CRISPR/Cas9 Editing for Gaucher Disease Modelling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
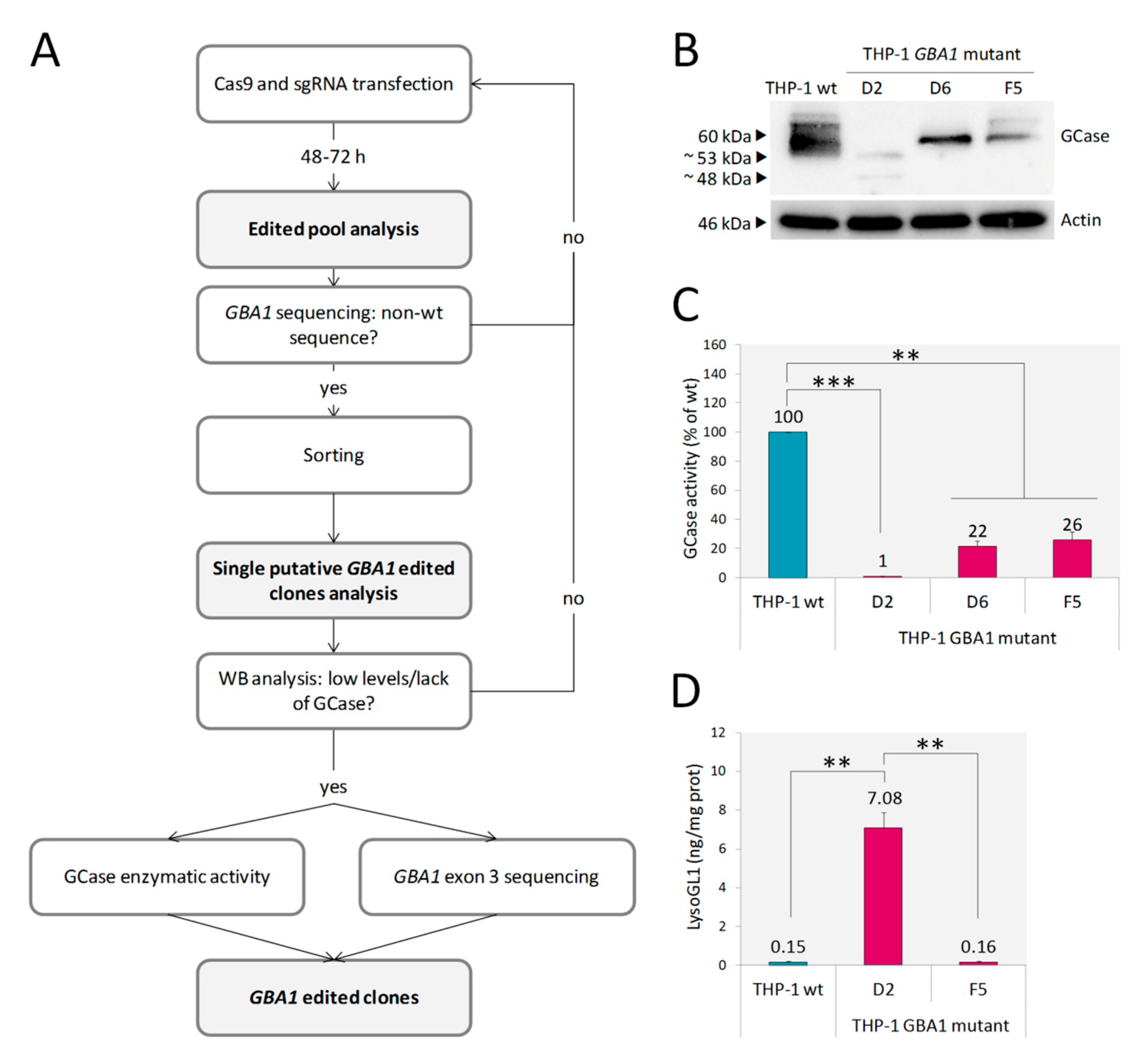
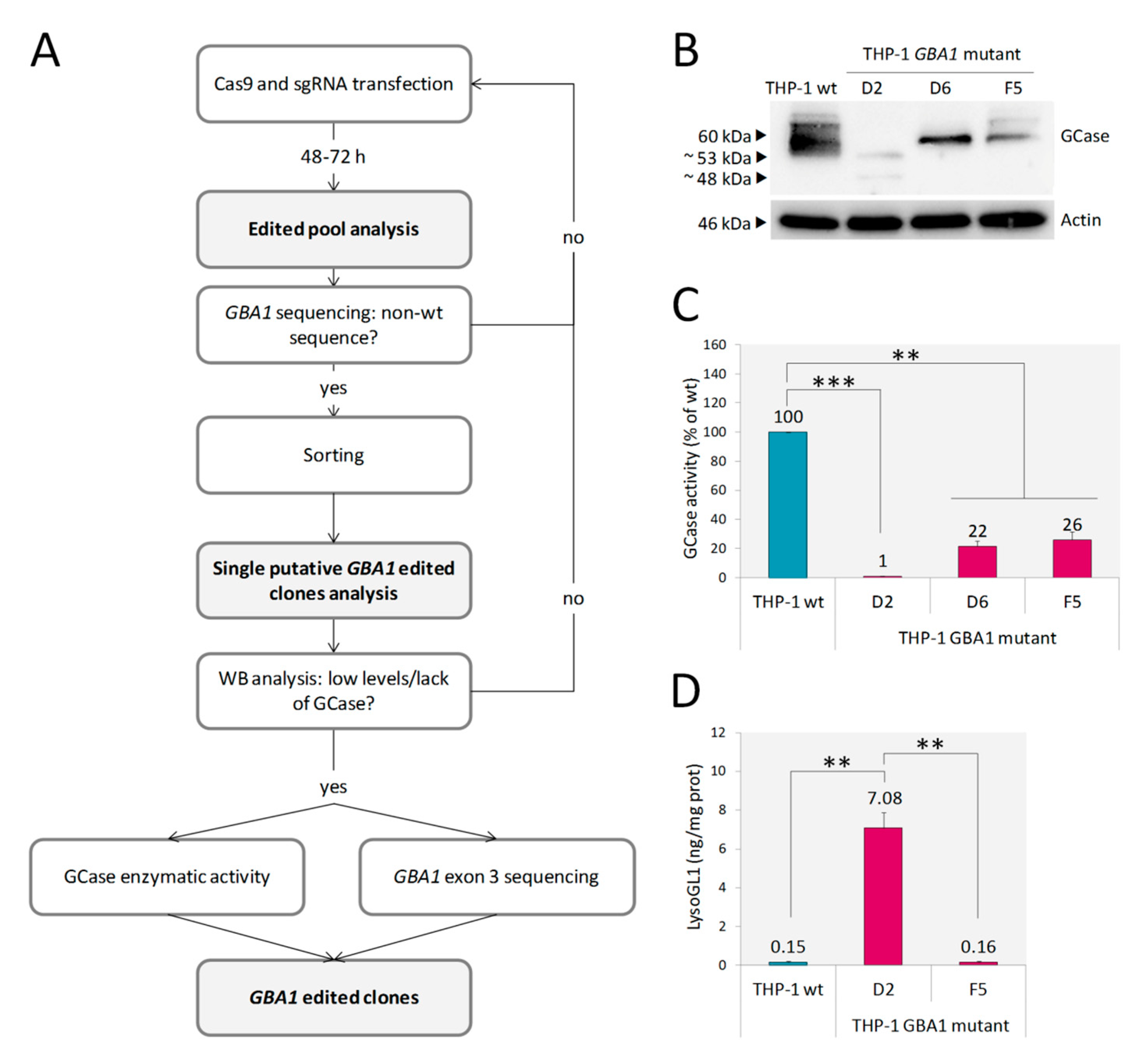
2.1. Monocytic GBA1 Mutant Cells: A Promising Cellular Model of GD
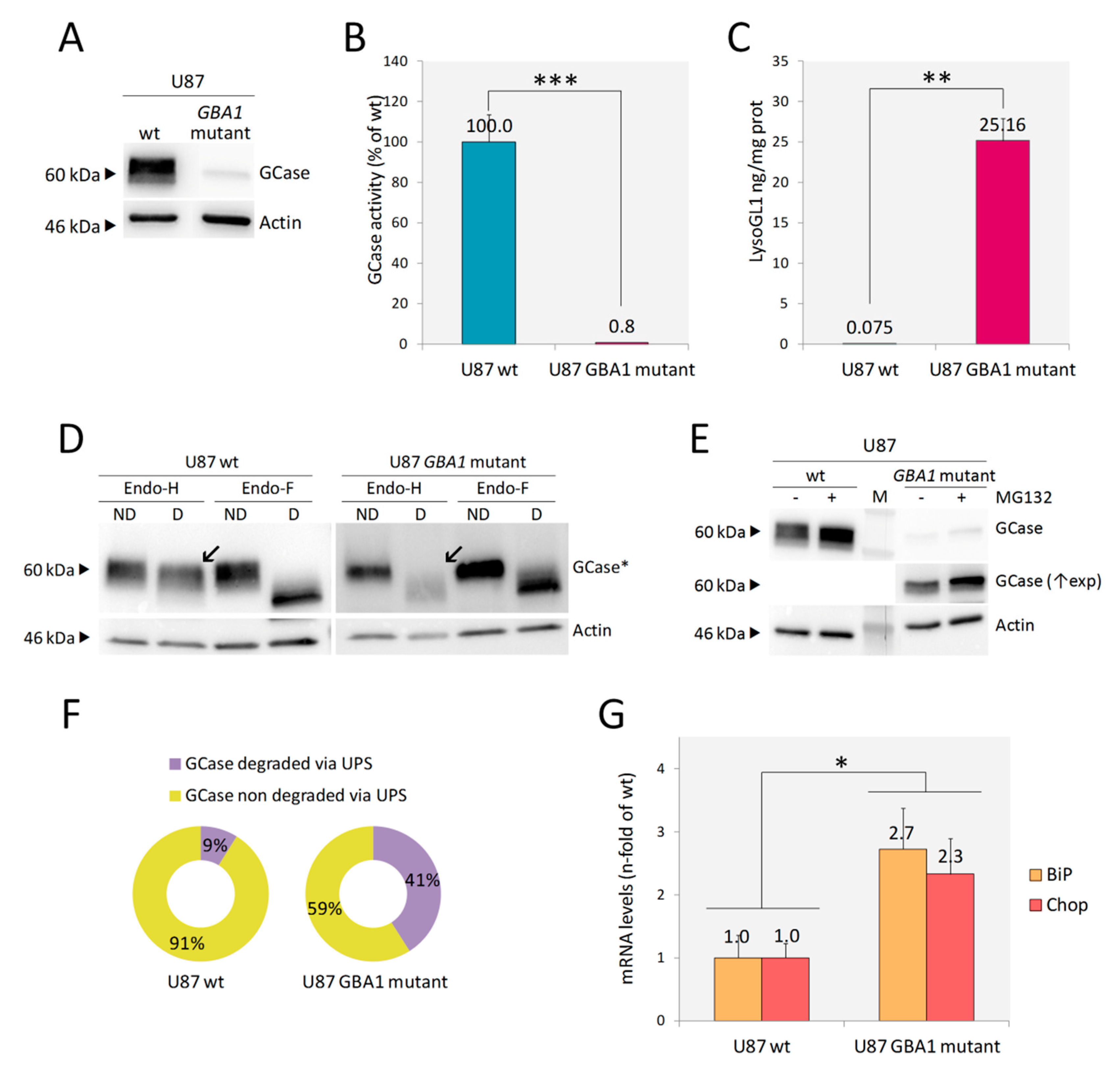
2.2. U87 GBA1 Mutant Cells: A Useful Tool to Study Neuroinflammation in GD
2.2.1. Endoplasmic Reticulum Associated Degradation (ERAD) and ER Stress
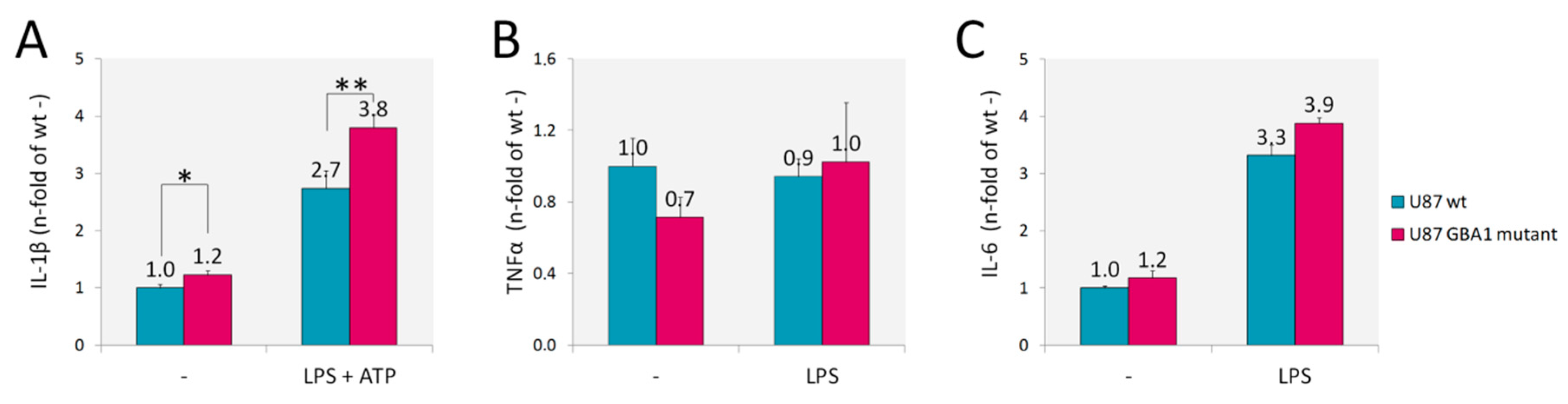
2.2.2. Inflammatory Markers
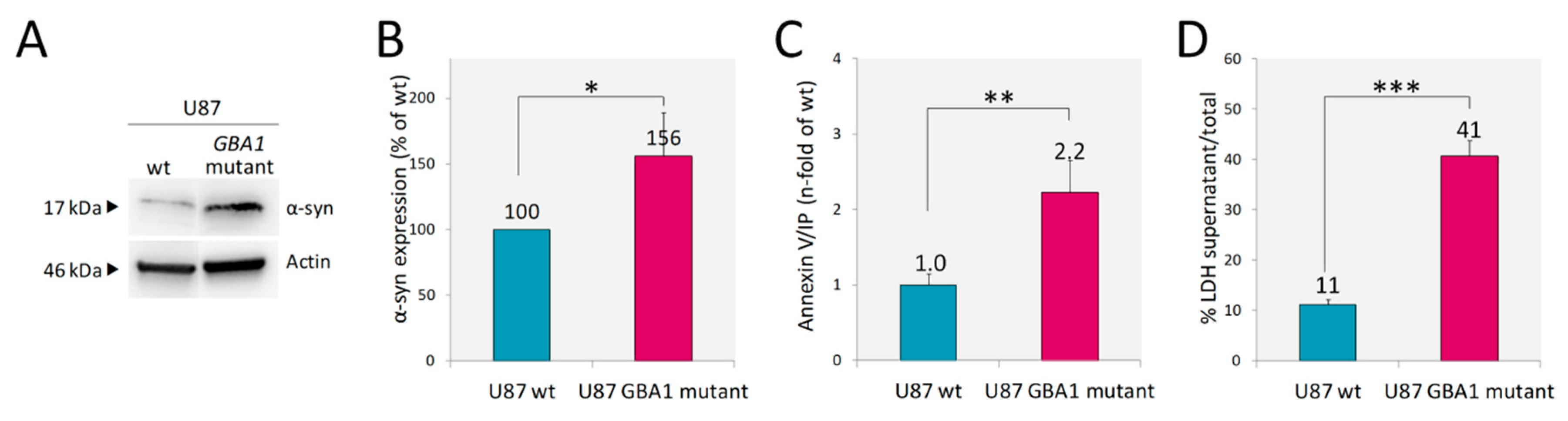
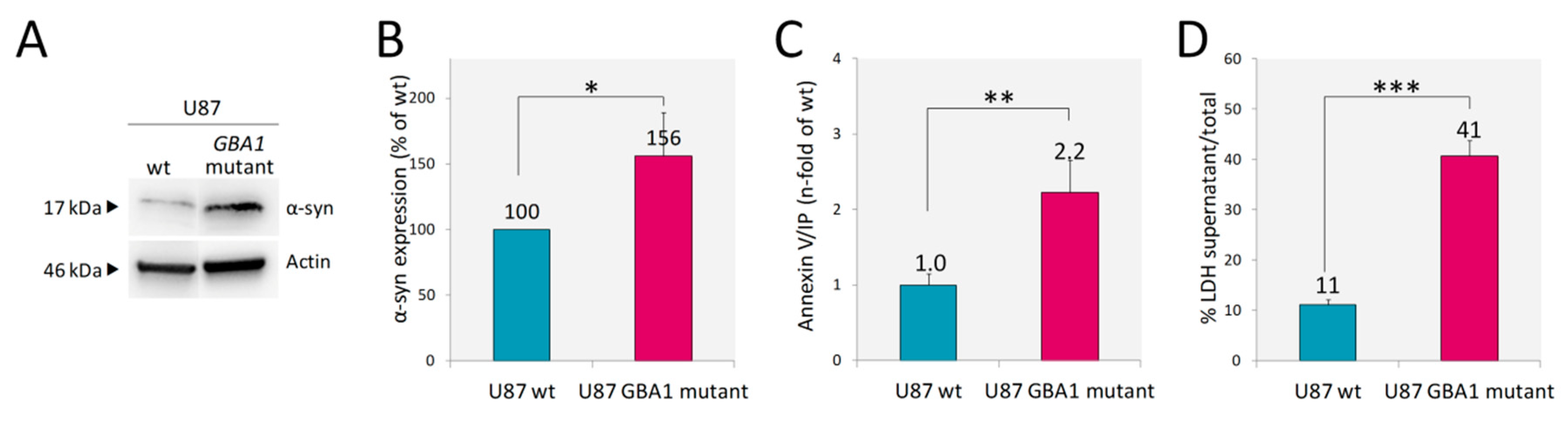
2.2.3. Accumulation of α-Synuclein and Cell Death
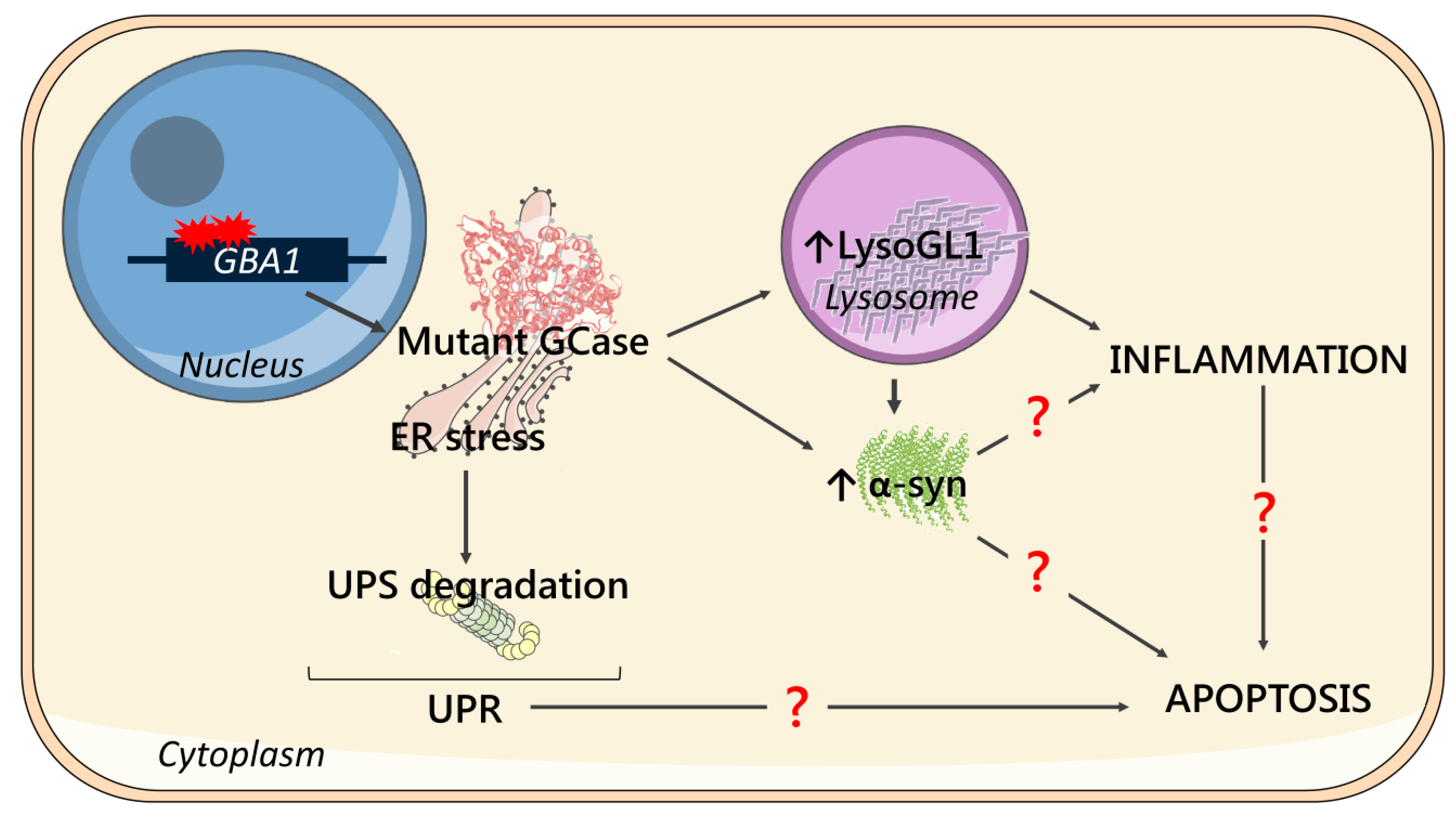
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MG132 Treatment
4.3. CRISPR/Cas9 GBA1 Editing
4.4. Off-Target Analysis
4.5. DNA Extraction, GBA1 Exon 3 Amplification and Sequencing
4.6. Protein Extraction and Western Blot
4.7. Endo-H and Endo-F Digestion
4.8. Enzymatic Activity
4.9. RNA Extraction, Reverse Transcription and Quantitative Real Time PCR
4.10. LysoGL1 Measurement
4.11. Cytokine Measurement
4.12. Annexin-V Staining
4.13. Lactate Dehydrogenase (LDH) Activity
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Endo-F | Endoglycase F |
| Endo-H | Endoglycase H |
| ER | Endoplasmic reticulum |
| ERAD | ER associated degradation |
| GCase | acid β-glucosidase |
| GD | Gaucher disease |
| GlcCer | Glucosylceramide |
| HEK | Human Embryonic Kidney cells 293 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| iPSC | Induced pluripotent stem cells |
| LDH | Lactate dehydrogenase |
| LysoGL1 | Glucosylsphingosine |
| MW | Molecular weight |
| nGD | Neuronopathic GD |
| PD | Parkinson’s disease |
| RNAi | RNA interference |
| THP-1 | Human monocytic cell line deriving from an acute monocytic leukemia patient |
| TNFα | Tumor necrosis factor α |
| U87 | Glioblastoma U87 cell line |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin-Proteasomal System |
| WB | Western Blot |
| α-syn | α-synuclein |
References
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of glucocerebroside. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M. Gaucher’s Disease. Pathological features. Baillieres Clin. Haematol. 1997, 10, 739–749. [Google Scholar] [CrossRef]
- Beutler, E.; Grabowski, G.A. Gaucher disease. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, A.L., Valle, D., Sly, W.S., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3635–3668. [Google Scholar]
- Linari, S.; Castaman, G. Clinical manifestations and management of Gaucher disease. Clin. Cases Miner. Bone Metab. 2015, 12, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Hughes, D.A. Gaucher Disease. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000. [Google Scholar]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.; Futerman, A.H. Neuronal forms of Gaucher disease. Handb. Exp. Pharmacol. 2013, 216, 405–419. [Google Scholar] [CrossRef]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Vitner, E.; Dekel, H.; Leshem, N.; Enquist, I.B.; Karlsson, S.; Futerman, A.H. No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Hum. Mol. Genet. 2009, 18, 1482–1488. [Google Scholar] [CrossRef]
- Michelakakis, H.; Spanou, C.; Kondyli, A.; Dimitriou, E.; Van Weely, S.; Hollak, C.E.; Van Oers, M.H.; Aerts, J.M. Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease. Biochim. Biophys. Acta 1996, 1317, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar]
- Hong, Y.B.; Kim, E.Y.; Jung, S.C. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef]
- Sung, K.; Jimenez-Sanchez, M. Autophagy in astrocytes and its implications in neurodegeneration. J. Mol. Biol. 2020, in press. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; Mcglinchey, R.P.; Mcmahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Borger, D.K.; Aflaki, E.; Sidransky, E. Applications of iPSC-derived models of Gaucher disease. Ann. Transl. Med. 2015, 3, 295. [Google Scholar] [CrossRef]
- Borger, D.K.; Sidransky, E.; Aflaki, E. New macrophage models of Gaucher disease offer new tools for drug development. Macrophage 2015, 2, e712. [Google Scholar]
- Santos, R.; Amaral, O. Advances in sphingolipidoses: CRISPR-Cas9 editing as an option for modelling and therapy. Int. J. Mol. Sci. 2019, 20, 5897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J. The human leukemia cell line, THP-1: A multifacetted model for the study of monocyte-macrophage differentiation. Experientia 1991, 47, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Wirsching, H.-G.; Galanis, E.; Weller, M. Glioblastoma. In Gliomas (Handbook of Clinical Neurology (Volume 134)); Berger, M.S., Weller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 381–397. [Google Scholar] [CrossRef]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shi, J.; Hachet, M.A.; Xue, C.; Bauer, R.C.; Jiang, H.; Li, W.; Tohyama, J.; Millar, J.; Billheimer, J.; et al. CRISPR/Cas9-Mediated Gene Editing in Human iPSC-Derived Macrophage Reveals Lysosomal Acid Lipase Function in Human Macrophages-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2156–2160. [Google Scholar] [CrossRef] [Green Version]
- Bendikov-Bar, I.; Horowitz, M. Gaucher disease paradigm: From ERAD to comorbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Miller, D.; Awad, O.; Bose, V.; Lun, Y.; Park, T.S.; Zambidis, E.T.; Sgambato, J.A.; Feldman, R.A. Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 2014, 32, 2338–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondar, C.; Ormazabal, M.; Crivaro, A.; Ferreyra-Compagnucci, M.; Delpino, M.; Rozenfeld, P.A.; Mucci, J.M. Osteocyte Alterations Induce Osteoclastogenesis in an In Vitro Model of Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Florer, J.; Mayhew, C.N.; Jia, Z.; Zhao, Z.; Xu, K.; Ran, H.; Liou, B.; Zhang, W.; Setchell, K.D.; et al. Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: Potential role in neuropathology. PLoS ONE 2015, 10, e0118771. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Kim, S.J.; Zhang, Z.; Tsai, P.C.; Wisniewski, K.E.; Mukherjee, A.B. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet. 2008, 17, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Mu, T.W.; Ong, D.S.; Wang, Y.J.; Balch, W.E.; Yates, J.R., III; Segatori, L.; Kelly, J.W. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Westbroek, W.; Gustafson, A.M.; Sidransky, E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 2011, 17, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Dammer, E.B.; Malovic, E.; Olsen, A.L.; Raza, S.A.; Gao, T.; Xiao, H.; Oliver, D.L.; Duong, D.; Joers, V.; et al. Molecular Signatures of Neuroinflammation Induced by αSynuclein Aggregates in Microglial Cells. Front. Immunol. 2020, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Crivaro, A.N.; Mucci, J.M.; Bondar, C.M.; Ormazabal, M.E.; Ceci, R.; Simonaro, C.; Rozenfeld, P.A. Efficacy of pentosan polysulfate in in vitro models of lysosomal storage disorders: Fabry and Gaucher Disease. PLoS ONE 2019, 14, e0217780. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavan, E.; Ormazabal, M.; Peruzzo, P.; Vaena, E.; Rozenfeld, P.; Dardis, A. CRISPR/Cas9 Editing for Gaucher Disease Modelling. Int. J. Mol. Sci. 2020, 21, 3268. https://doi.org/10.3390/ijms21093268
Pavan E, Ormazabal M, Peruzzo P, Vaena E, Rozenfeld P, Dardis A. CRISPR/Cas9 Editing for Gaucher Disease Modelling. International Journal of Molecular Sciences. 2020; 21(9):3268. https://doi.org/10.3390/ijms21093268
Chicago/Turabian StylePavan, Eleonora, Maximiliano Ormazabal, Paolo Peruzzo, Emilio Vaena, Paula Rozenfeld, and Andrea Dardis. 2020. "CRISPR/Cas9 Editing for Gaucher Disease Modelling" International Journal of Molecular Sciences 21, no. 9: 3268. https://doi.org/10.3390/ijms21093268
APA StylePavan, E., Ormazabal, M., Peruzzo, P., Vaena, E., Rozenfeld, P., & Dardis, A. (2020). CRISPR/Cas9 Editing for Gaucher Disease Modelling. International Journal of Molecular Sciences, 21(9), 3268. https://doi.org/10.3390/ijms21093268