-crizotinib reduces gastric cancer growth through oxidative DNA damage and triggers pro-survival akt signal. Cell Death Dis. 2018, 9, 660. [Google Scholar] [CrossRef]
- Van der Waals, L.M.; Laoukili, J.; Jongen, J.M.J.; Raats, D.A.; Borel Rinkes, I.H.M.; Kranenburg, O. Differential anti-tumour effects of MTH1 inhibitors in patient-derived 3D colorectal cancer cultures. Sci. Rep. 2019, 9, 819. [Google Scholar] [CrossRef]
- Samaranayake, G.J.; Huynh, M.; Rai, P. MTH1 as a chemotherapeutic target: The elephant in the room. Cancers 2017, 9, 47. [Google Scholar] [CrossRef]
- Einarsdottir, B.O.; Karlsson, J.; Söderberg, E.M.V.; Lindberg, M.F.; Funck-Brentano, E.; Jespersen, H.; Brynjolfsson, S.F.; Olofsson Bagge, R.; Carstam, L.; Scobie, M.; et al. A patient-derived xenograft pre-clinical trial reveals treatment responses and a resistance mechanism to karonudib in metastatic melanoma. Cell Death Dis. 2018, 9, 810. [Google Scholar] [CrossRef]
- Hua, X.; Sanjiv, K.; Gad, H.; Pham, T.; Gokturk, C.; Rasti, A.; Zhao, Z.; He, K.; Feng, M.; Zang, Y.; et al. Karonudib is a promising anticancer therapy in hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919866960. [Google Scholar] [CrossRef]
- Morland, I.; Luna, L.; Gustad, E.; Seeberg, E.; Bjørås, M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair 2005, 4, 381–387. [Google Scholar] [CrossRef]
- Mahajan, T.R.; Ytre-Arne, M.E.; Strøm-Andersen, P.; Dalhus, B.; Gundersen, L.-L. Synthetic routes to N-9 alkylated 8-oxoguanines; weak inhibitors of the human DNA glycosylase OGG1. Molecules 2015, 20, 15944–15965. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Visnes, T.; Homan, E.J.; Seashore-Ludlow, B.; Hedenström, M.; Wiita, E.; Vallin, K.; Paulin, C.B.J.; Zhang, J.; Wallner, O.; et al. Computational and experimental druggability assessment of human DNA glycosylases. ACS Omega 2019, 4, 11642–11656. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Zaika, E.; Fernandes, A.S.; Zharkov, D.O.; Miller, H.; Grollman, A.P. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 2003, 2, 581–591. [Google Scholar] [CrossRef]
- Taricani, L.; Shanahan, F.; Pierce, R.H.; Guzi, T.J.; Parry, D. Phenotypic enhancement of thymidylate synthetase pathway inhibitors following ablation of Neil1 DNA glycosylase/lyase. Cell Cycle 2010, 9, 4876–4883. [Google Scholar] [CrossRef][Green Version]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabó, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [CrossRef]
- Touati, E.; Michel, V.; Thiberge, J.-M.; Avé, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef]
- Li, G.; Yuan, K.; Yan, C.; Fox, J., III; Gaid, M.; Breitwieser, W.; Bansal, A.K.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, A.; Aguilera-Aguirre, L.; Szczesny, B.; Radak, Z.; Hazra, T.K.; Sur, S.; Ba, X.; Boldogh, I. Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair 2013, 12, 18–26. [Google Scholar] [CrossRef]
- Kunisada, M.; Yogianti, F.; Sakumi, K.; Ono, R.; Nakabeppu, Y.; Nishigori, C. Increased expression of versican in the inflammatory response to UVB- and reactive oxygen species-induced skin tumorigenesis. Am. J. Pathol. 2011, 179, 3056–3065. [Google Scholar] [CrossRef]
- Yogianti, F.; Kunisada, M.; Nakano, E.; Ono, R.; Sakumi, K.; Oka, S.; Nakabeppu, Y.; Nishigori, C. Inhibitory effects of dietary Spirulina platensis on UVB-induced skin inflammatory responses and carcinogenesis. J. Invest. Dermatol. 2014, 134, 2610–2619. [Google Scholar] [CrossRef]
- Ye, Y.; Lin, P.; Zhang, W.; Tan, S.; Zhou, X.; Li, R.; Pu, Q.; Koff, J.L.; Dhasarathy, A.; Ma, F.; et al. DNA repair interacts with autophagy to regulate inflammatory responses to pulmonary hyperoxia. J. Immunol. 2017, 198, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Hajas, G.; Aguilera-Aguirre, L.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Sur, S.; Hazra, T.K.; Mitra, S. Activation of Ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 2012, 287, 20769–20773. [Google Scholar] [CrossRef] [PubMed]
- German, P.; Szaniszlo, P.; Hajas, G.; Radak, Z.; Bacsi, A.; Hazra, T.K.; Hegde, M.L.; Ba, X.; Boldogh, I. Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair 2013, 12, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1–KRAS–NF-κB pathway. J. Immunol. 2014, 193, 4643–4653. [Google Scholar] [CrossRef]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor κB-driven gene expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Abbasi, A.A.; Boldogh, I.; Ba, X. OGG1-DNA interactions facilitate NF-κB binding to DNA targets. Sci. Rep. 2017, 7, 43297. [Google Scholar] [CrossRef]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef]
- Li, H.; Endutkin, A.V.; Bergonzo, C.; Fu, L.; Grollman, A.P.; Zharkov, D.O.; Simmerling, C. DNA deformation-coupled recognition of 8-oxoguanine: Conformational kinetic gating in human DNA glycosylase. J. Am. Chem. Soc. 2017, 139, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Kennedy, L.; Evans, E.; Chen, C.-M.; Craven, L.; Detloff, P.J.; Ennis, M.; Shelbourne, P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003, 12, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Møllersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Goula, A.-V.; Berquist, B.R.; Wilson, D.M., III; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009, 5, e1000749. [Google Scholar] [CrossRef]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Peña, S.; McMurray, C.T. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. 2015, 11, e1005267. [Google Scholar] [CrossRef]
- Bobola, M.S.; Kolstoe, D.D.; Blank, A.; Chamberlain, M.C.; Silber, J.R. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front. Oncol. 2012, 2, 176. [Google Scholar] [CrossRef]
- Fishel, M.L.; Seo, Y.R.; Smith, M.L.; Kelley, M.R. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003, 63, 608–615. [Google Scholar]
- Rinne, M.; Caldwell, D.; Kelley, M.R. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol. Cancer Ther. 2004, 3, 955–967. [Google Scholar]
- Fishel, M.L.; He, Y.; Smith, M.L.; Kelley, M.R. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin. Cancer Res. 2007, 13, 260–267. [Google Scholar] [CrossRef]
- Tang, J.-b.; Svilar, D.; Trivedi, R.N.; Wang, X.-H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase β modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011, 13, 471–486. [Google Scholar] [CrossRef]
- Song, S.; Xing, G.; Yuan, L.; Wang, J.; Wang, S.; Yin, Y.; Tian, C.; He, F.; Zhang, L. N-methylpurine DNA glycosylase inhibits p53-mediated cell cycle arrest and coordinates with p53 to determine sensitivity to alkylating agents. Cell Res. 2012, 22, 1285–1303. [Google Scholar] [CrossRef][Green Version]
- Leguisamo, N.M.; Gloria, H.C.; Kalil, A.N.; Martins, T.V.; Azambuja, D.B.; Meira, L.B.; Saffi, J. Base excision repair imbalance in colorectal cancer has prognostic value and modulates response to chemotherapy. Oncotarget 2017, 8, 54199–54214. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.L.; Mi, L.-J.; Chaung, W.; Teebor, G.; Boorstein, R.J. 5-Chloro-2’-deoxyuridine cytotoxicity results from base excision repair of uracil subsequent to thymidylate synthase inhibition. Mutat. Res. 2000, 459, 161–169. [Google Scholar] [CrossRef]
- Turner, D.P.; Cortellino, S.; Schupp, J.E.; Caretti, E.; Loh, T.; Kinsella, T.J.; Bellacosa, A. The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 2006, 66, 7686–7693. [Google Scholar] [CrossRef] [PubMed]
- Kunz, C.; Focke, F.; Saito, Y.; Schuermann, D.; Lettieri, T.; Selfridge, J.; Schär, P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009, 7, e91. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Emura, T.; Fukushima, M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int. J. Oncol. 2011, 39, 263–270. [Google Scholar] [CrossRef]
- Paik, J.; Duncan, T.; Lindahl, T.; Sedgwick, B. Sensitization of human carcinoma cells to alkylating agents by small interfering RNA suppression of 3-alkyladenine-DNA glycosylase. Cancer Res. 2005, 65, 10472–10477. [Google Scholar] [CrossRef]
- Allan, J.M.; Engelward, B.P.; Dreslin, A.J.; Wyatt, M.D.; Tomasz, M.; Samson, L.D. Mammalian 3-methyladenine DNA glycosylase protects against the toxicity and clastogenicity of certain chemotherapeutic DNA cross-linking agents. Cancer Res. 1998, 58, 3965–3973. [Google Scholar]
- Sorribes, I.C.; Handelman, S.K.; Jain, H.V. Mitigating temozolomide resistance in glioblastoma via DNA damage-repair inhibition. J. R. Soc. Interface 2020, 17, 20190722. [Google Scholar] [CrossRef]
- Goellner, E.M.; Grimme, B.; Brown, A.R.; Lin, Y.-C.; Wang, X.-H.; Sugrue, K.F.; Mitchell, L.; Trivedi, R.N.; Tang, J.-B.; Sobol, R.W. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 2011, 71, 2308–2317. [Google Scholar] [CrossRef]
- Lorenzi, P.L.; Landowski, C.P.; Brancale, A.; Song, X.; Townsend, L.B.; Drach, J.C.; Amidon, G.L. N-methylpurine DNA glycosylase and 8-oxoguanine DNA glycosylase metabolize the antiviral nucleoside 2-bromo-5,6-dichloro-1-(β-D-ribofuranosyl)benzimidazole. Drug Metab. Dispos. 2006, 34, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Hjelmgren, T.; Lindahl, T. Induction of a DNA glycosylase for N-methylated purines is part of the adaptive response to alkylating agents. Nature 1982, 296, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Bjelland, S.; Bjørås, M.; Seeberg, E. Excision of 3-methylguanine from alkylated DNA by 3-methyladenine DNA glycosylase I of Escherichia coli. Nucleic Acids Res. 1993, 21, 2045–2049. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bjelland, S.; Seeberg, E. Purification and characterization of 3-methyladenine DNA glycosylase I from Escherichia coli. Nucleic Acids Res. 1987, 15, 2787–2801. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Athar, A.; Ahmed, Z.; Lali, S.M.; Sohail, A. DNA glycosylase enzymes induced during chemical adaptation of M. luteus. Nucleic Acids Res. 1987, 15, 6607–6624. [Google Scholar] [CrossRef][Green Version]
- Roy, R.; Brooks, C.; Mitra, S. Purification and biochemical characterization of recombinant N-methylpurine-DNA glycosylase of the mouse. Biochemistry 1994, 33, 15131–15140. [Google Scholar] [CrossRef]
- Tudek, B.; VanZeeland, A.A.; Kusmierek, J.T.; Laval, J. Activity of Escherichia coli DNA-glycosylases on DNA damaged by methylating and ethylating agents and influence of 3-substituted adenine derivatives. Mutat. Res. 1998, 407, 169–176. [Google Scholar] [CrossRef]
- Drohat, A.C.; Kwon, K.; Krosky, D.J.; Stivers, J.T. 3-methyladenine DNA glycosylase I is an unexpected helix-hairpin-helix superfamily member. Nat. Struct. Biol. 2002, 9, 659–664. [Google Scholar] [CrossRef]
- Rajesh, S.S.; Sivaraman, T. Cheminformatic designing of de novo inhibitors to 3-methyl adenine DNA glycosylase I (LiTagA) from Leptospira interrogans serovar lai strain 56601. Med. Chem. Res. 2013, 22, 3434–3443. [Google Scholar] [CrossRef]
- Stivers, J.T. 2-Aminopurine fluorescence studies of base stacking interactions at abasic sites in DNA: Metal-ion and base sequence effects. Nucleic Acids Res. 1998, 26, 3837–3844. [Google Scholar] [CrossRef]
- Stivers, J.T.; Pankiewicz, K.W.; Watanabe, K.A. Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1999, 38, 952–963. [Google Scholar] [CrossRef]
- Maksimenko, A.; Ishchenko, A.A.; Sanz, G.; Laval, J.; Elder, R.H.; Saparbaev, M.K. A molecular beacon assay for measuring base excision repair activities. Biochem. Biophys. Res. Commun. 2004, 319, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Mirbahai, L.; Kershaw, R.M.; Green, R.M.; Hayden, R.E.; Meldrum, R.A.; Hodges, N.J. Use of a molecular beacon to track the activity of base excision repair protein OGG1 in live cells. DNA Repair 2010, 9, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Long, Y.; Liu, B.; Xiang, D.; Zhu, H. Real time monitoring uracil excision using uracil-containing molecular beacons. Anal. Chim. Acta 2014, 819, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, M.-H.; Li, Y.; Tang, B.; Zhang, C.-Y. Simultaneous sensitive detection of multiple DNA glycosylases from lung cancer cells at the single-molecule level. Chem. Sci. 2018, 9, 712–720. [Google Scholar] [CrossRef]
- Sauvaigo, S.; Guerniou, V.; Rapin, D.; Gasparutto, D.; Caillat, S.; Favier, A. An oligonucleotide microarray for the monitoring of repair enzyme activity toward different DNA base damage. Anal. Biochem. 2004, 333, 182–192. [Google Scholar] [CrossRef]
- Gines, G.; Saint-Pierre, C.; Gasparutto, D. On-bead fluorescent DNA nanoprobes to analyze base excision repair activities. Anal. Chim. Acta 2014, 812, 168–175. [Google Scholar] [CrossRef]
- Gines, G.; Saint-Pierre, C.; Gasparutto, D. A multiplex assay based on encoded microbeads conjugated to DNA NanoBeacons to monitor base excision repair activities by flow cytometry. Biosens. Bioelectron. 2014, 58, 81–84. [Google Scholar] [CrossRef]
- Flaender, M.; Costa, G.; Nonglaton, G.; Saint-Pierre, C.; Gasparutto, D. A DNA array based on clickable lesion-containing hairpin probes for multiplexed detection of base excision repair activities. Analyst 2016, 141, 6208–6216. [Google Scholar] [CrossRef]
- Hölz, K.; Pavlic, A.; Lietard, J.; Somoza, M.M. Specificity and efficiency of the uracil DNA glycosylase-mediated strand cleavage surveyed on large sequence libraries. Sci. Rep. 2019, 9, 17822. [Google Scholar] [CrossRef]
- Wang, L.-J.; Ma, F.; Tang, B.; Zhang, C.-Y. Base-excision-repair-induced construction of a single quantum-dot-based sensor for sensitive detection of DNA glycosylase activity. Anal. Chem. 2016, 88, 7523–7529. [Google Scholar] [CrossRef]
- Xiang, Y.; Lu, Y. Expanding targets of DNAzyme-based sensors through deactivation and activation of DNAzymes by single uracil removal: Sensitive fluorescent assay of uracil-DNA glycosylase. Anal. Chem. 2012, 84, 9981–9987. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, Y.; Wang, L.; Zhu, J.; Jiang, W. A DNA machine-based fluorescence amplification strategy for sensitive detection of uracil-DNA glycosylase activity. Biosens. Bioelectron. 2015, 68, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-J.; Hu, D.-P.; Deng, Q.; Wang, Z.-Y.; Huang, B.-H.; Fang, Y.-X.; Zhang, K.; Wong, W.-L. Sensitive and selective detection of uracil-DNA glycosylase activity with a new pyridinium luminescent switch-on molecular probe. Analyst 2015, 140, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, K.; Liu, H.; Xia, K.; Wang, K.; Wang, J. Label-free fluorescence turn-on detection of uracil DNA glycosylase activity based on G-quadruplex formation. Talanta 2016, 160, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, D.; Tang, H.; Liang, M.; Jiang, J. A sensitive, homogeneous fluorescence assay for detection of thymine DNA glycosylase activity based on exonuclease-mediated amplification. Chem. Commun. 2013, 49, 5874–5876. [Google Scholar] [CrossRef]
- Wang, X.; Hou, T.; Lu, T.; Li, F. Autonomous exonuclease III-assisted isothermal cycling signal amplification: A facile and highly sensitive fluorescence DNA glycosylase activity assay. Anal. Chem. 2014, 86, 9626–9631. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, Y.; Kong, R.; Zhang, L.; Yang, W.; Zhao, S. Tungsten disulfide nanosheet and exonuclease III co-assisted amplification strategy for highly sensitive fluorescence polarization detection of DNA glycosylase activity. Anal. Chim. Acta 2015, 887, 216–223. [Google Scholar] [CrossRef]
- Wu, Y.; Yan, P.; Xu, X.; Jiang, W. A unique dual recognition hairpin probe mediated fluorescence amplification method for sensitive detection of uracil-DNA glycosylase and endonuclease IV activities. Analyst 2016, 141, 1789–1795. [Google Scholar] [CrossRef]
- Wang, L.-J.; Wang, Z.-Y.; Zhang, Q.; Tang, B.; Zhang, C.-Y. Cyclic enzymatic repairing-mediated dual-signal amplification for real-time monitoring of thymine DNA glycosylase. Chem. Commun. 2017, 53, 3878–3881. [Google Scholar] [CrossRef]
- Song, J.; Yin, F.; Li, X.; Dong, N.; Zhu, Y.; Shao, Y.; Chen, B.; Jiang, W.; Li, C.-Z. Sensitive detection of formamidopyrimidine-DNA glycosylase activity based on target-induced self-primed rolling circle amplification and magnetic nanoprobes. Analyst 2018, 143, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Liu, S.; Wang, H.; Zhang, X.; Song, X.; Huang, J. Base excision repair initiated rolling circle amplification-based fluorescent assay for screening uracil-DNA glycosylase activity using Endo IV-assisted cleavage of AP probes. Analyst 2018, 143, 3951–3958. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Li, J.; Xiao, F.; Yu, R.; Jiang, J. A label-free and highly sensitive strategy for uracil-DNA glycosylase activity detection based on stem-loop primer-mediated exponential amplification (SPEA). Anal. Chim. Acta 2017, 991, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Ren, M.; Zhang, Q.; Tang, B.; Zhang, C.-Y. Excision repair-initiated enzyme-assisted bicyclic cascade signal amplification for ultrasensitive detection of uracil-DNA glycosylase. Anal. Chem. 2017, 89, 4488–4494. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, M.; Wei, J.; Liu, X.; Wang, F. A C-HCR assembly of branched DNA nanostructures for amplified uracil-DNA glycosylase assays. Chem. Commun. 2017, 53, 12878–12881. [Google Scholar] [CrossRef] [PubMed]










































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).